

Abstract
Many insects depend on ancient associations with intracellular bacteria to perform essential metabolic functions. These endosymbionts exhibit striking examples of convergence in genome architecture, including a high degree of structural stability that is not typical of their free-living counterparts. However, the recently sequenced genome of the obligate whitefly endosymbiont Portiera revealed features that distinguish it from other ancient insect associates, such as a low gene density and the presence of perfectly duplicated sequences. Here, we report the comparative analysis of Portiera genome sequences both within and between host species. In one whitefly lineage (Bemisia tabaci), we identify large-scale structural polymorphisms in the Portiera genome that exist even within individual insects. This variation is likely mediated by recombination across identical repeats that are maintained by gene conversion. The complete Portiera genome sequence from a distantly related whitefly host (Trialeurodes vaporarium) confirms a history of extensive genome rearrangement in this ancient endosymbiont. Using gene-order-based phylogenetic analysis, we show that the majority of rearrangements have occurred in the B. tabaci lineage, coinciding with an increase in the rate of nucleotide substitutions, a proliferation of short tandem repeats (microsatellites) in intergenic regions, and the loss of many widely conserved genes involved in DNA replication, recombination, and repair. These results indicate that the loss of recombinational machinery is unlikely to be the cause of the extreme structural conservation that is generally observed in obligate endosymbiont genomes and that large, repetitive intergenic regions are an important substrate for genomic rearrangements.
Keywords: genome reduction, indel spectrum, insect endosymbionts, inversions, mutators
Introduction
The genomes of bacteria that maintain an obligately intracellular lifestyle share a number of commonalities, including an accelerated rate of sequence evolution, biased nucleotide composition, and reductions in total size and gene content (Andersson and Kurland 1998; Moran et al. 2008; McCutcheon and Moran 2011). In addition, obligate or “primary” endosymbionts in insects exhibit an unusually high degree of stability in genome structure. This was first observed in the aphid endosymbiont Buchnera aphidicola when complete genomes were sequenced from a pair of host species with a fossil-based divergence time of more than 50 Myr (Shigenobu et al. 2000; Tamas et al. 2002). Despite extensive divergence in sequence and numerous gene losses, these genomes retain perfectly conserved synteny. Subsequent studies of Buchnera in more anciently diverged aphid hosts and endosymbionts in other insect groups confirmed that genomic stability is common in these bacteria (table 1). In addition, organelle genomes in many eukaryotic lineages exhibit similar stability, suggesting that structural conservation is a recurrent phenomenon in anciently derived endosymbionts. For example, plastid genome structure is essentially identical in most flowering plants, indicating that it has been maintained since the most recent common ancestor of this group (Raubeson and Jansen 2005). Similarly, most vertebrate mitochondrial genomes share the exact same gene order (Boore 1999).
Table 1.
Obligate Insect Endosymbionts for Which Multiple Complete Genomes Have Been Sequenced
| Symbiont | Host Group | Genomesa | Inversions | Phylogenetic Historyb | References |
|---|---|---|---|---|---|
| Blattabacterium | Cockroaches | 5 | 2 | >600 Myr | Lopez-Sanchez et al. (2009); Sabree et al. (2009, 2012); Neef et al. (2011); and Huang et al. (2012) |
| Blochmannia | Carpenter ants | 3 | 0 | >40 Myr | Gil et al. (2003); Degnan et al. (2005); and Williams and Wernegreen (2010) |
| Buchnera | Aphids | 7 | 1 | >400 Myr | Shigenobu et al. (2000)z; Tamas et al. (2002); van Ham et al. (2003); Perez-Brocal et al. (2006); Degnan et al. (2011); and Lamelas et al. (2011) |
| Carsonella | Psyllids | 6 | 0 | >300 Myr | Nakabachi et al. (2006) and Sloan and Moran (2012a) |
| Sulcia | Auchenorrhyncha | 4 | 0 | >600 Myr | McCutcheon and Moran (2007, 2010); McCutcheon et al. (2009); and Woyke et al. (2010) |
| Wigglesworthia | Tsetse flies | 2 | 1 | >100 Myr | Akman et al. (2002) and Rio et al. (2012) |
| Portiera | Whiteflies | 2 | 17 | <200 Myr | Sloan and Moran (2012b) and present study |
aCounts exclude cases in which multiple genomes have been sequenced from the same insect host species.
bPhylogenetic history represents total branch length in the insect host phylogeny.
Notably, the recent genome sequencing of the obligate endosymbiont from the whitefly Bemisia tabaci identified multiple structural features that distinguish it from other obligate insect endosymbionts (Sloan and Moran 2012b). In particular, the genome of this species (Candidatus Portiera aleyrodidarum, hereafter referred to as Portiera) contained two pairs of perfectly duplicated dispersed repeats (172 and 349 bp) and exhibited unusually large intergenic regions, with intact gene sequences accounting for only 69.4% of the genome. The rarity of large intergenic regions in bacteria has been explained by an observed excess of deletion mutations relative to insertions, which purge nonfunctional sequence from the genome (Mira et al. 2001). Therefore, cases of low gene density in bacteria have been interpreted as the result of either a relaxation of this deletion bias (Kuo and Ochman 2009) or a rapid rate of gene inactivation that exceeds the rate at which pseudogenes are deleted from the genome (Toh et al. 2006).
Assembly of the Portiera genome also revealed evidence of structural variation within the pooled DNA sample used for sequencing (fig. 1), raising the possibility of a more dynamic history of structural evolution than typically observed in obligate insect endosymbionts (Sloan and Moran 2012b). Here, we take advantage of the availability of additional Portiera genomes that were independently sequenced from different populations and biotypes in the B. tabaci species complex (Santos-Garcia et al. 2012; Jiang et al. 2013). We also report the complete Portiera genome from the distantly related host species Trialeurodes vaporarium. We show that the Portiera genome has experienced a number of changes in gene content and genome architecture in the B. tabaci lineage. These changes have been associated with a dramatic increase in the rate of structural rearrangements, providing insight into the mechanisms responsible for the high level of genomic stability that is typically observed in ancient endosymbionts.
Fig. 1.—

Genome maps of Portiera BT-B and TV. Colored lines on the outer ring represent blocks of conserved synteny between the two genomes. Blue and yellow bands on the inner ring represent annotated genes on the forward and revere strands, respectively. The black bars on the inner ring reflect the proportion of microsatellite sequence in that region. The internal lines indicate paired-end conflicts, in which the forward and reverse sequencing reads map to locations that are inconsistent with the library insert size (∼450 bp). Clusters of read pair conflicts associated with identical repeats in the Portiera BT-B genome are highlighted in red, and the location of the corresponding repeats are indicated by short red lines.
Materials and Methods
Portiera Genome Sequences
Previously sequenced Portiera genomes from the B. tabaci host species complex were obtained from GenBank (Santos-Garcia et al. 2012; Sloan and Moran 2012b; Jiang et al. 2013), including two sequences from B. tabaci biotype B (Portiera BT-B) and two from biotype Q (Portiera BT-Q). Two additional whitefly collections were made in June 2012. Trialeurodes vaporarium was collected from the Yale University Marsh Botanical Garden greenhouse (New Haven, CT), and a new collection of B. tabaci biotype B was taken from the Yale University West Campus greenhouse (West Haven, CT). Whitefly species identity was confirmed by sequencing a portion of the mitochondrial COI locus, and populations from each collection were maintained in laboratory colonies growing on cowpea (Vigna unguiculata).
The T. vaporarium colony was used to sequence the complete Portiera genome (Portiera TV), using methods described previously (Sloan and Moran 2012a, 2012b). In brief, total insect DNA was extracted from a pool of multiple individuals and used for 2 × 76 bp paired-end sequencing on an Illumina HiSeq2000 at the Yale Center for Genome Analysis. Sequencing reads from the Portiera genome were identified and assembled into a closed circle, using a combination of Velvet v1.1.06 (Zerbino and Birney 2008), MIRA v3.4.0 (Chevreux et al. 1999), and SOAP v2.21 (Li et al. 2009). Annotation was performed independently with the JGI IMG-ER pipeline (Markowitz et al. 2009) and the IGS Annotation Engine (Galens et al. 2011). Annotation results were then manually curated, and the finished Portiera TV genome sequence was submitted to GenBank (CP004358).
Analysis of Structural Variation in the Portiera BT Genome
Three methods were used to analyze structural variation within the Portiera BT genome. First, paired-end Illumina reads from the original Portiera BT-B assembly (Sloan and Moran 2012b) were mapped to the genome with SOAP v2.21 (Li et al. 2009) to identify conflicting read pairs that were consistent with intragenomic recombination between repeats. Second, polymerase chain reaction (PCR) primers were designed in regions flanking repeat sequences and used to amplify all possible products that could be generated by repeat-mediated recombination. Amplification was performed with total insect DNA from individual whiteflies (2012 West Haven collection), using standard PCR techniques. Third, DNA gel blots (i.e., Southern blots) were generated with total insect DNA and hybridized with probes located adjacent to repeats in the Portiera BT genome. DNA was extracted from individual insects and pooled samples (all from the aforementioned B. tabaci biotype B laboratory colony), digested with restriction enzymes (or left uncut), resolved on 0.8% agarose gels, and transferred to positively charged nylon membranes (Roche) by capillary blotting. Digoxigenin probe labeling, hybridization, and chemiluminescent detection were performed with the DIG High Prime DNA Labeling and Detection Starter Kit II (Roche). Blots were imaged with an ImageQuant LAS 4000 (GE Healthcare). Primer sequences used to amplify probes are provided in supplementary table S1, Supplementary Material online. Because of the large gap between the 5.1-kb and 21.2-kb bands in the Roche DIG-labeled ladder (fig. 2), we ran this ladder side-by-side with an unlabeled Invitrogen 1 kb plus ladder on the same gel used for blotting. This portion of gel was cut off and stained with ethidium bromide to allow for more accurate interpolation of fragment sizes in the DNA gel blot.
Fig. 2.—
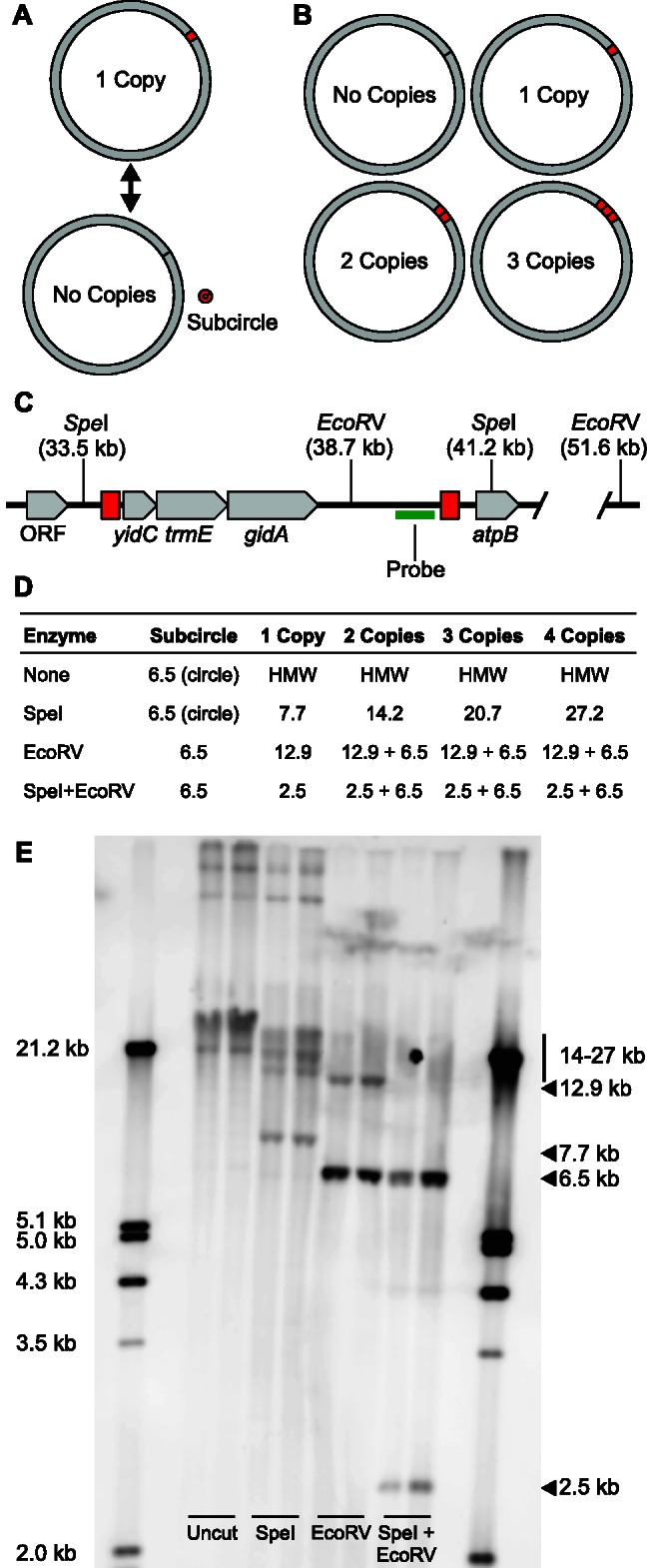
Evidence that a 6.1-kb sequence exists in a variable number of tandem copies in Portiera BT genomes. Two alternative structural models are consistent with the observed read pair conflicts (fig. 1). (A) The 6.1-kb sequence (red) flanked by identical 349-bp repeats (black lines) could interconvert between an integrated form and a separate subcircle form via repeat-mediated recombination. (B) Alternatively, the sequence could exist in a variable number of tandem copies, falsely implying the existence of a subcircular form. (C) Map of target region within the Portiera BT-B genome as originally reported with a single integrated copy of the 6.1-kb sequence flanked by 349-bp repeats (red boxes). (D) Predicted fragment sizes resulting from hybridizing digested Portiera DNA with a probe located within the 6.1-kb sequence. “HMW” indicates high molecular weight. (E) DNA gel blot hybridization, probing for sequence within the 6.1 sequence. In conflict with model A, uncut and SpeI samples show no evidence of the predicted 6.5-kb circular molecule that would consist of the 6.1-kb sequence and one copy of the 349-bp repeat. Instead, the SpeI digests show a range of fragment sizes that are consistent with the expectations for the coexistence of chromosomes with anywhere from one to four (or more) tandem copies of this sequence, supporting model B. Inferred sizes of key bands are indicated on the right.
Measurement of Indel Spectrum
To estimate the relative frequency of insertions versus deletion mutations in Portiera BT, intergenic regions from the four sequenced genomes were aligned with MUSCLE v3.7 (Edgar 2004), and indels that were unique to only one of the four genomes were identified. Because we used two reciprocally monophyletic pairs of Portiera genomes (two from B. tabaci biotype B and two from biotype Q), we were able to apply a simple parsimony assumption to predict that these unique indels represent recent mutations (i.e., the derived state). We excluded any indels that overlapped with indels in other Portiera BT genomes because of ambiguity in inferring the ancestral state in these cases. Extraction of intergenic regions and detection of indels were performed with custom Perl scripts and BioPerl modules (Stajich et al. 2002).
Identification of Tandem Repeats
Phobos v3.3.12 was used to identify all tandemly repeating sequences with a total length of at least 18 bp and a unit size of up to 40 bp in the genomes of Portiera and other representative insect endosymbionts. The Phobos analysis was conducted in “exact” search mode.
Gene-Order Phylogeny and Relative Rates of Sequence and Structural Evolution
To examine the history of genome rearrangement in Portiera, a set of 145 protein genes with orthologs in Portiera BT, Portiera TV, and some related species of Gammaproteobacteria (Candidatus Carsonella ruddii, Chromohalobacter salexigens, Halomonas elongata, Pseudomonas aeruginosa, and Escherichia coli) was identified with reciprocal Basic Local Alignment Search Tool (BLAST) searches. Three different phylogenetic reconstruction methods were used to infer the relationships among these species on the basis of the order and orientation of these genes within each genome. First, the program MGR v2.01 was used to identify the topology requiring the fewest number of inversions to explain structural differences among the set of circular genomes (Bourque and Pevzner 2002). Second, the gene order data were analyzed with TIBA, using a neighbor-joining algorithm and 100 bootstrap replicates (Lin et al. 2011). Finally, a Bayesian analysis was implemented in BADGER v1.02b with five independent search chains of 50 million cycles each and a sample interval of 1,000 cycles (Larget et al. 2005). The first 20,000 sampled trees from each chain were discarded as burn-in.
To test for differences in the frequency of rearrangements in Portiera genomes from B. tabaci and T. vaporarium, we applied a relative rate test (Sarich and Wilson 1973) with the outgroup Ca. Carsonella ruddii (Nakabachi et al. 2006) and pairwise inversion distances calculated with GRIMM v2.01 (Tesler 2002). Significance was assessed by generating 10,000 simulated rearrangement data sets with the same number of genes and total inversions as observed in our analysis. Comparisons of the rate of amino sequence evolution between Portiera BT and Portiera TV were conducted using Tajima’s relative rate test (Tajima 1993) with the outgroup C. salexigens. This outgroup was chosen instead of the more closely related Ca. Carsonella ruddii, because its slower rate of amino acid substitution has resulted in less overall sequence divergence.
Results
Structural Polymorphisms Exist within Individual Insects in the Portiera BT Genome
Comparisons among four published Portiera BT genomes (Santos-Garcia et al. 2012; Sloan and Moran 2012b; Jiang et al. 2013) found that they differed with respect to the presence of a 6.1-kb sequence that contains three genes (yidC, trmE, and gidA). This variation was not associated with host biotype, because the sequence was reported in one of the two published Portiera genomes from each of the B. tabaci biotypes. The 6.1-kb sequence is flanked on either side by copies of a 349-bp repeat that was previously reported to be associated with paired-end read conflicts, suggesting the coexistence of alternative genome structures (Sloan and Moran 2012b). By performing PCR with pairwise combinations of primers from regions flanking these repeats, we found evidence for the existence of all four possible structural conformations within individual insects (supplementary fig. S1, Supplementary Material online). Because repeat-mediated recombination can be a PCR artifact (Alverson et al. 2011), we also used DNA gel blot hybridizations as an amplification-free method to verify the existence of alternative structures in both individual and pooled DNA samples (fig. 2 and supplementary fig. S2, Supplementary Material online). All three methods (PCR, DNA gel blot hybridizations, and analysis of paired-end sequencing conflicts) also identified alternative structures associated with a second pair of (172 bp) repeats that are found in the ancient paralogs prfA and prfB (fig. 1 and supplementary figs. S1 and S2, Supplementary Material online).
In both cases, repeat pairs occur in the same (i.e., direct) orientation within the genome. Therefore, recombination between repeats could potentially result in interconversion between a main circular chromosome and sets of subgenomic circles, similar to the multipartite model of genome organization described decades ago in plant mitochondria (Palmer and Shields 1984). In the case of the repeats flanking the 6.1-kb sequence, DNA gel blot hybridizations identified a strong 6.5-kb band in samples digested with EcoRV, which has a single recognition sequence in this region. Although this result is consistent with the expectations for a linearized subcircle containing the 6.1-kb sequence and one copy of the flanking 349-bp repeat, we found no evidence of a 6.5-kb circular molecule in uncut DNA samples or samples digested with SpeI, which lacks any recognition sequences within the 6.1-kb region. Instead, this sequence appears to exist predominantly or exclusively in an integrated form within the main chromosome, often occurring in tandem arrays. This model is consistent with the EcoRV digest results, because tandem repeats map as circles. It is also supported by the detection of SpeI fragments that are consistent with expectations for anywhere from a single copy to four (or more) tandem copies of this sequence (fig. 2).
Interestingly, in spite of the evidence that the 6.1-kb sequence often exists as part of a tandem repeat, shotgun sequencing of this genome (Sloan and Moran 2012b) produced a very similar average read depth in this region (265×) when compared with the rest of the genome (255×). This can be explained by the fact that the majority of chromosomes do not have any copies of the 6.1-kb region, roughly balancing the chromosomes with multiple copies (supplementary fig. S2, Supplementary Material online). Conversely, the majority of copies of the 6.1-kb sequence exist on chromosomes carrying more than one copy (fig. 2). In addition to the evidence from relative band intensities in hybridizations, these inferences are supported by the large percentage of read pairs (81.3%) that conflict with a genome conformation that contains a single integrated copy of the 6.1-kb sequence. Therefore, the structural differences between Portiera BT genomes reported in the literature (Sloan and Moran 2012b; Jiang et al. 2013) appear to reflect different chromosomal forms that co-occur within insects rather than differences among the particular strains that were chosen for sequencing.
Because the prfA/prfB repeats are separated by a distance of more than 150 kb, we did not attempt to confirm whether the two large genomic subcircles predicted to result from intramolecular recombination actually occur in vivo. However, both hybridization band intensity (supplementary fig. S2, Supplementary Material online) and read pair conflicts indicate that the chimeric genes resulting from recombination between prfA and prfB are rare relative to the “normal” conformation. Only 6.4% of read pairs support the chimeric structures.
Ancient and Ongoing Gene Conversion in Portiera
The existence of identical repeat sequences in Portiera is evidence of active gene conversion in these endosymbionts. As noted earlier, all four sequenced Portiera BT genomes contain a pair of 172-bp repeats in homologous regions of prfA and prfB, which encode enzymes that terminate translation by recognizing UAR and URA stop codons, respectively. Despite extensive divergence between these ancient paralogs, their distant sequence homology appears to have provided a template for gene conversion (fig. 3a and b). The location of the repeats corresponds to a highly conserved region (fig. 3c), containing a GGQ tripeptide that interacts with the ribosome to mediate polypeptide release (Mora et al. 2003). The amino acid motifs that determine stop codon specificity (Ito et al. 2000) are found outside the region affected by gene conversion in Portiera, suggesting that both paralogs retain their ancestral function. The history of gene conversion between prfA and prfB appears to be relatively ancient, preceding the divergence of the Bemisia and Trialeurodes host lineages, as the Portiera TV genome contains a pair of identical 56-bp repeats in the same region (fig. 3c). Gene conversion has also remained active in these genomes on recent timescales, as indicated by the occurrence of identical substitutions in both paralogs in one of the Portiera BT-Q genomes, maintaining perfect sequence identity between the repeat copies in each genome (fig. 3b).
Fig. 3.—
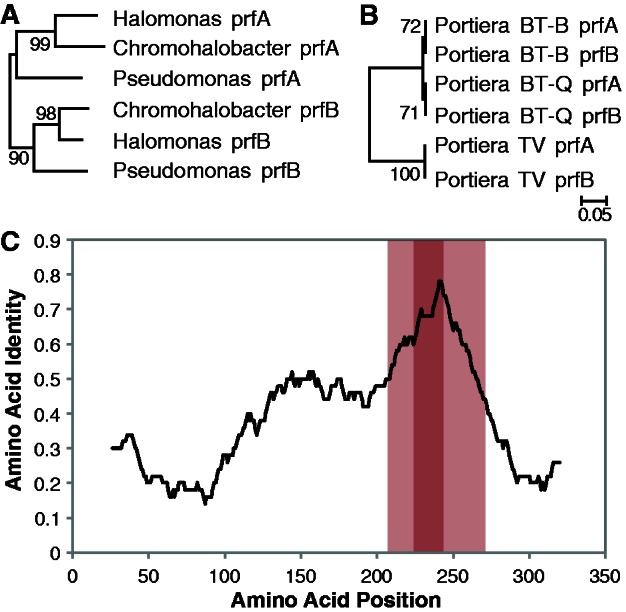
Gene conversion between peptide release factor genes prfA and prfB. In bacteria, these sequences typically cluster by gene rather than by species (A), reflecting the ancient origin of the paralogs by gene duplication. However, in Portiera, prfA and prfB contain a region of perfect sequence identity, presumably arising by gene conversion and concerted evolution (B). Both trees were inferred by neighbor joining with 1,000 bootstrap replicates, using the portion of the gene that has undergone gene conversion in Portiera. This region coincides with the part of the gene that has experienced the highest degree of sequence conservation in other species (C). The plot shows the amount of amino acid sequence identity between aligned prfA and prfB sequences in Escherichia coli with a sliding window of 50 amino acids. The Portiera gene conversion region is highlighted in red, with the lighter shading representing the regions that are identical in Portiera BT but not in Portiera TV.
Extensive Genomic Rearrangements in Portiera
The Portiera genome sequences from two divergent host lineages, B. tabaci and T. vaporarium, share similar gene complements (table 2) but are highly rearranged with respect to each other (fig. 4). A minimum of 17 inversion events is required to explain the differences in gene order between these genomes. The rearrangements have accumulated asymmetrically between the two host lineages with the majority occurring in B. tabaci (fig. 5d) (relative rate test; P < 0.01). The acceleration in the rate of structural evolution in the Portiera BT genome appears to have coincided with an increased substitution rate (Thao and Baumann 2004). Relative rate tests identified 69 protein-coding genes with a significantly higher rate of amino acid substitutions in Portiera BT than in Portiera TV (at an uncorrected significance level of α = 0.05), whereas the reverse was true for only one gene. The structural rate analysis was performed with Ca. Carsonella ruddii, the obligate endosymbiont of psyllids, as an outgroup. Phylogenetic inferences based on gene-order support earlier sequence-based analyses that suggested that these two insect endosymbionts form a monophyletic group (fig. 5a–c) (Spaulding and von Dohlen 1998; Thao and Baumann 2004; Sloan and Moran 2012b).
Table 2.
Genomic Features of Portiera BT and TV
Fig. 4.—
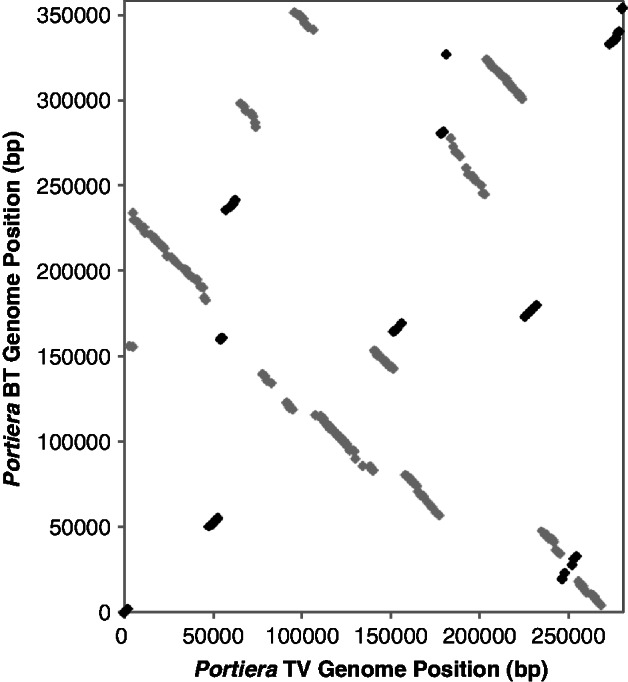
Extensive rearrangements in the Portiera genome since the divergence of the Bemisia tabaci and Trialeurodes vaporarium host lineages. Black and gray dots indicate genes shared between the two genomes in forward and reverse orientation, respectively.
Fig. 5.—
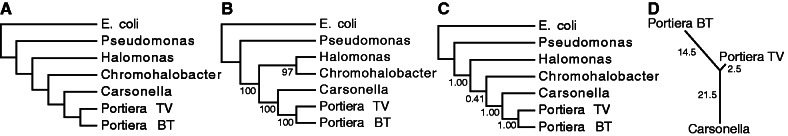
Phylogenetic relationships among select Gammaproteobacteria as inferred from gene order and orientation with MGR (A), TIBA (B), and BADGER (C). Bipartition support is indicated with bootstrap values and Bayesian posterior probabilities from the TIBA and BADGER analyses, respectively. A relative rate test using Carsonella as the closest outgroup to Portiera found that inversions had accumulated asymmetrically in the Portiera BT genome (D).
Proliferation of Short Tandem Repeats and the Indel Spectrum in Portiera BT Intergenic Regions
Despite containing a slightly larger number of genes, the Portiera TV genome is approximately 20% smaller in size than its counterparts in B. tabaci (table 2). The size difference reflects the presence of much longer intergenic sequences in Portiera BT (fig. 6), which may have resulted from a relaxation of the typical bias in the indel mutation spectrum. In contrast to the pattern observed in other bacteria (Mira et al. 2001; Kuo and Ochman 2009), recent intergenic indels in Portiera BT do not exhibit an excess of deletions (fig. 7). Notably, the Portiera BT indel spectrum is highly enriched for 7-bp changes (fig. 7). All these indels occur in the context of short tandem repeats, and the distribution of tandem repeat lengths in the Portiera BT genome exhibits a dramatic spike at 7 bp that is not found in other insect endosymbionts, including Portiera TV (fig. 8). These repeats are concentrated in intergenic regions (fig. 1) and spatially correlated with structural variation in the Portiera BT genome identified by paired-end read conflicts (r = 0.45; P < 0.0001). In contrast, tandem repeats in the Portiera TV genome are rare and not significantly correlated with paired-end read conflicts (r = 0.03; P = 0.43).
Fig. 6.—
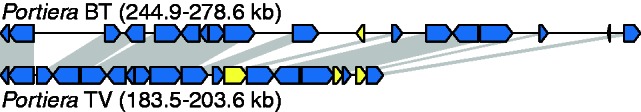
Abnormally large intergenic regions in the Portiera BT genome. Gray shading connects orthologous genes that are shared between strains in a region of conserved synteny. Yellow blocks represent genes without an intact ortholog in the other strain.
Fig. 7.—
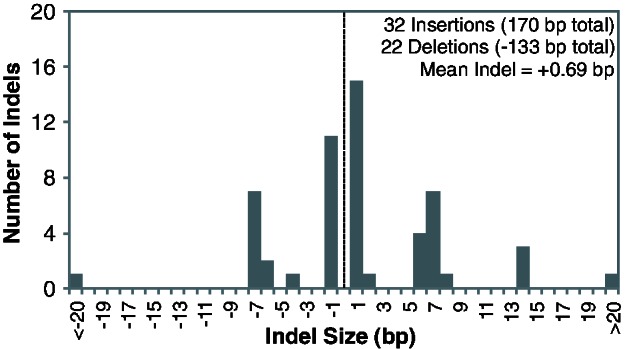
Size distribution of indels in Portiera BT.
Fig. 8.—
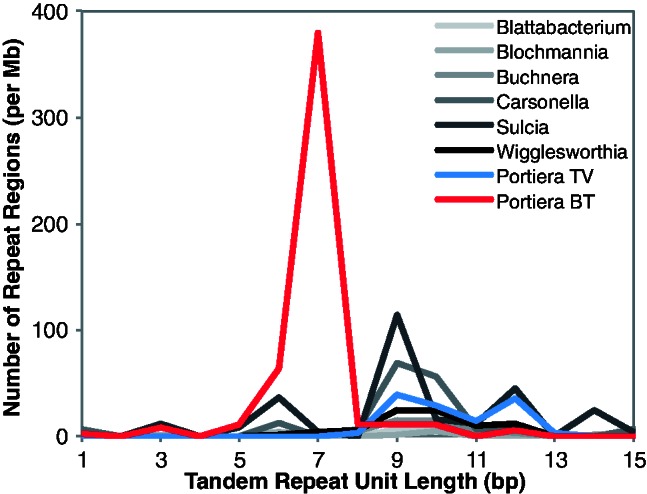
Frequency of short tandem repeats in genomes of obligate insect endosymbionts.
Accurately characterizing an indel spectrum requires high-quality genome sequences and an appropriate model for inferring ancestral states. In identifying indels that were unique among the four Portiera BT genomes, we found that the two genomes from B. tabaci biotype B exhibited a higher degree of similarity than the two from biotype Q. Although we identified numerous indels between the two Portiera BT-B genomes, these only occurred in structurally variable regions that were not conserved with Portiera BT-Q and, therefore, had to be excluded from the analysis. We found that 23 of 28 of the short indels (<4 bp) that were unique to one of the two Portiera BT-Q genomes were in or adjacent to single-nucleotide repeat regions (homopolymers). By itself, this is not surprising because homopolymers are prone to high rates of indel mutations, but we unexpectedly found that a clear majority (18 of 23) of these indels occurred in one of the two genomes (GenBank accession CP003835). Although this genome may have experienced a higher rate of structural change, it is also possible that some of the homopolymer-associated indels in this genome represent sequencing errors, because it was generated largely with the Roche 454 platform, a technology that is known to produce imprecise estimates of homopolymer length (Santos-Garcia et al. 2012). Therefore, sequencing errors probably added noise to our estimate of the indel spectrum. Although these inaccuracies should not pertain to the large number of indels found in microsatellite regions, short tandem repeats are prone to recurrent mutations (homoplasy) and, therefore, may violate our parsimony assumption for inferring ancestral states. Consequently, although we find no evidence of a deletion bias in Portiera BT, a more definitive characterization of the indel spectrum will require additional data.
Loss of DNA Replication, Recombination, and Repair Machinery in Portiera BT
The Portiera BT and TV genomes share 277 genes in common. Notably, these shared genes include those encoding enzymes for the biosynthesis of essential amino acids and of carotenoids (Sloan and Moran 2012b), indicating that provisioning of these compounds to hosts is an ancient function of Portiera that has been preserved through purifying selection. After excluding loci that are annotated only as hypothetical proteins, there are 27 genes that are present in Portiera TV but have been lost from Portiera BT and only four genes for which the reverse is true. The set that has been recently lost from Portiera BT is highly enriched for genes involved in DNA replication, recombination, and repair (table 3).
Table 3.
DNA Replication, Recombination, and Repair Genes that Are Present in Portiera TV but Have Been Lost from the Portiera BT Lineage
| Gene ID | Product Description |
|---|---|
| dnaN | DNA polymerase III subunit beta |
| dnaQ | DNA polymerase III subunit epsilon |
| dnaX | DNA polymerase III subunit gamma and tau |
| holA | DNA polymerase III subunit delta |
| holB | DNA polymerase III subunit delta' |
| mutL | DNA mismatch repair protein MutL |
| ruvC | Crossover junction endodeoxyribonuclease RuvC |
| ssb | Single-stranded DNA-binding protein |
Discussion
The history of structural rearrangements and the presence of large, repetitive intergenic regions in the Portiera BT genome stand in obvious contrast with observations in other obligate insect endosymbionts, including Portiera strains from other whitefly hosts. Therefore, the evolution of genome architecture in Portiera provides an opportunity to understand the mechanisms responsible for the genomic stability that typically characterizes ancient endosymbionts. One potential explanation holds that ancient endosymbionts experience an unusually low rate of structural mutations because pervasive loss of genes required for DNA recombination and repair has left them without the molecular machinery necessary to generate chromosomal inversions (Tamas et al. 2002; Silva et al. 2003). This hypothesis was devised to explain the observed chromosomal stability in the aphid endosymbiont Buchnera, which lacks the widely conserved recombination gene recA. However, subsequent studies of insect endosymbionts that have retained recA (e.g., Blattabacterium, Carsonella, and Wigglesworthia) revealed similar levels of genome stability, undermining this hypothesis (table 1). Our results cast further doubt on the role of gene loss in promoting structural stability. Portiera BT contains one of the most reduced sets of DNA replication, recombination, and repair genes ever identified in a cellular genome, consisting of only the DNA polymerase III alpha subunit gene dnaE, the replicative DNA helicase gene dnaB, and the mismatch repair gene mutS. In spite of its paucity of recombination-related genes, however, the Portiera BT genome has experienced numerous inversions. In addition, the evidence for gene conversion (fig. 3) and the coexistence of alternative structures (fig. 2 and supplementary fig. S2, Supplementary Material online) indicate that at least some recombination mechanisms are active in Portiera BT.
An alternative explanation for the atypical pattern of structural evolution in the Portiera BT genome is that the presence of large and repetitive intergenic regions promotes rearrangements. Intergenic regions are generally small in bacteria, and, in some obligate insect endosymbionts, gene density is so high that protein-coding sequences often overlap (Nakabachi et al. 2006; McCutcheon et al. 2009). In addition, obligate endosymbionts with an ancient history of vertical transmission generally lack large repeats arising from mobile sequences such as insertion elements and phage, which are often found in free-living bacteria (McCutcheon and Moran 2011). The expanded intergenic regions in Portiera BT could accelerate structural evolution through at least two mechanisms. First, repetitive content in these regions may directly increase the rate of structural mutations. Dispersed repeats can generate inversions via recombination, whereas tandem-repeat-rich sequences may be a source of replication errors and DNA breakage. Second, the reduced density of functional elements in intergenic regions may make them more likely to tolerate rearrangements even if structural mutations do not arise at a higher rate. At this point, it is not clear whether the effects of expanded intergenic regions might be mediated through changes in selection, mutation rate, or a combination of both.
Although the presence of large intergenic regions and repetitive content is unusual in the genomes of obligate endosymbionts, it is not unprecedented. The recent sequencing of the genome of Candidatus Tremblaya princeps, the primary endosymbiont of mealybugs, revealed a number of parallels with the Portiera BT genome, including reduced gene density and the presence of perfect repeats and a polymorphic inversion (McCutcheon and von Dohlen 2011). Paradoxically, the reduced gene complements involved in DNA replication, recombination, and repair in both Tremblaya and Portiera BT appear to be insufficient to mediate the recombinational processes that are clearly active in their respective genomes. Tremblaya holds the remarkable distinction of harboring another bacterial species (Candidatus Moranella endobia) inside its own cells, and this nested endosymbiont likely compensates for the loss of some metabolic pathways and genetic machinery in Tremblaya (McCutcheon and von Dohlen 2011). In contrast, there is no evidence of a nested endosymbiont within Portiera (although other bacterial symbionts can be found within whitefly cells), raising the possibility that host-encoded genes mediate some fundamental genetic processes in Portiera.
The loss of dnaQ from the Portiera BT genome is particularly striking. This gene encodes the epsilon subunit of DNA polymerase III and is responsible for 3′ to 5′ exonuclease proofreading during DNA replication. It is nearly universal in bacteria and has been maintained even in the most extreme examples of endosymbiotic genome reduction (e.g., Tremblaya, Hodgkinia, and Carsonella) (Moran et al. 2008). Mutations that reduce the catalytic activity of this enzyme have strong mutator effects, and dnaQ null mutants have severe, often lethal, growth phenotypes, resulting from the extreme rate of replication errors (Lifsics et al. 1992; Fijalkowska and Schaaper 1996). Amino acids substitutions in this gene have also been shown to increase the instability of microsatellite regions (Zahra et al. 2007). Therefore, the loss of dnaQ and other DNA replication and repair genes in Portiera BT likely contributed to the observed increases in nucleotide substitution rate and microsatellite content in this lineage and suggests that gene loss can play a central role in shaping the evolution of genome architecture.
Most whitefly species are confined to woody plants and restricted geographic ranges, and the B. tabaci complex is unusual in colonizing a wide range of herbaceous plants throughout all warm areas of the world (Martin and Mound 2007). The complex may comprise as many as 30 distinct species in 11 major lineages, and it shows considerable geographic structure and mitochondrial sequence divergence, suggesting an age of at least several million years (de Barro et al. 2011). Whether a connection exists between the unusual chromosome instability of Portiera-BT and the unusual biology of the B. tabaci complex might be clarified through further characterization of Portiera genomes from different whitefly groups.
The maintenance of structural polymorphisms in the Portiera BT genome raises important functional questions. We confirmed that the polymorphisms are shared among geographically isolated populations and that they are even present within individual insects. Thus, the recent suggestion that the different Portiera chromosomal arrangements may distinguish different host biotypes (Jiang et al. 2013) appears not to be supported, at least not for the B biotype that we studied. Given that endosymbiont genomes often exist at very high copy numbers (Komaki and Ishikawa 1999; Woyke et al. 2010), it is also likely that these structural variants coexist within individual bacterial cells. Because one of the repeat pairs occurs within paralogous protein-coding genes, recombination between these repeats generates chimeric prfA/prfB open reading frames. However, it is not clear whether these chimeras are expressed (and functionally relevant) or whether they are simply generated as a nonadaptive byproduct of recombination between identical repeats. We also found that Portiera BT chromosomes contain anywhere from zero to four (or more) tandem copies of a 6.1-kb sequence that is flanked by 349-bp repeats. Because this sequence contains multiple genes, it is likely that the observed copy number variation results in functional differences among alternative chromosomes. Identifying differences in functional content and replicative ability among coexisting chromosomes would provide an opportunity to dissect the effects of natural selection acting on the hierarchical levels that exist within endosymbiotic systems (e.g., at the levels of individual chromosomes, bacteria, and insects) and to determine the extent to which conflicting selection pressures act to maintain the observed structural polymorphisms.
Supplementary Material
Supplementary figures S1 and S2 and table S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Kim Hammond for collecting whiteflies and maintaining laboratory colonies, Eli Powell for assistance with Southern blots, and Ryuichi Koga and the Moran/Ochman laboratory group for helpful discussion of data. We are also grateful for annotation data provided by the Joint Genome Institute’s IMG-ER pipeline and the Institute for Genome Sciences Annotation Engine Service at the University of Maryland School of Medicine. This work was supported by Yale University and a National Institutes of Health Postdoctoral Fellowship (1F32GM099334).
Literature Cited
- Akman L, et al. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat Genet. 2002;32:402–407. doi: 10.1038/ng986. [DOI] [PubMed] [Google Scholar]
- Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD. The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One. 2011;6:e16404. doi: 10.1371/journal.pone.0016404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SG, Kurland CG. Reductive evolution of resident genomes. Trends Microbiol. 1998;6:263–268. doi: 10.1016/s0966-842x(98)01312-2. [DOI] [PubMed] [Google Scholar]
- De Barro PJ, Liu SS, Boykin LM, Dinsdale AB. Bemisia tabaci: a statement of species status. Ann Rev Entomol. 2011;56:1–19. doi: 10.1146/annurev-ento-112408-085504. [DOI] [PubMed] [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque G, Pevzner PA. Genome-scale evolution: reconstructing gene orders in the ancestral species. Genome Res. 2002;12:26–36. [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, Suhai S. Genome sequence assembly using trace signals and additional sequence information. Computer science and biology: Proceedings of the German Conference on Bioinformatics (GCB) Abstract. 1999;99:45–56. 1999 Oct 4–6; Hanover, Germany. [Google Scholar]
- Degnan PH, Lazarus AB, Wernegreen JJ. Genome sequence of Blochmannia pennsylvanicus indicates parallel evolutionary trends among bacterial mutualists of insects. Genome Res. 2005;15:1023–1033. doi: 10.1101/gr.3771305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan PH, Ochman H, Moran NA. Sequence conservation and functional constraint on intergenic spacers in reduced genomes of the obligate symbiont Buchnera. PLoS Genet. 2011;7:e1002252. doi: 10.1371/journal.pgen.1002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska IJ, Schaaper RM. Mutants in the exo I motif of Escherichia coli dnaQ: defective proofreading and inviability due to error catastrophe. Proc Natl Acad Sci U S A. 1996;93:2856–2861. doi: 10.1073/pnas.93.7.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galens K, et al. The IGS standard operating procedure for automated prokaryotic annotation. Stand Genomic Sci. 2011;4:244–251. doi: 10.4056/sigs.1223234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil R, et al. The genome sequence of Blochmannia floridanus: comparative analysis of reduced genomes. Proc Natl Acad Sci U S A. 2003;100:9388–9393. doi: 10.1073/pnas.1533499100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Sabree ZL, Moran NA. Genome sequence of Blattabacterium sp. strain BGIGA, endosymbiont of the Blaberus giganteus cockroach. J Bacteriol. 2012;194:4450–4451. doi: 10.1128/JB.00789-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Uno M, Nakamura Y. A tripeptide “anticodon” deciphers stop codons in messenger RNA. Nature. 2000;403:680–684. doi: 10.1038/35001115. [DOI] [PubMed] [Google Scholar]
- Jiang ZF, et al. Comparison of the genome sequences of “Candidatus Portiera aleyrodidarum” primary endosymbionts of the whitefly Bemisia tabaci B and Q biotypes. Appl Environ Microbiol. 2013;79: 1757–1759. doi: 10.1128/AEM.02976-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaki K, Ishikawa H. Intracellular bacterial symbionts of aphids possess many genomic copies per bacterium. J Mol Evol. 1999;48:717–722. doi: 10.1007/pl00006516. [DOI] [PubMed] [Google Scholar]
- Kuo CH, Ochman H. Deletional bias across the three domains of life. Genome Biol Evol. 2009;1:145–152. doi: 10.1093/gbe/evp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamelas A, Gosalbes MJ, Moya A, Latorre A. New clues about the evolutionary history of metabolic losses in bacterial endosymbionts, provided by the genome of Buchnera aphidicola from the aphid Cinara tujafilina. Appl Environ Microbiol. 2011;77:4446–4454. doi: 10.1128/AEM.00141-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larget B, Simon DL, Kadane JB, Sweet D. A Bayesian analysis of metazoan mitochondrial genome arrangements. Mol Biol Evol. 2005;22:486–495. doi: 10.1093/molbev/msi032. [DOI] [PubMed] [Google Scholar]
- Li R, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Lifsics MR, Lancy ED, Maurer R. DNA replication defect in Salmonella typhimurium mutants lacking the editing (epsilon) subunit of DNA polymerase III. J Bacteriol. 1992;174:6965–6973. doi: 10.1128/jb.174.21.6965-6973.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Rajan V, Moret BM. Fast and accurate phylogenetic reconstruction from high-resolution whole-genome data and a novel robustness estimator. J Comput Biol. 2011;18:1131–1139. doi: 10.1089/cmb.2011.0114. [DOI] [PubMed] [Google Scholar]
- Lopez-Sanchez MJ, et al. Evolutionary convergence and nitrogen metabolism in Blattabacterium strain Bge, primary endosymbiont of the cockroach Blattella germanica. PLoS Genet. 2009;5:e1000721. doi: 10.1371/journal.pgen.1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, et al. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- Martin JH, Mound LA. An annotated check list of the world's whiteflies (Insecta: Hemiptera: Aleyrodidae) Zootaxa. 2007;1492:1–84. [Google Scholar]
- McCutcheon JP, McDonald BR, Moran NA. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc Natl Acad Sci U S A. 2009;106:15394–15399. doi: 10.1073/pnas.0906424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc Natl Acad Sci U S A. 2007;104:19392–19397. doi: 10.1073/pnas.0708855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol Evol. 2010;2:708–718. doi: 10.1093/gbe/evq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2011;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- McCutcheon JP, von Dohlen CD. An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Curr Biol. 2011;21:1366–1372. doi: 10.1016/j.cub.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. doi: 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- Mora L, et al. The essential role of the invariant GGQ motif in the function and stability in vivo of bacterial release factors RF1 and RF2. Mol Microbiol. 2003;47:267–275. doi: 10.1046/j.1365-2958.2003.03301.x. [DOI] [PubMed] [Google Scholar]
- Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- Nakabachi A, et al. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- Neef A, et al. Genome economization in the endosymbiont of the wood roach Cryptocercus punctulatus due to drastic loss of amino acid synthesis capabilities. Genome Biol Evol. 2011;3:1437–1448. doi: 10.1093/gbe/evr118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JD, Shields CR. Tripartite structure of the Brassica campestris mitochondrial genome. Nature. 1984;307:437–440. [Google Scholar]
- Perez-Brocal V, et al. A small microbial genome: the end of a long symbiotic relationship? Science. 2006;314:312–313. doi: 10.1126/science.1130441. [DOI] [PubMed] [Google Scholar]
- Raubeson LA, Jansen RK. Chloroplast genomes of plants. In: Henry RJ, editor. Plant diversity and evolution: genotypic and phenotypic variation in higher plants. Wallingford (United Kingdom): CABI; 2005. pp. 45–68. [Google Scholar]
- Rio RV, et al. Insight into the transmission biology and species-specific functional capabilities of tsetse (Diptera: Glossinidae) obligate symbiont Wigglesworthia. MBio. 2012;3:e00240–11. doi: 10.1128/mBio.00240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabree ZL, et al. Genome shrinkage and loss of nutrient-providing potential in the obligate symbiont of the primitive termite Mastotermes darwiniensis. Appl Environ Microbiol. 2012;78:204–210. doi: 10.1128/AEM.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabree ZL, Kambhampati S, Moran NA. Nitrogen recycling and nutritional provisioning by Blattabacterium, the cockroach endosymbiont. Proc Natl Acad Sci U S A. 2009;106:19521–19526. doi: 10.1073/pnas.0907504106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Garcia D, et al. Complete genome sequence of “Candidatus Portiera aleyrodidarum” BT-QVLC, an obligate symbiont that supplies amino acids and carotenoids to Bemisia tabaci. J Bacteriol. 2012;194:6654–6655. doi: 10.1128/JB.01793-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarich VM, Wilson AC. Generation time and genomic evolution in primates. Science. 1973;179:1144–1147. doi: 10.1126/science.179.4078.1144. [DOI] [PubMed] [Google Scholar]
- Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- Silva FJ, Latorre A, Moya A. Why are the genomes of endosymbiotic bacteria so stable? Trends Genet. 2003;19:176–180. doi: 10.1016/S0168-9525(03)00041-6. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Moran NA. Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol Biol Evol. 2012a;29:3781–3792. doi: 10.1093/molbev/mss180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Moran NA. Endosymbiotic bacteria as a source of carotenoids in whiteflies. Biol Lett. 2012b;8:986–989. doi: 10.1098/rsbl.2012.0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding AW, von Dohlen CD. Phylogenetic characterization and molecular evolution of bacterial endosymbionts in psyllids (Hemiptera: Sternorrhyncha) Mol Biol Evol. 1998;15:1506–1513. doi: 10.1093/oxfordjournals.molbev.a025878. [DOI] [PubMed] [Google Scholar]
- Stajich JE, et al. The BioPerl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics. 1993;135:599–607. doi: 10.1093/genetics/135.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamas I, et al. 50 million years of genomic stasis in endosymbiotic bacteria. Science. 2002;296:2376–2379. doi: 10.1126/science.1071278. [DOI] [PubMed] [Google Scholar]
- Tesler G. Efficient algorithms for multichromosomal genome rearrangements. J Comp Syst Sci. 2002;65:587–609. [Google Scholar]
- Thao ML, Baumann P. Evolutionary relationships of primary prokaryotic endosymbionts of whiteflies and their hosts. Appl Environ Microbiol. 2004;70:3401–3406. doi: 10.1128/AEM.70.6.3401-3406.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh H, et al. Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res. 2006;16:149–156. doi: 10.1101/gr.4106106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham RC, et al. Reductive genome evolution in Buchnera aphidicola. Proc Natl Acad Sci U S A. 2003;100:581–586. doi: 10.1073/pnas.0235981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LE, Wernegreen JJ. Unprecedented loss of ammonia assimilation capability in a urease-encoding bacterial mutualist. BMC Genomics. 2010;11:687. doi: 10.1186/1471-2164-11-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyke T, et al. One bacterial cell, one complete genome. PLoS One. 2010;5:e10314. doi: 10.1371/journal.pone.0010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahra R, Blackwood JK, Sales J, Leach DR. Proofreading and secondary structure processing determine the orientation dependence of CAG x CTG trinucleotide repeat instability in Escherichia coli. Genetics. 2007;176:27–41. doi: 10.1534/genetics.106.069724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.