

Abstract
Ganglioneuromas (GNs) arising from neural crest sympathogonia are rare benign neurogenic tumors. The most commonly affected sites are the posterior mediastinum, the retroperitoneum and the adrenal gland. GNs often present as a solitary, painless and slow-growing mass, and multiple occurrences in the cervical region are extremely rare. Here, we report a case of massive multiple cervical GN in a 4-year-old girl, and review cases of cervical GN that have been reported in the past 10 years. The results demonstrated that cervical GN, compared to other sites, is seldom secretory. The signs and symptoms of cervical GN are unspecific; the ultimate diagnosis of GN depends on pathological examination. Fine-needle aspiration biopsy has limited value in diagnosis. Surgical excision is the treatment of choice and the prognosis is excellent even in cases where complete excision cannot be achieved. Furthermore, GNs should be considered in patients with multiple masses in the neck.
Keywords: neurogenic tumor, ganglioneuroma, children, cervical
Introduction
Peripheral neuroblastic tumors (pNTs), including neuroblastomas (NB), ganglioneuroblastomas (GNBs) and ganglioneuromas (GNs), are rare and constitute only 6% of tumors in children. GNs are the most differentiated and benign form of pNTs and the majority of GNs display as slow-growing, solitary lesions that may or may not have an effect on neighboring structures. The most commonly affected sites are the posterior mediastinum, the retroperitoneum and the adrenal gland, which affect 41.5, 37.5 and 21% of cases, respectively. Few GNs occur in the cervical region (8%), of which the majority are in single form, and to the best of our knowledge, only one case of multiple GN in one side of the neck has been reported (1). Here, we report a case of massive multiple GN located in the neck of a 4-year-old girl who was successfully treated using a surgical approach. We also reviewed cases of GN located in the head and neck region that have been reported in the English-language literature over the past 10 years. The clinically related features, differential diagnosis, management and pathological findings are discussed.
The study was approved by the ethic committee of The General Hospital of the People’s Liberation Army, Beijing, China. The patient’s guardians consented to the publication of the study.
Case report
A 4-year-old girl was admitted to the General Hospital of the People’s Liberation Army, Beijing, China, with a slow-growing, painless mass in the right lateral neck region, which had been present since birth. In the past year, the mass had demonstrated accelerated growth and tenderness, and an increasingly heavy snore during sleep was noticed by her parents. There was no growth retardation and no specific treatments were conducted.
The physical examination revealed two firm solid masses extending from the left mastoid process to the clavicle, with the frontal border of the mass extending across the midline where throbbing of the carotid artery was observed. The overlying skin was normal with no adhesion to the masses. No symptoms of Horner’s syndrome, including ptosis, myosis, ipsilateral facial anhidrosis and flushing, were observed. Additionally, routine laboratory tests, including quantitative analysis of homovanillic acid (HVA) and vanillylmandelic acid (VMA), revealed no abnormality.
Computed tomography (CT) images identified two ovoid and well-circumscribed masses on the right side of the neck. The first mass extended from the scull base to the clavicle level in the prevertebral region and across the midline and measured 10×6.4×5.7 cm. The other mass was located in the paravertebral region from C4 to C7 level and measured 4.1×2.6×5 cm (Fig. 1A and B). A working diagnosis of neurilemmoma was established.
Figure 1.
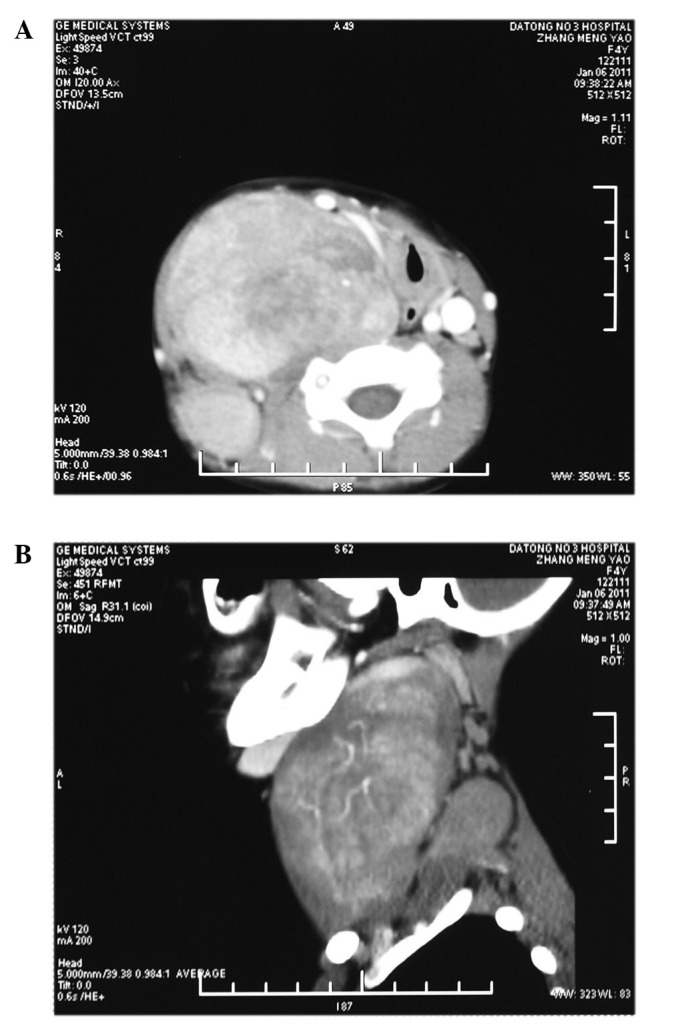
Contrast-enhanced CT scans. (A) Two masses in the paravertebral and prevertebral region are well-circumscribed with sparse dot-like calcifications. The medially displaced trachea and esophagus, the common carotid artery, internal jugular vein and vagus nerve are displaced anteromedially to the near surface of neck. (B) The bigger mass extends from the skull base to clavicle level, which is anterior to the smaller mass. CT, computed tomography.
Complete surgical excision of the masses located beneath the sternocleidomastoid and trapezius muscles was performed under general anesthesia using a T-shaped cervical approach. Upon macroscopy, the two masses were thick and well-defined with no adhesion to the surrounding tissues. The common carotid artery and internal jugular vein were displaced anteromedially, and two round and thick neurogenic pedicles were observed to originate from underneath the prevertebral fascia (Fig. 2). Once the common carotid artery, internal jugular vein, phrenic nerve and cranial nerves X–XII were identified and carefully protected, the masses were circumferentially separated using blunt dissection and the pedicles were ligated and transected. Both masses were completely excised.
Figure 2.

Preoperative view of GN with thick neurogenic pedicles in the neck. GN, ganglioneuroma.
The excised mass was smooth and well-encapsulated. The gross sections revealed a homogeneous tan-white internal appearance. Microscopically, the tumor was composed of large mature ganglion cells, schwann cells and tangled masses of neurites in bundles of schwannian stroma. Additionally, slight immaturity of the ganglion cells was observed in part of the tumor (Fig. 3). Positivity was observed for S-100, which is consistent with the neurogenic nature of the neoplasm. A pathological diagnosis of GN was established for both masses.
Figure 3.
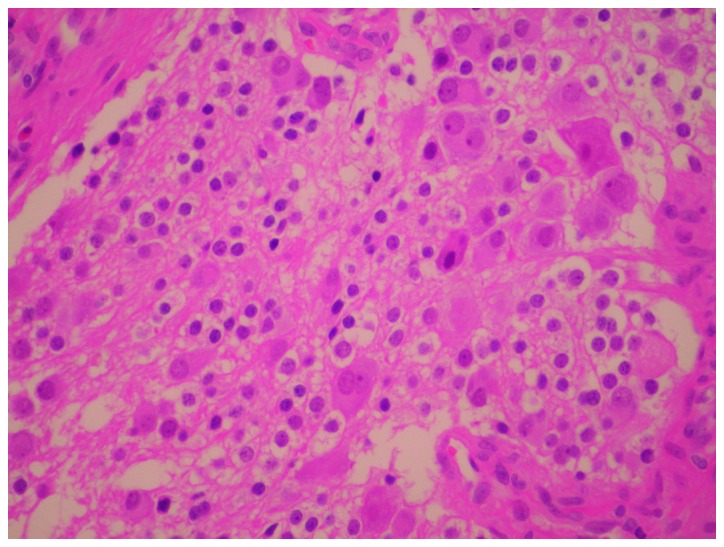
Representative histopathological image of GN stained with H&E. GN, ganglioneuroma; H&E, hematoxylin and eosin.
The postoperative period was uneventful with no observed symptoms of Horner’s syndrome with the exception of myosis in the right eye. The diameter of the right pupil was 1.5 mm, approximately one third of the left pupil, although it recovered to 2.5 mm by the time the patient was discharged on the 10th postoperative day. No specific adjuvant therapy was performed and no signs of recurrence were observed during a 1.5-year follow-up.
Discussion
GN is a rare, benign, non-invasive and neurogenic tumor. We reviewed cases of GN located in the head and neck region that were reported in the English-language literature during the past 10 years. A total of 16 papers and 26 cases were included (2–17). The epidemiology and clinical characteristics are summarized in Table I. At presentation, 18 cases were under 18 years, the mean age was 20.6 years and females demonstrated a slight preponderance, accounting for 61.5% of all cases.
Table I.
Characteristics of GN located in the head and neck region reported in the English-language literature over the past 10 years.
| Characteristics | No. of cases | % |
|---|---|---|
| Gender | ||
| Female | 16 | 61.5 |
| Male | 9 | 34.6 |
| Location | ||
| Parapharyngeal space | 6 | 23.1 |
| Retropharyngeal space | 3 | 11.5 |
| Spine | 2 | 7.7 |
| Mandible | 1 | 3.8 |
| Superior orbit | 1 | 3.8 |
| Internal auditory canal | 1 | 3.8 |
| Other | 13 | 50.0 |
| Form | ||
| Solitary | 23 | 88.5 |
| Multiple | 3 | 11.5 |
| Unilateral mass | 23 | 88.5 |
| Bilateral mass | 3 | 11.5 |
| Symptoms | ||
| Asymptomatic mass | 15 | 57.7 |
| Dysphagia snoring | 5 | 19.2 |
| Pain | 2 | 7.7 |
| Proptosis of the right eye | 2 | 7.7 |
| Spinal cord compression symptoms | 2 | 7.7 |
| Hearing loss | 1 | 3.8 |
| Mandibular asymmetry | 1 | 3.8 |
| VMA/HVA abnormality | 0/5 | 0.0 |
| Treatment | ||
| Complete excision | 23 | 88.5 |
| Partial excision | 3 | 11.5 |
| Postoperative Horner’s syndrome | 10 | 38.5 |
| Recurrence | 1 | 3.8 |
GN, ganglioneuroma. VMA, vanillylmandelic acid; HVA, homovanillic acid.
GN in the head and neck region usually presents as a slowly enlarging mass, predominantly in single form. Only one case of the multiple form in one side of the neck has been reported; thus, it is extremely rare (1). Of the 26 reviewed cases of GN, 3 demonstrated bilateral masses in the neck, 2 of which were presented in the spine (2,3). The majority of cases reviewed were primary lesions (34.5%), occurring in the parapharyngeal and retropharyngeal space. The cervical sympathetic chain is the most frequent structure of origin in the neck. Other sites of origin include the larynx, pharynx and ganglion nodosum of the vagus nerve. In rare circumstances, GN evolves from differentiating NBs or GNBs, either spontaneously or due to treatment. One case of GN resulted from metastasis of NB in the adrenal gland (5), which extends the scope of differential diagnosis of masses in the head and neck region.
Most GN cases we reviewed in the head and neck region presented as an asymptomatic mass (57.7%), and only 2 cases experienced pain (7.7%). The present case also demonstrated tenderness. When masses compress the pharyngeal wall, cranial nerves, eustachian tubes or vascular structures, dysphagia, dyspnea, snoring and obstructive sleep apnea may be reported (19.2%). Symptoms of Horner’s syndrome may ensue from the compression of the cervical sympathetic chain. In this review, no symptoms of Horner’s syndrome at presentation were reported; however, 1 case in the internal auditory canal presented hearing loss (6).
It has been reported that GNs are secretory in up to 39% of patients releasing catecholamines (18). The elevated catecholamines increase the levels of VMA or HMA in the plasma or urine, causing hypertension, diarrhea, sweating, flushing, renal acidosis and other symptoms of catecholamine excess. However, in our review, including the present case, no secretory function-related symptoms were observed, and none of the VMA/VHA tests identified any abnormality. Therefore, quantitative analysis of VMA/VHA is not indispensable for the differential diagnosis of GN in this region.
Due to the scarcity of GN and the lack of specific signs and symptoms, it is often difficult to reach a definite diagnosis prior to pathological examination. CT and magnetic resonance imaging (MRI) provide valuable information on the size, location, composition of the mass and its relationship to adjacent significant structures, which is of great aid in determining a surgical plan. GN usually appears as a defined oval or irregular mass with a hypodense appearance. Punctate and coarse calcification could also be observed in certain cases in a disseminated sprinkled pattern, and administration of contrast media could lead to low or moderate enhancement. MRI is superior to CT in defining the intraspinal involvement; low intensity on T1-weighted images (T1WI), marked high intensity on T2-weighted images (T2WI) and gradual increasing enhancement on dynamic MR images are typical appearances of GN (19).
With these image characteristics it remains difficult to discriminate GN from other lesions in the cervical region, including salivary gland tumors (pleomorphic adenoma), other neurogenic tumors (neurolemmoma, neurofibroma and fibrosarcomas) and soft tissue lesions (fibrosarcomas, rhabdomyosarcomas and malignant lymphomas). When GN is in close proximity to the thyroid gland, these lesions may be mistaken for thyroid swellings (7). Recently, fine-needle aspiration biopsy (FNAB) has been conducted in the diagnosis of GN (7–11). It is a rapid, cost-effective and safe diagnostic procedure, particularly for the pediatric age group; however, it is not always reliable. In certain cases, it was inconclusive and even misleading (7,8,11). Mature ganglion cells are characteristic of GN, and if FNA fails to take the ganglion cells the diagnosis of other neurogenic neoplasms, including schwannomas and neurofibromas, may be mistakenly made. Moreover, GNBs may only contain small foci of neuroblasts and FNA may easily miss this immature component, leading to the wrong diagnosis. Thus, the results of FNA should be taken cautiously (9).
Peripheral neuroblastic tumors represent a spectrum of diseases from undifferentiated and malignant NB to well-differentiated and benign GN (8). According to the international neuroblastoma pathology classification (the Shimada System), neuroblastic tumors are classified into 4 groups: NB (schwannian stroma-poor), intermixed GNB (schwannian stroma-rich), nodular GNB (schwannian stroma-rich/stroma-dominant and stroma-poor) and GN (schwannian stroma-dominant), which is further divided into 2 subtypes (maturing and mature). Typical GN is composed of mature ganglion cells and schwannian stroma; however, complete maturation of ganglion cells is rare (approximately 7%) and slightly atypical ganglion cells could be detected in the present case. Immaturity of ganglion cells did not influence the diagnosis of GN (20).
Complete surgical excision is the treatment of choice, in which a transcervical or transoral approach may be employed depending on the location of the tumor. The risks are mainly related to the intraoperative sacrifice of the neural structures and the vasculature associated with the tumor. The prognosis of GN is usually good. Complete excision was achieved in 23 (88.5%) cases in this review and no recurrences were reported in those patients. In the 3 cases of partial excision, 2 cases demonstrated no growth in 2–4 years and recurrence was reported in 1 case. Symptoms of Horner’s syndrome are often detected following surgery, but these symptoms are usually completely resolved within several months. Further therapy, chemotherapy or radiotherapy, is not usually required, even for cases with partial excision. Retrosi et al (21) studied 10 patients that underwent incomplete excision, and after 33.5±40 months’ follow-up, no tumor progression or recurrence occurred. However, if slight immaturity is present in GN, close follow-up is recommended.
References
- 1.Shotton JC, Milton CM, Allen JP. Multiple ganglioneuroma of the neck. J Laryngol Otol. 1992;106:277–278. doi: 10.1017/s0022215100119267. [DOI] [PubMed] [Google Scholar]
- 2.Miyakoshi N, Hongo M, Kasukawa Y, Misawa A, Shimada Y. Bilateral and symmetric C1–C2 dumbbell ganglioneuromas associated with neurofibromatosis type 1 causing severe spinal cord compression. Spine J. 2010;10:e11–e15. doi: 10.1016/j.spinee.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 3.Bacci C, Sestini R, Ammannati F, Bianchini E, Palladino T, Carella M, Melchionda S, Zelante L, Papi L. Multiple spinal ganglioneuromas in a patient harboring a pathogenic NF1 mutation. Clin Genet. 2010;77:293–297. doi: 10.1111/j.1399-0004.2009.01292.x. [DOI] [PubMed] [Google Scholar]
- 4.Zebing Z, Jianwei S, Yan C, Yan G. Clinicopathological characteristics of neck ganglioneuroma. Oral Med Pathol. 2008;12:131–134. [Google Scholar]
- 5.Patterson AR, Barker CS, Loukota RA, Spencer J. Ganglioneuroma of the mandible resulting from metastasis of neuroblastoma. Int J Oral Maxillofac Surg. 2009;38:196–198. doi: 10.1016/j.ijom.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Ozluoglu LN, Yilmaz I, Cagici CA, Bal N, Erdogan B. Ganglioneuroma of the internal auditory canal: a case report. Audiol Neurootol. 2007;12:160–164. doi: 10.1159/000099018. [DOI] [PubMed] [Google Scholar]
- 7.Leonardis M, Sperb D, Alster C, Campisi C, Herter NT. Ganglioneuroma of the neck, masquerading as a goiter. Eur J Surg Oncol. 2003;29:929–930. doi: 10.1016/j.ejso.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Cannady SB, Chung BJ, Hirose K, Garabedian N, Van Den Abbeele T, Koltai PJ. Surgical management of cervical ganglioneuromas in children. Int J Pediatr Otorhinolaryngol. 2006;70:287–294. doi: 10.1016/j.ijporl.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 9.Ponce-Camacho MA, Diaz de Leon-Medina R, Miranda-Maldonado I, Garza-Guajardo R, Hernandez-Salazar J, Barboza-Quintana O. A 5-year-old girl with a congenital ganglioneuroma diagnosed by fine needle aspiration biopsy: a case report. Cytojournal. 2008;5:5. doi: 10.1186/1742-6413-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolte SS. Ganglioneuroma presenting as a neck mass diagnosed by fine needle aspiration cytology. Cytopathology. 2011;22:205–206. doi: 10.1111/j.1365-2303.2010.00794.x. [DOI] [PubMed] [Google Scholar]
- 11.Müller A, Förster G, Behrendt W, Kosmehl H. Headache as an unusual presenting symptom of retropharyngeal ganglioneuroma. Acta Otolaryngol. 2002;122:565–568. doi: 10.1080/00016480260092426. [DOI] [PubMed] [Google Scholar]
- 12.Kaufman MR, Rhee JS, Fliegelman LJ, Costantino PD. Ganglioneuroma of the parapharyngeal space in a pediatric patient. Otolaryngol Head Neck Surg. 2001;124:702–704. doi: 10.1177/019459980112400625. [DOI] [PubMed] [Google Scholar]
- 13.Choi HY, Lee JH, Park JM, Shin MK. Orbital ganglioneuroma in a young healthy person. Arch Ophthalmol. 2009;127:223–225. doi: 10.1001/archophthalmol.2008.573. [DOI] [PubMed] [Google Scholar]
- 14.Katilmis H, Ozturkcan S, Adadan I, Ozdemir I, Algin H, Tunakan M. Cervical ganglioneuroma. Int J Pediatr Otorhinolaryngeal Extra. 2006;1:157–159. [Google Scholar]
- 15.Gary C, Robertson H, Ruiz B, Zuzukin V, Walvekar RR. Retropharyngeal ganglioneuroma presenting with neck stiffness: report of a case and review of literature. Skull Base. 2010;20:371–374. doi: 10.1055/s-0030-1253580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cannon TC, Brown HH, Hughes BM, Wenger AN, Flynn SB, Westfall CT. Orbital ganglioneuroma in a patient with chronic progressive proptosis. Arch Ophthalmol. 2004;122:1712–1714. doi: 10.1001/archopht.122.11.1712. [DOI] [PubMed] [Google Scholar]
- 17.Friedlander PL, Hunt JP, Palacios E. Ganglioneuroma of the neck. Ear Nose Throat J. 2002;81:435. [PubMed] [Google Scholar]
- 18.Geoerger B, Hero B, Harms D, Grebe J, Scheidhauer K, Berthold F. Metabolic activity and clinical features of primary ganglioneuromas. Cancer. 2001;91:1905–1913. doi: 10.1002/1097-0142(20010515)91:10<1905::aid-cncr1213>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 19.Ichikawa T, Ohtomo K, Araki T, Fujimoto H, Nemoto K, Nanbu A, Onoue M, Aoki K. Ganglioneuroma: computed tomography and magnetic resonance features. Br J Radiol. 1996;69:114–121. doi: 10.1259/0007-1285-69-818-114. [DOI] [PubMed] [Google Scholar]
- 20.Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, Stram DO, Gerbing RB, Lukens JN, Matthay KK, Castleberry RP. The international neuroblastoma pathology classification (the Shimada system) Cancer. 1999;86:364–372. [PubMed] [Google Scholar]
- 21.Retrosi G, Bishay M, Kiely EM, Sebire NJ, Anderson J, Elliott M, Drake DP, Coppi P, Eaton S, Pierro A. Morbidity after ganglioneuromaexcision: is surgery necessary? Eur J Pediatr Surg. 2011;21:33–37. doi: 10.1055/s-0030-1263195. [DOI] [PubMed] [Google Scholar]