

Abstract
To investigate the immunopathogenesis of inflammation-associated fibrosis we analyzed the chronic colitis and late-developing fibrosis occurring in BALB/c mice administered weekly doses of intra-rectal trinitrobenzene sulfonic acid (TNBS). We showed first in this model that an initial Th1 response involving IL-12p70 and IFN-γ subsides after three weeks to be supplanted by an IL-23/IL-25 response beginning after 4–5 weeks. This evolution is followed by gradually increasing production of IL-17 and cytokines ordinarily seen in a Th2 response, particularly IL-13, which reaches a plateau at 8–9 weeks. We then show that IL-13 production results in the induction of an IL-13 receptor formerly thought to function only as a decoy receptor, IL-13Rα2, and this receptor is critical to the production of TGF-β1 and the onset of fibrosis. Thus, if IL-13 signaling through this receptor is blocked by administration of soluble IL-13Rα2-Fc, or by administration of IL-13Rα2–specific siRNA, TGF-β1 is not produced and fibrosis does not occur. These studies show that in chronic TNBS colitis, fibrosis is dependent on the development of an IL-13 response that acts through a novel cell-surface-expressed IL-13 receptor to induce TGF-β1.
The end stage of many chronic inflammatory diseases is often characterized by extensive tissue fibrosis and its associated untoward effects on organ function. This is seen quite clearly in the two major forms of inflammatory bowel disease (IBD) (1–5). Thus, the main feature of Crohn’s Disease (CD) is a transmural, granulomatous inflammation that frequently leads to mechanical obstruction of the intestinal lumen due to extensive local tissue fibrosis (3, 5). In addition, in ulcerative colitis (UC) one sees a more superficial and ulcerative inflammation that nevertheless results in a stiff, fibrotic large bowel unable to carry out peristalsis. Thus, in both forms of IBD, a fibrotic reaction supervenes that becomes a dominant part of the pathologic picture.
The Chronic TNBS-Colitis Model
To investigate the immunologic events underlying the evolution of a chronic colitis we analyzed the chronic TNBS- colitis occurring in BALB/c mice administered weekly doses of intra-rectal trinitrobenzene sulfonic acid (TNBS) (6, 7). Mice treated in this way initially develop intense colitis associated with severe weight loss and considerable mortality. However, about three weeks after the initiation of this form of TNBS-colitis, the colitis moderates and, while the mice do not exhibit the weight gain of control mice, they regain their lost weight. This period of moderate colitis lasts about four weeks. Importantly, the termination phase of this inflammatory cycle is accompanied by the development of fibrotic cycle. Thus, four-five weeks after initiation of chronic TNBS-colitis, the mice develop steadily increasing fibrosis of the colonic lamina propria. As shown in Figure 1, this increase in intestinal fibrosis can be demonstrated by measuring the collagen content of the colon.
Figure 1.
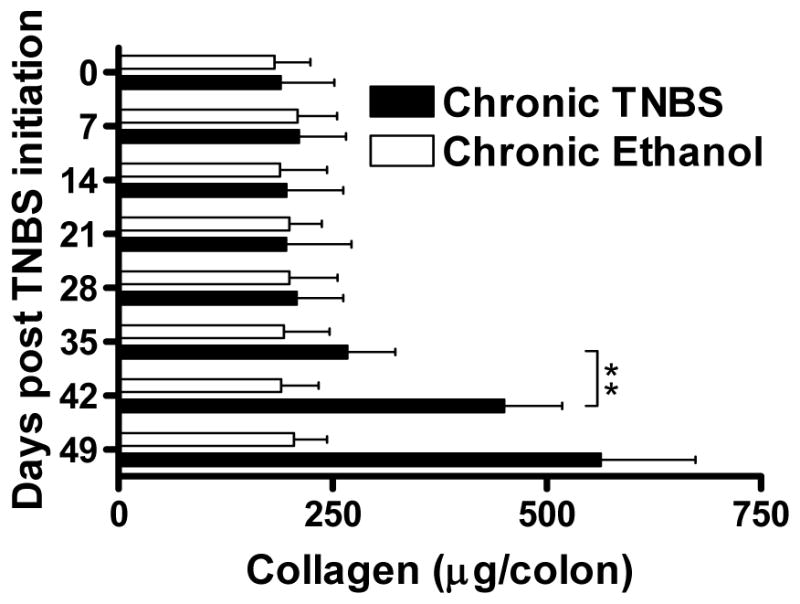
Collagen content of the colon. Collagen content was determined during chronic TNBS-colitis and chronic Ethanol administration by a Sircol assay. Data shown are mean values ± SD and are derived from at least five mice per group; **, p < 0.01.
Cytokine Responses in Chronic TNBS-Colitis
To understand the immunologic basis of this complex series of events we determined the cytokines produced by lamina propria cells during the various stages of the inflammation. As established as an overview in Figure 2, we noted that the production of Th1 cytokines, IL-12p70 and IFN-γ by extracted cells were greatly elevated on day 7 after initiation of TNBS administration. However, surprisingly, the production of these cytokines gradually decreased over the next 2 weeks, returning to baseline levels by day 21 and then gradually declining to levels increasingly below that observed at baseline.
Figure 2.

Cytokine production of colonic lamina propria mononuclear cells at weekly time points during chronic TNBS-colitis in BALB/c mice. Lamina propria mononuclear cells were extracted from the lamina propria and cultured for 48h in the presence of T cell or APC stimulants. Cytokine concentrations in the culture supernatants were determined by cytokine-specific ELISA. Data shown represent an overview of the cytokine production during the time course of the animal model.
Interestingly, an increased production of an alternative set of pro-inflammatory cytokines, namely IL-23 and IL-17, accompanied the decline in the production of IL-12p70 and IFN-γ. Increased IL-23 production above baseline was noted first on day 21, and thereafter increased further until it reached a plateau on day 42. Increased IL-17 production was noted shortly after the increase of IL-23 production. The expression levels of both cytokines increased further until they reached a plateau on day 42. Somewhat surprisingly, increased production of IL-23 and IL-17 coincided with increased production of cytokines usually considered a Th2 cytokines, namely, IL-25 and IL-13.
Previous studies have shown that IL-25, a member of the IL-17 family of proteins (and designated IL-17E), is associated with expression of Th2 cytokines, at least when it is over-expressed in transgenic mice (8). On day 35 a massive increase in IL-25 secretion appeared and thereafter IL-25 secretion further increased, reaching its highest level on day 49. At about the same time, the secretion of IL-13 was initiated and the level of this cytokine also increased until it reached a plateau on day 49.
Mechanisms of Intestinal Fibrosis
Coming now to the molecular events during intestinal fibrogenesis, in previous studies we showed that IL-13 induces TGF-β1 production by signaling through cell-surface IL-13Rα2, an IL-13 receptor formerly thought to function only as a soluble decoy receptor (9–12). Since IL-13 is produced in the later phase of chronic TNBS-colitis we reasoned that this pathway might be involved in the development of fibrosis occurring during this phase. To explore this possibility we first determined the expression of both IL-13Rα1 and IL-13Rα2 in extracts of colonic LPMC during the course of chronic TNBS colitis. As shown in Figure 3, IL-13Rα1 was constitutively expressed throughout the time course of the animal model. In contrast, IL-13Rα2 was an induced receptor since it was not evident in naïve mice or in the first few weeks of chronic TNBS-colitis, but could be detected initially at a low concentration on day 35 and later at a high concentration on days 42 and 49 after initiation of colitis.
Figure 3.
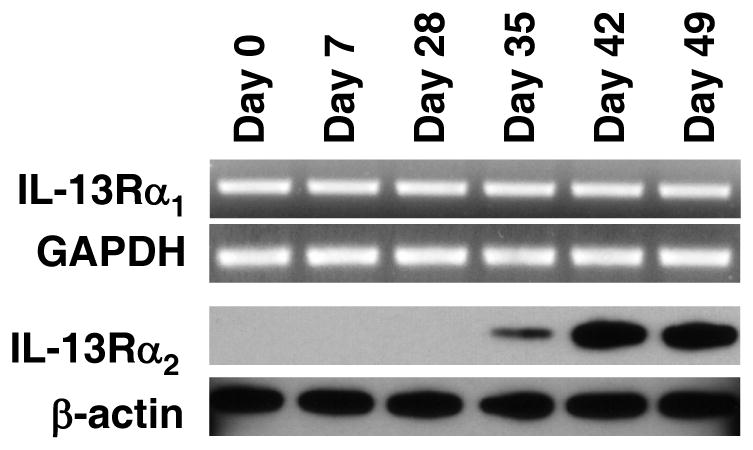
IL-13Rα1 and IL-13Rα2 expression in BALB/c mice during the course of chronic TNBS-colitis. IL-13Rα1 expression is constitutive whereas IL-13Rα2 expression is induced on day 35 during chronic TNBS-colitis. IL-13Rα1 mRNA expression was determined by RT-PCR of RNA extracted from colonic LPMC and IL-13Rα2 protein expression was determined by Western blot analysis analyzed of colonic LPMC lysates.
The above time course of IL-13Rα2 expression correlated with that of IL-13 and was thus consistent with the possibility that the latter was an inducing cytokine. To address this question more directly we next blocked IL-13 activity by administration of a plasmid (pCI-sIL-13Rα2-Fc) encoding a soluble IL-13Rα2-Fc fusion protein (pCI-sIL-13Rα2-Fc) or a control plasmid (pCl empty vector). The administration of the pCl-sIL-13Rα2-Fc plasmid by this route, but not the control plasmid, led to a reduction in LPMC production of IL-13. This effect is consistent with that noted previously in a study by Chiaramonte et al where it was shown that mice with IL-13Rα2 receptor deficiency have reduced IL-13 production and is a possible result of the fact that autocrine IL-13 signaling via this receptor is necessary for optimal IL-13 production (9). In addition, as shown in Figure 4, administration pCI-sIL-13Rα2-Fc led to virtually complete loss of IL-13Rα2 expression in extracts of colonic tissue obtained at the conclusion of the study (day 49). In contrast, neutralization of TGF-β1 by administration of anti-TGF-β1 had no effect on receptor expression or signaling; this was expected since there is no evidence to suggest that TGF-β1 induces up-regulation of IL-13Rα2. Taken together, these results strongly suggest that IL-13 production occurring in the late phase of chronic TNBS-colitis is indeed responsible for the expression of IL-13Rα2.
Figure 4.
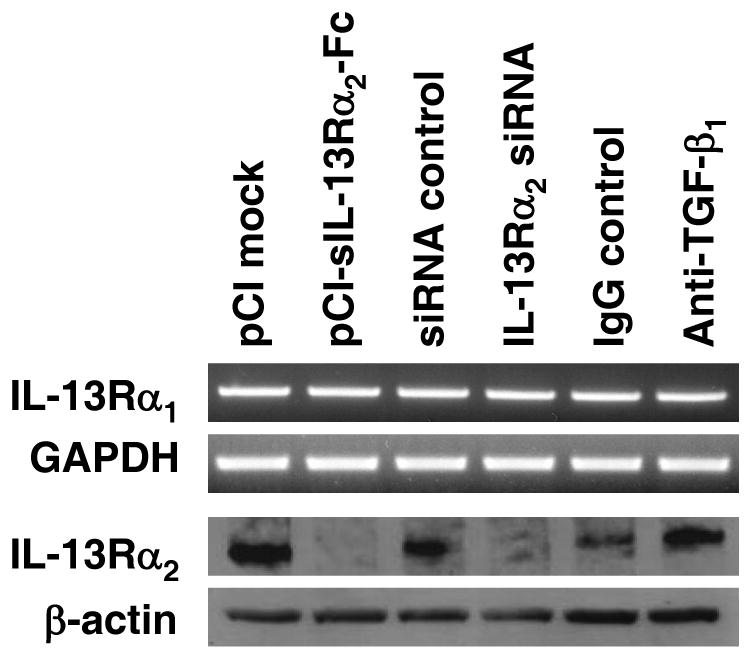
IL-13Rα1 and IL-13Rα2 expression after administration of pCI-sIL-13Rα2-Fc, IL-13Rα2-specific siRNA, or anti-TGF-β1-antibody. IL-13Rα2 expression (but not IL-13Rα1) is reduced after treatment of mice with chronic TNBS-colitis with pCI-sIL-13Rα2-Fc and IL-13Rα2-specific siRNA. IL-13Rα1 mRNA expression was determined by RT-PCR of RNA extracted from colonic LPMC and IL-13Rα2 protein expression was determined by Western blot analysis analyzed of colonic LPMC lysates.
In previous studies we have shown that intra-rectal administration of NF-κB decoy oligonucleotides on days 35 and 42 abrogates the inflammation and the accompanying cytokine response characteristic of the chronic inflammation at this stage of the inflammation, including the high levels of IL-23, IL-17 and IL-13 (6). In addition, such treatment prevented the production of TGF-β1 and the development of intestinal tissue fibrosis. However, while these data established that the IL-13 and TGF-β1 responses as well as the collagen deposition were an outgrowth of the inflammation, they did not address the question of whether or not IL-13 and/or TGF-β1 were the cause of such deposition.
To address the question of whether IL-13 and TGF-β1 are responsible for the collagen deposition, in the present study we determined the effect of in vivo inhibition of either IL-13 signaling or TGF-β1 activity on the generation of tissue fibrosis. In these studies we inhibited IL-13 signaling in two ways: 1) by intra-nasal administration of the plasmid described above encoding a soluble IL-13Rα2-Fc fusion protein (pCI-sIL-13Rα2-Fc) or a control plasmid (pCl empty vector) to block the receptor with the use of a decoy; and 2) by intra-rectal administration of IL-13Rα2-specific siRNA encapsulated in HVJ-E or a control siRNA to down-regulate the receptor by gene silencing. Both the sIL-13Rα2-Fc plasmid and the siRNA were administered every other day starting on day 35 after the initiation of the chronic inflammation. On the other hand, for study of the effects of inhibition of TGF-β1 we administered anti-TGF-β-antibody or isotype control Ig by an intra-peritoneal route over the same time period. The effects of these treatments on IL-13 and TGF-β1 levels were subsequently determined at the termination of the study on day 49. For this purpose, LPMC extracted from the colons of sacrificed mice were cultured for 48h with anti-CD3/anti-CD28 and their secretion of IL-13 and TGF-β1 into the culture supernatant measured by ELISA.
Neutralization of IL-13 signaling by fusion protein or gene-silencing of IL-13Rα2 by siRNA as well as neutralization of TGF-β1 by antibody was accompanied by only a small, non-significant increase in body weight and no measurable change in colitis score. In addition, these treatments were not accompanied by changes in inflammatory cytokine secretion. Thus, LPMC extracted from the colons of sacrificed mice on day 49 and evaluated for secretion of cytokines secreted equal amounts of IL-12p70, IFN-γ, TNF-α, IL-23, IL-17, IL-25 and IL-4 whether or not they were subjected to neutralization of IL-13 signaling or TGF-β1. In contrast, a very different picture was obtained with respect to TGF-β1 secretion. In this case, the cells extracted from the colons of mice subjected to neutralization of IL-13 signaling by fusion protein or IL-13Rα2 downregulation by siRNA exhibited greatly reduced production of TGF-β1. In addition, as shown in Figure 5, those mice subjected to neutralization of IL-13 signaling as well as those subjected to TGF-β1 neutralization displayed basal collagen levels seen in naïve mice whereas those mice not subjected to such neutralization displayed undiminished collagen levels as compared to other mice with chronic TNBS-colitis. Overall, these data show quite clearly that neutralization of IL-13 signaling via IL-13Rα2 does not alter the course of the inflammation of chronic TNBS-colitis but does block the development of fibrosis; in addition, they show that such signaling results in fibrosis via its effect on the induction of TGF-β1.
Figure 5.
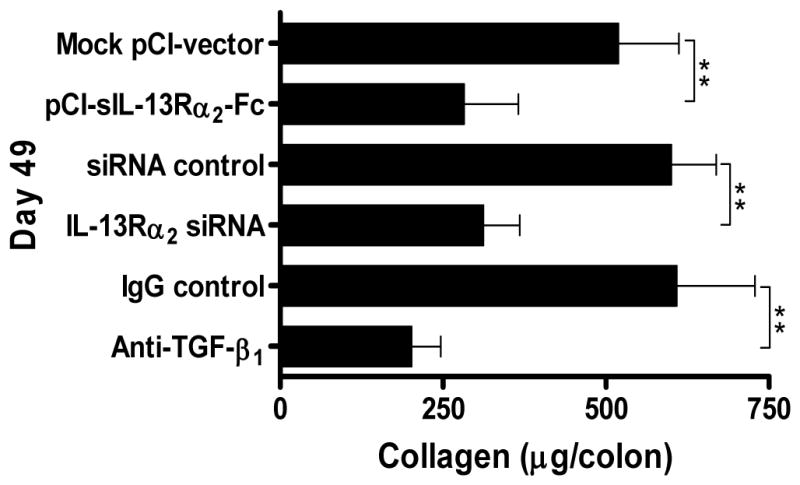
Collagen content of the colon after administration of pCI-sIL-13Rα2-Fc, IL-13Rα2-specific siRNA, or anti-TGF-β1-antibody. Collagen content was determined during chronic TNBS-colitis by a Sircol assay. Data shown are mean values ± SD and are derived from at least five mice per group; **, p < 0.01.
In summary, these findings suggest that chronic inflammation is orchestrated by a succession of cytokines that ultimately result in IL-13 and TGF-β1 production. Both cytokines is then responsible for the development of intestinal fibrosis. In addition, blockade of this cytokine interaction can prevent intestinal fibrosis by selectively interfering with the pro-fibrotic program.
Footnotes
Disclosure
The authors claim no financial interest.
References
- 1.Beck PL, Podolsky DK. Growth factors in inflammatory bowel disease. Inflamm Bowel Dis. 1999;5:44–60. doi: 10.1097/00054725-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nature reviews. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 3.Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- 4.Harper PH, V, Fazio W, Lavery IC, Jagelman DG, Weakley FL, Farmer RG, Easley KA. The long-term outcome in Crohn’s disease. Dis Colon Rectum. 1987;30:174–179. doi: 10.1007/BF02554332. [DOI] [PubMed] [Google Scholar]
- 5.Podolsky DK. Inflammatory bowel disease. The New England journal of medicine. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 6.Fichtner-Feigl S, I, Fuss J, Preiss JC, Strober W, Kitani A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF-kappaB decoy oligonucleotides. J Clin Invest. 2005;115:3057–3071. doi: 10.1172/JCI24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawrance IC, Wu F, Leite AZ, Willis J, West GA, Fiocchi C, Chakravarti S. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology. 2003;125:1750–1761. doi: 10.1053/j.gastro.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 8.Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R, Muchamuel T, Hurst SD, Zurawski G, Leach MW, Gorman DM, Rennick DM. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 9.Chiaramonte MG, Mentink-Kane M, Jacobson BA, Cheever AW, Whitters MJ, Goad ME, Wong A, Collins M, Donaldson DD, Grusby MJ, Wynn TA. Regulation and function of the interleukin 13 receptor alpha 2 during a T helper cell type 2-dominant immune response. The Journal of experimental medicine. 2003;197:687–701. doi: 10.1084/jem.20020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha(2) receptor is involved in induction of TGF-beta(1) production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 11.Mentink-Kane MM, Cheever AW, Thompson RW, Hari DM, Kabatereine NB, Vennervald BJ, Ouma JH, Mwatha JK, Jones FM, Donaldson DD, Grusby MJ, Dunne DW, Wynn TA. IL-13 receptor alpha 2 down-modulates granulomatous inflammation and prolongs host survival in schistosomiasis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:586–590. doi: 10.1073/pnas.0305064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wynn TA, Hesse M, Sandler NG, Kaviratne M, Hoffmann KF, Chiaramonte MG, Reiman R, Cheever AW, Sypek JP, Mentink-Kane MM. P-selectin suppresses hepatic inflammation and fibrosis in mice by regulating interferon gamma and the IL-13 decoy receptor. Hepatology. 2004;39:676–687. doi: 10.1002/hep.20102. [DOI] [PubMed] [Google Scholar]