

Abstract
Purpose of Review
Autoimmune Lymphoproliferative Syndrome (ALPS) is a disorder of disrupted lymphocyte homeostasis, resulting from mutations in the Fas apoptotic pathway. Clinical manifestations include lymphadenopathy, splenomegaly, and autoimmune cytopenias. A number of new insights have improved the understanding of the genetics and biology of ALPS. These will be discussed in this review.
Recent Findings
A number of key observations have been made recently that better define the pathophysiology of ALPS, including the characterization of somatic FAS variant ALPS, the identification of haploinsufficiency as a mechanism of decreased Fas expression, and the description of multiple genetic hits in FAS in some families that may explain the variable penetrance of the disease. In addition, ALPS has been shown to be a more common condition, as patients diagnosed with other disorders, including Evans syndrome and common variable immune deficiency have been found to have ALPS. Finally, the treatment of the disease has changed as splenectomy and rituximab have been shown to have unexpected ALPS specific toxicities, and mycophenolate mofetil and sirolimus have been demonstrated to have marked activity against the disease.
Summary
Based on novel advances the diagnostic algorithm and recommended treatment for ALPS have changed significantly, improving quality of life for many patients.
Keywords: Autoimmune Lymphoproliferative Syndrome, cytopenias, secondary malignancy, FAS, RAS, Evans syndrome, mTOR
Introduction
Autoimmune Lymphoproliferative Syndrome (ALPS) is a disorder of abnormal lymphocyte survival caused by dysregulation of the FAS apoptotic pathway. Patients with ALPS develop chronic non-malignant lymphoproliferation, autoimmune disease, and secondary malignancies. ALPS was first described in the 1990s, and since its discovery a number of significant advances have been made that have changed diagnosis and treatment of ALPS and have better defined its pathophysiology. ALPS was originally thought to be extremely rare but may be more common as more cases are being diagnosed with increasing clinician awareness. This article reviews the diagnosis and treatment of ALPS, focusing on recent scientific advances.
Pathophysiology and Genetics
ALPS is a syndrome defined by a defect in the Fas apoptotic pathway (Figure 1).[1] In order to downregulate the normal immune response, activated B and T lymphocytes increase Fas expression, and activated T lymphocytes increase expression of Fas-ligand.[2] Fas and Fas-ligand interact through the Fas-activating death domain (FADD), triggering the caspase cascade, culminating in DNA degradation, proteolysis and apoptosis.[3] Defective apoptosis can lead to lymphoproliferation, autoimmunity, and cancer.[4,5] Over 70% of patients with ALPS have identifiable mutations in Fas pathway genes. Most patients have germline (60–70% of patients) or somatic mutations (10% of patients) in FAS (TNFRSF6). Rarely, patients have mutations in FASL (TNFSF6, <1% of patients) and CASP10 (2–3% of patients).[6] Twenty to 30% of patients have no identifiable mutation.[6] Recently, an international consensus conference at the NIH redefined ALPS nomenclature, using a gene-based classification (Table 1).[7]
Figure 1. Fas apoptotic pathway.

ALPS is caused by defective Fas-mediated apoptosis. Normally, as part of the down-regulation of the immune response, activated B and T lymphocytes increase Fas expression, and activated T lymphocytes increase expression of Fas ligand. Fas and Fas ligand interact which activates the Fas-associated death domain (FADD) and triggers the caspase cascade, culminating in cellular apoptosis. Fas-mediated signaling is part of the extrinsic apoptotic pathway, because it is activated through the interactiong of cell surface death receptors. In contrast, the intrinsic apoptotic pathway is activated by cellular stressors that lead to decreased mitochondrial membrane permeability with release of apoptosis-inducing substances. © Sue Seif, MA (used with permission).
Table 1.
Revised classification of ALPS and ALPS-related diseases. (Adapted with permission from [7])
| ALPS types | ||
|---|---|---|
| New | Old | Gene |
| ALPS-FAS | ALPS Ia | FAS (germline) |
| ALPS-sFAS | ALPS Im (or ALPS Is) | FAS (somatic) |
| ALPS-FASL | ALPS Ib | FASL |
| ALPS-CASP10 | ALPS IIa | CASP10 |
| ALPS-U | ALPS III | Unknown/Undefined |
| ALPS-related diseases (formally classified as ALPS) | ||
| New | Old | Gene |
| CEDS | ALPS IIb | CASP8 |
| RALD | ALPS IV | NRAS, KRAS |
CEDS: Caspase 8 deficiency state; RALD: Ras-associated leukoproliferative disease
Germline mutations in FAS are usually inherited in an autosomal dominant fashion.[8] Most often FAS defects are dominant negative heterozygous missense mutations in the intracellular death domain (exons 7–9), with the mutated allele inhibiting the function of the wild-type allele. Approximately 30% of ALPS causative mutations affect the extracellular domain. Recently, Keuhn and colleagues established these are mostly nonsense or frameshift mutations resulting in haploinsufficiency.[9] Dominant negative mutations typically lead to absence of FAS activity, whereas haploinsufficient mutations usually lead to decreased activity. As such, disease penetrance appears to be much higher in families with dominant negative intracellular mutations compared with haploinsufficient extracellular mutations.[9–11] ALPS mutations have highly variable expressivity, and mutation type is not predictive of disease manifestations or laboratory biomarkers. The only reported association is a higher incidence of secondary lymphomas in patients with dominant negative intracellular mutations.[9] Of note, the total number of reported patients with ALPS-associated lymphoma is small, and this observed difference may result from selection bias.
A significant subset of ALPS patients have somatic FAS mutations, primarily limited to the non-thymic double negative T cell (DNT) compartment. DNTs (phenotype, CD3+, CD4−, CD8−, TCRα/β+) are a subset of T cells normally found at small percentages in the peripheral blood that are markedly elevated in patients with ALPS. The elevated DNTs in ALPS were originally considered an epiphenomenon; however, they may drive abnormal B cell activity and subsequent autoimmunity.[12] The recent description of somatic ALPS is a strong argument that DNTs play a role in ALPS pathogenesis. ALPS patients with germline and somatic-variant FAS mutations are phenotypically similar in both disease manifestations and laboratory abnormalities.[13,14] Predictably, the primary difference is that patients with germline FAS mutations typically present at a younger age than their somatic counterparts. Causative somatic mutations in CASP10 and FASL have not been described.
FAS mutations have variable penetrance, as ALPS patients often have family members with the same genetic alterations and an absent or very mild clinical phenotype [15,16]. This suggests a second “hit” is required for disease, such as additional genetic mutations and/or environmental triggers. Recently, Magerus-Chatinet and colleagues observed that disease penetrance can be explained in some families as a consequence of multiple FAS mutations.[17] They identified seven ALPS patients who inherited heterozygous FAS mutations and also acquired a somatic alteration in the second FAS allele. Family members with only germline FAS mutations were asymptomatic. Multiple abnormalities in FAS may explain the variable penetrance in some families; however, patients may acquire mutations in other cooperating genes as the second event.
Clinical Manifestations and Diagnosis
Lymphoproliferation is the most common clinical manifestation in ALPS, presenting as lymphadenopathy, splenomegaly, and/or hepatomegaly.[6,18–20] Most patients develop lymphoproliferation at a young age (median 11.5 months) that varies from mild to severe enough to compromise vital organs. [21] Autoimmunity is the second most common finding and is the most likely to require medical intervention. Over 70% of patients develop autoimmune disease, most commonly immune-mediated cytopenias, which can affect erythrocytes (autoimmune hemolytic anemia), platelets (immune thrombocytopenia) or neutrophils (autoimmune neutropenia).[15] Severity ranges from asymptomatic laboratory abnormalities to severe, chronic, and debilitating destruction of multiple cell lineages.[6] Other autoimmune manifestations are less frequent, although ALPS autoimmunity can affect virtually any organ, and patients can develop nephritis, gastritis, hepatitis, urticaria, arthritis, colitis, and pulmonary fibrosis, similar to systemic lupus erythematosis.[10,22,23] While autoimmune neurologic disease is a rare manifestation of ALPS, we have diagnosed severe autoimmune neurologic complications in three patients, including autoantibody-positive autoimmune cerebellar ataxia, transverse myelitis, and Guillain-Barre syndrome (unpublished observation). All three patients responded to systemic immune suppression.
ALPS patients have an increased risk of secondary malignancies, most commonly EBER+ non-Hodgkin lymphoma.[10,15] The risk is estimated to be 10–20% and is most prevalent in FAS-mutant ALPS.[16] Unaffected family members with FAS mutations are also predisposed to malignancy, consistent with observations that somatic mutations in FAS family genes are highly prevalent in lymphomas in the general population.[24] Identifying malignancy can be difficult as clinical manifestations of ALPS mirror lymphoma. No imaging modality, including FDG-PET, can accurately distinguish benign from malignant lymphoproliferation because rapidly proliferating cells in ALPS have high FDG uptake.[25] Unlike carcinomas and other solid tumors, early diagnosis of lymphoma does not change clinical outcome. Accordingly, based on the very high false-positive rate, our group does not perform routine serial imaging of ALPS patients. Rather, we investigate for malignancy if patients develop constitutional symptoms or have a significant change in disease pattern. ALPS lymphadenopathy consists primarily of polyclonal expansion of abnormal cells. Progression to lymphoma is associated with monoclonal expansion of malignant lymphocytes. Testing for clonality in lymphocyte subsets can sometimes help distinguish benign from malignant disease.
Until 2010, diagnosis of ALPS required meeting three mandatory criteria: (1) chronic (> 6 months) non-malignant lymphoproliferation; (2) elevated peripheral blood DNTs; and (3) defective in vitro Fas-mediated apoptosis (Table 2). The first two criteria remain mandatory for diagnosis. DNTs are assessed by flow cytometry of peripheral blood or lymphoid tissue. DNTs should only be tested in an experienced clinical laboratory and must include testing for the TCRα/β receptor, as other non-pathogenic lymphocyte subsets are CD3+/CD4−/CD8−. DNT test results must be interpreted carefully, as the normal range can vary between laboratories. Finally, lymphopenic patients and those on high dose steroids or sirolimus should not undergo DNT testing due to risks of false positives or negatives, respectively.
Table 2.
Diagnostic Criteria for ALPS. (Adapted with permission from [7])
| Old criteria |
| Required |
| 1. Chronic non-malignant lymphoproliferation |
| 2. Elevated peripheral blood DNTs |
| 3. Defective in vitro Fas mediated apoptosis |
| New criteria |
| Required |
| 1. Chronic non-malignant lymphoproliferation (<6 months lymphadenopathy and/or splenomegaly) |
| 2. Elevated peripheral blood DNTs |
| Accessory |
| Primary |
| 1. Defective in vitro Fas mediated apotosis (in 2 separate assays) |
| 2. Somatic or germline mutation in ALPS causative gene (FAS, FASL, CASP10) |
| Secondary |
1. Elevated biomarkers (Any of following)
|
| 2. Immunohistochemical findings consistent with ALPS as determined by experienced histopathologist |
| 3. Autoimmune cytopenias AND polyclonal hypergammaglobulinemia |
| 4. Family history of ALPS or nonmalignant lymphoproliferation |
|
Definitive Diagnosis: Required plus one primary accessory criteria Probable Diagnosis: Required plus one secondary accessory criteria Of note, probable and definitive ALPS should be treated the same in the clinic |
In order to test for defective in vitro Fas-mediated apoptosis, a patient’s peripheral blood mononuclear cells are isolated, activated with mitogen, and expanded with interleukin-2 (IL-2) in culture for 10–28 days.[4] Normally, mitogen activation and T cell expansion upregulate the Fas pathway. Subsequent exposure of normal T cells to anti-Fas immunoglobulin M (IgM) monoclonal antibody in vitro leads to rapid apoptosis.[26] ALPS patients’ T cells do not die after exposure.[27] This is an elegant but expensive and labor-intensive assay, requiring samples from patients and unaffected controls. DNTs do not survive in routine culture; this assay only identifies Fas defects in non-DNT T cells. Patients with somatic-variant ALPS or FASL mutations usually have normal apoptosis assays.
A number of recent studies have identified reliable biomarkers for ALPS, including elevated serum vitamin B12, soluble Fas ligand, IL-10, and IL-18.[13,28] These biomarkers are very specific for ALPS in patients with lymphoproliferation and elevated DNTs and are most predictive for FAS-variant (germline or somatic) ALPS. Our group established that a combination of autoimmune cytopenias and hypergammaglobulinemia are very predictive of ALPS in patients with lymphoproliferation and elevated DNTs.[29]
The recent international consensus conference led to a new diagnostic algorithm that includes the three previously mandatory criteria but also allows for the diagnosis of ALPS with genetic testing, biomarkers, family history, and/or histopathology (Table 2).[7] The new algorithm makes a nomenclature distinction between definitive versus probable diagnoses; however, the published consensus statement recommends that patients with definitive and probable ALPS be treated the same clinically. A number of clinical laboratories can perform specialized testing for DNTs, FAS and other gene mutations, and biomarkers, including IL-10 and sFASL. The Diagnostic Immunology and Molecular Genetics laboratories at Cincinnati Children’s Hospital (Cincinnati, OH) provide the most comprehensive assortment of testing. Other laboratories that offer a number of specialized tests, include the Children’s Hospital of Philadelphia (Philadelphia, PA), and GeneDX (Gaithersburg, MD).
Differential Diagnosis
ALPS patients have highly heterogeneous phenotypes with clinical findings that overlap with malignant, infectious, autoimmune, and rheumatologic conditions. Several lymphoproliferative disorders, including Castleman disease, Rosai-Dorfman disease, X-linked lymphoproliferative disease (XLP), Dianzani Autoimmune Lymphoproliferative Disease (DALD), Kikuchi-Fujimoto disease, Caspase 8 deficiency syndrome (CEDS), and Ras-associated leukoproliferative disorder (RALD) can present with clinical features similar to ALPS. As these disorders are often distinguishable by histopathology, most patients should undergo tissue biopsy (bone marrow and/or lymph node) at initial presentation.
Patients with mutations in CASP8 were originally classified as having ALPS because caspase 8 and caspase 10 have similar functions, and CASP8 mutant patients present with lymphadenopathy and defective Fas-mediated apoptosis [30]. While patients with ALPS primarily have apoptotic defects in T lymphocytes, patients with CASP8 mutations have profound apoptotic defects in B, T, and NK lymphocytes [30]. Patients with CASP8 mutations are predisposed to significant mucocutaneous infections with herpes virus [30]. Thus, patients with CASP8 mutations are now considered to have a distinct disease termed CEDS.
Rosai-Dorfman (RD) disease is a histiocytic disorder with considerable phenotypic overlap with ALPS. RD is diagnosed by histopathology. Emperipolesis (lymphophagocytosis) was thought to be pathognomonic for RD; however, lymph node biopsies from ALPS patients can demonstrate emperipolesis, as well as other RD features.[31] Accordingly, testing for ALPS should be considered in any child diagnosed with RD.
X-linked lymphoproliferative disorder (XLP) is a rare disorder characterized by a dysregulated T and NK cell-mediated immune response to EBV infection, most commonly as a consequence loss of SLAM-associated protein (SAP) because of mutations in SH2D1A. Patients with XLP can present with fulminant mononucleosis, hematophagocytic syndrome, aplastic anemia and/or aggressive lymphoproliferative disease. Recently, SAP-deficient T cells collected from XLP patients were demonstrated to have defective restimulation-induced apoptosis.[32] Thus, XLP can be classified as an ALPS-like disorder of abnormal lymphocyte apoptosis.
Ras-associated leukoproliferative disorder (RALD) is a newly described lymphoproliferative disorder characterized by impaired cytokine withdrawal-induced apoptosis in T cells due to gain of function somatic mutations in RAS family genes (KRAS and NRAS).[33–35] Fewer than 10 patients with RALD have been reported, resulting in limited knowledge of the clinical manifestations of the disease. Common features include lymphoproliferation, autoimmune cytopenias, and hypergammaglobulinemia. DNTs may be mildly elevated in RALD but are usually normal. As in juvenile myelomonocytic leukemia (JMML; a myeloproliferative disorder characterized by RAS mutations), myeloid cells from these patients may demonstrate hypersensitivity to GM-CSF. Recent literature suggests RALD is a non-malignant disease based on its indolent nature; however, no published studies have described whether the abnormal cells in RALD are clonal. Secondary cancers and unexplained sudden death have been described in RALD and somatic gain-of-function mutations in RAS family genes are extremely common in cancer. Thus, more studies are needed to determine if RALD is a benign lymphoproliferative disorder, a pre-malignant condition, or a malignant state.
Patients with common variable immunodeficiency (CVID) can present with lymphadenopathy and autoimmune disease mirroring ALPS.[36] In addition, a small subset of ALPS patients have co-morbid CVID. Specific testing for ALPS, including DNT analysis, apoptosis assays, and genetic testing, may aid in distinguishing the conditions. Arguably, any patient with CVID and secondary autoimmune cytopenias should be tested for ALPS.
Evans syndrome (ES), defined by autoimmune destruction of at least two hematologic cell types, can also have a similar presentation to ALPS [37]. Recently, our group demonstrated in an multi-institutional trial that a significant percentage of children diagnosed with ES have ALPS.[26,29] There was likely selection bias, as not all ES patients were captured at each participating institution, and a similar prevalence of ALPS may not be found among ES patients in other populations. A recent comprehensive analysis of children with autoimmune hemolytic anemia in France, including a large cohort of ES patients, did not find a high incidence of ALPS.[38] ALPS patients were identified in the study but at a lower rate than in US studies. Of note, not all patients were tested for ALPS, and only ES patients with autoimmune hemolytic anemia were included.
Treatment
While some ALPS patients require no treatment, many require immunosuppression, particularly to treat cytopenias. Some patients develop organ compromise from lymphoproliferation, requiring medical intervention. Most patients respond to short corticosteroid pulses. [39] While appropriate for periodic disease flares, corticosteroids are too toxic for use in chronic disease. As recently as five years ago, medication choices for ALPS autoimmunity did not differ from other autoimmune conditions. Recent studies have established that some therapies commonly used in other conditions are relatively contraindicated, while other less common therapies are very effective in ALPS.
Rituximab and splenectomy are often the treatments of choice in refractory autoimmune cytopenias in children. Rituximab can lead to prolonged, clinically significant hypogammaglobulinemia when used in ALPS. Thus, alternative immune suppressants should be tried before using rituximab, if possible.[40,41] Rituximab is active in many ALPS patients, and can be used when other agents are ineffective or not tolerated. Patients should be advised that they may require prolonged IVIgG replacement therapy. Patients with ALPS have a very high risk of developing post-splenectomy sepsis, even with antibiotic prophylaxis and vaccination.[42,43] Accordingly, splenectomy should be avoided except in the case of uncontrolled hypersplenism that fails other medical management.
The two best-studied and most effective non-steroid agents used in children with ALPS are mycophenolate mofetil (Cellcept, MMF) and sirolimus (rapamycin). MMF inactivates inosine monophosphate, a key enzyme in purine synthesis required for lymphocyte proliferation [44,45]. Over thirty patients treated with MMF have been reported, and over 80% of these patients demonstrated measurable improvements in autoimmune disease [44,46,47]. MMF does not improve lymphoproliferation or reduce DNTs. Many of these patients had only partial responses, and some relapsed. MMF is a well-tolerated medication with side effects including neutropenia and diarrhea. In Europe, another purine synthesis inhibitor, mercaptopurine, is commonly used instead of MMF.
Sirolimus (rapamycin), a mammalian target of rapamycin (mTOR) inhibitor, has also been studied extensively in ALPS. We hypothesized that targeting the PI3K/mTOR/Akt signaling pathway may be effective in ALPS (Figure 2). In support, the mTOR signaling pathway is upregulated in murine ALPS (our unpublished results), and sirolimus is very active in this disease model, demonstrating superior efficacy than other therapies including MMF.[48] We subsequently opened a clinical trial that has examined 13 patients thus far, and continues to enroll (NCT00392951).[49,50] Our early results indicate that sirolimus is uniquely active in ALPS with most patients showing rapid, complete responses (unpublished data).[49] The majority of patients had failed other treatments, often involving multiple agents, including corticosteroids and MMF. In these patients, sirolimus inhibited both autoimmune disease and lymphoproliferation, and in most patients, eliminated the abnormal DNTs. These results have been confirmed in independent studies.[51,52] Based on our observation that the mTOR signaling pathway is abnormally activated in ALPS, treating ALPS patients with sirolimus is a form of targeted therapy that is well tolerated with little toxicity. Side effects include hypercholesterolemia, hypertension, and mucositis.[53] Of note, we continue to offer MMF as a first line for chronic treatment, as sirolimus requires therapeutic drug monitoring. For patients with more aggressive disease or in those with symptomatic lymphoproliferation, we use sirolimus as first line.
Figure 2. Hypothetical Model of Dysregulation of PI3K/Akt/mTOR Signaling in ALPS.
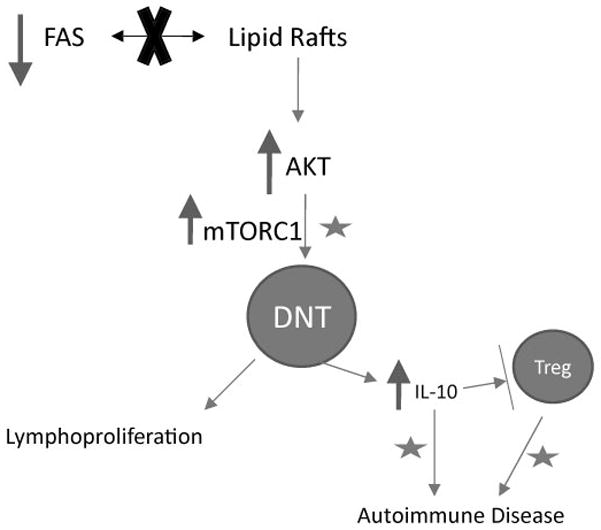
ALPS is a defined by defects in the Fas apoptotic pathway; however, individuals with causative mutations do not always manifest disease symptoms. Fas must be recruited into lipid rafts in order to stimulate apoptosis. We hypothesize that in ALPS the defects in the Fas pathway lead to decreased distribution of Fas receptor into lipid rafts, thereby activating the serine threonine kinase Akt. In support, Akt activity and distribution of Fas into lipid rafts are inversely correlated in some lymphocyte subsets.[54] Constitutive activation of Akt stimulates the mTOR pathway, causing uncontrolled proliferation of the double negative T cells (DNTs) with consequent lymphadenopathy and splenomegaly. These DNTs also secrete IL-10, causing autoimmunity.High levels of IL-10 down-regulate TGF-β producing Tregs, a subset of T lymphocytes that can regulate immune tolerance.[55,56] We observe abnormally low Treg numbers in ALPS (unpublished). Moreover, low levels of TGF-β producing T-cells can cause an “ALPS-like” disease in mice.[57] Thus, the Treg dysregulation may exacerbate the systemic autoimmunity. Accordingly, mTOR inhibitors, including sirolimus may alleviate ALPS through combined mechanisms indicated by stars. First, mTOR inhibitors can directly target the dysregulated PI3K/Akt/mTOR signaling axis and reduce accumulation of abnormal DNTs. Second, mTOR inhibitors may block signaling through IL-10.[58,59] Finally, in contrast to most immunosuppressants, mTOR inhibitors can spare Tregs, and thereby help to reset the immune imbalance.
Conclusion
In summary, over the past decade a number of remarkable insights have been made that improve understanding of the underlying pathogenesis of ALPS. Through international collaborations, the diagnosis and treatment of ALPS has changed significantly in a short time, making it easier for physicians to diagnosis the disease and improving quality of life for patients.
Key Points.
The diagnostic algorithm for ALPS has recently changed with less focus on a functional research based apoptosis assay and more reliance on genetic testing and biomarkers.
A combination of multiple mutations (acquired and inherited) in ALPS causative genes may help explain low disease penetrance in families
Autoimmune disease and lymphoproliferation are treated differently in ALPS than similar disorders. Rituximab and splenectomy should be avoided. Mycophenolate mofetil and sirolimus should be considered in patients with chronic disease.
Acknowledgments
Support: This work was supported by grants from the United States Immunodeficiency Network (USIDNET, N01-A1-30070) (DTT), a Foerderer-Murray Award (DTT), the Goldman Philanthropic Partnerships and the Rockefeller Brothers Fund (DTT), Partnership for Cures (DTT), The Longest Day of Golf (DTT), Larry and Helen Hoag Foundation Clinical Translational Research Career Development Award (DTT). No author has competing financial interests to declare.
References
- 1.Rieux-Laucat F, Le Deist F, Fischer A. Autoimmune lymphoproliferative syndromes: genetic defects of apoptosis pathways. Cell Death Differ. 2003;10:124–133. doi: 10.1038/sj.cdd.4401190. [DOI] [PubMed] [Google Scholar]
- 2.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 3.Sneller MC, Straus SE, Jaffe ES, Jaffe JS, Fleisher TA, Stetler-Stevenson M, Strober W. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest. 1992;90:334–341. doi: 10.1172/JCI115867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober W, Lenardo MJ, Puck JM. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 5.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 6.Bleesing JJ. Autoimmune lymphoproliferative syndrome: A genetic disorder of abnormal lymphocyte apoptosis. Immunology and Allergy Clinics of North America. 2002;22:339–349. doi: 10.1016/s0031-3955(05)70272-8. [DOI] [PubMed] [Google Scholar]
- 7*.Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, Rieux-Laucat F, Siegel RM, Su HC, Teachey DT, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116:e35–40. doi: 10.1182/blood-2010-04-280347. This article is an international consensus statement from experts in ALPS that defines a new diagnostic algorithm for ALPS and revises the nomenclature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teachey DT, Seif AE, Grupp SA. Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS) Br J Haematol. 2010;148:205–216. doi: 10.1111/j.1365-2141.2009.07991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Kuehn HS, Caminha I, Niemela JE, Rao VK, Davis J, Fleisher TA, Oliveira JB. FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome. J Immunol. 186:6035–6043. doi: 10.4049/jimmunol.1100021. This article establishes that FAS haploinsufficiency is an important disease mechanism in ALPS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, Dale JK, Fleisher TA, Middelton LA, Sneller MC, et al. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet. 1999;64:1002–1014. doi: 10.1086/302333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rieux-Laucat F, Blachere S, Danielan S, De Villartay JP, Oleastro M, Solary E, Bader-Meunier B, Arkwright P, Pondare C, Bernaudin F, et al. Lymphoproliferative syndrome with autoimmunity: A possible genetic basis for dominant expression of the clinical manifestations. Blood. 1999;94:2575–2582. [PubMed] [Google Scholar]
- 12.Ohga S, Nomura A, Takahata Y, Ihara K, Takada H, Wakiguchi H, Kudo Y, Hara T. Dominant expression of interleukin 10 but not interferon gamma in CD4(−)CD8(−) alphabetaT cells of autoimmune lymphoproliferative syndrome. Br J Haematol. 2002;119:535–538. doi: 10.1046/j.1365-2141.2002.03848.x. [DOI] [PubMed] [Google Scholar]
- 13*.Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, Neven B, Schaffner C, Ducrot N, Arkwright PD, Bader-Meunier B, Barbot J, Blanche S, et al. FAS-L, IL-10, and double-negative CD4− CD8− TCR alpha/beta+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood. 2009;113:3027–3030. doi: 10.1182/blood-2008-09-179630. This article provides evidence that biomarkers can be used to predict ALPS. [DOI] [PubMed] [Google Scholar]
- 14.Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, Blanche S, Bartunkova J, Vilmer E, Fischer A, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351:1409–1418. doi: 10.1056/NEJMoa040036. [DOI] [PubMed] [Google Scholar]
- 15.Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med. 1999;130:591–601. doi: 10.7326/0003-4819-130-7-199904060-00020. [DOI] [PubMed] [Google Scholar]
- 16.Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rosen-Wolff A, Peters AM, Sneller MC, Hallahan CW, Wang J, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. doi: 10.1182/blood.v98.1.194. [DOI] [PubMed] [Google Scholar]
- 17**.Magerus-Chatinet A, Neven B, Stolzenberg MC, Daussy C, Arkwright PD, Lanzarotti N, Schaffner C, Cluet-Dennetiere S, Haerynck F, Michel G, et al. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest. 121:106–112. doi: 10.1172/JCI43752. This article establishes that a combination of multiple mutations (acquired and inherited) in ALPS causative genes may help explain low disease penetrance in families. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Werff ten Bosch J. Autoimmune lymphoproliferative syndrome: etiology, diagnosis, and management. Paediatr Drugs. 2003;5:185–193. doi: 10.2165/00128072-200305030-00005. [DOI] [PubMed] [Google Scholar]
- 19.Bleesing JJ. Autoimmune lymphoproliferative syndrome (ALPS) Curr Pharm Des. 2003;9:265–278. doi: 10.2174/1381612033392107. [DOI] [PubMed] [Google Scholar]
- 20.Wei A, Cowie T. Rituximab responsive immune thrombocytopenic purpura in an adult with underlying autoimmune lymphoproliferative syndrome due to a splice-site mutation (IVS7+2 T>C) affecting the Fas gene. Eur J Haematol. 2007;79:363–366. doi: 10.1111/j.1600-0609.2007.00924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson CE, Puck JM. Autoimmune lymphoproliferative syndrome, a disorder of apoptosis. Curr Opin Pediatr. 1999;11:521–527. doi: 10.1097/00008480-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 22.Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, et al. Clincial, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341–1348. [PubMed] [Google Scholar]
- 23.Worth A, Thrasher AJ, Gaspar HB. Autoimmune lymphoproliferative syndrome: molecular basis of disease and clinical phenotype. Br J Haematol. 2006;133:124–140. doi: 10.1111/j.1365-2141.2006.05993.x. [DOI] [PubMed] [Google Scholar]
- 24.Poppema S, Maggio E, van den Berg A. Development of lymphoma in Autoimmune Lymphoproliferative Syndrome (ALPS) and its relationship to Fas gene mutations. Leuk Lymphoma. 2004;45:423–431. doi: 10.1080/10428190310001593166. [DOI] [PubMed] [Google Scholar]
- 25.Rao VK, Carrasquillo JA, Dale JK, Bacharach SL, Whatley M, Dugan F, Tretler J, Fleisher T, Puck JM, Wilson W, et al. Fluorodeoxyglucose positron emission tomography (FDG-PET) for monitoring lymphadenopathy in the autoimmune lymphoproliferative syndrome (ALPS) Am J Hematol. 2006;81:81–85. doi: 10.1002/ajh.20523. [DOI] [PubMed] [Google Scholar]
- 26.Teachey DT, Manno CS, Axsom KM, Andrews T, Choi JK, Greenbaum BH, McMann JM, Sullivan KE, Travis SF, Grupp SA. Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS) Blood. 2005;105:2443–2448. doi: 10.1182/blood-2004-09-3542. [DOI] [PubMed] [Google Scholar]
- 27.Sneller MC, Dale JK, Straus SE. Autoimmune lymphoproliferative syndrome. Curr Opin Rheumatol. 2003;15:417–421. doi: 10.1097/00002281-200307000-00008. [DOI] [PubMed] [Google Scholar]
- 28*.Caminha I, Fleisher TA, Hornung RL, Dale JK, Niemela JE, Price S, Davis J, Perkins K, Dowdell KC, Brown MR, et al. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2010;125:946–949. e946. doi: 10.1016/j.jaci.2009.12.983. This article provides evidence that biomarkers can predict ALPS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Seif AE, Manno CS, Sheen C, Grupp SA, Teachey DT. Identifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: a multi-institutional study. Blood. 2010;115:2142–2145. doi: 10.1182/blood-2009-08-239525. This article demonstrates that ALPS is underdiagnosed as patients with presumed idiopathic autoimmune cytopenia syndromes may have ALPS. [DOI] [PubMed] [Google Scholar]
- 30.Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–399. doi: 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- 31.Maric I, Pittaluga S, Dale JK, Niemela JE, Delsol G, Diment J, Rosai J, Raffeld M, Puck JM, Straus SE, et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am J Surg Pathol. 2005;29:903–911. doi: 10.1097/01.pas.0000157997.61177.08. [DOI] [PubMed] [Google Scholar]
- 32.Snow AL, Marsh RA, Krummey SM, Roehrs P, Young LR, Zhang K, van Hoff J, Dhar D, Nichols KE, Filipovich AH, et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest. 2009;119:2976–2989. doi: 10.1172/JCI39518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niemela JE, Lu L, Fleisher TA, Davis J, Caminha I, Natter M, Beer LA, Dowdell KC, Pittaluga S, Raffeld M, et al. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2010;117:2883–2886. doi: 10.1182/blood-2010-07-295501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliveira JB, Bidere N, Niemela JE, Zheng L, Sakai K, Nix CP, Danner RL, Barb J, Munson PJ, Puck JM, et al. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 2007;104:8953–8958. doi: 10.1073/pnas.0702975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takagi M, Shinoda K, Piao J, Mitsuiki N, Matsuda K, Muramatsu H, Doisaki S, Nagasawa M, Morio T, Kasahara Y, et al. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2010;117:2887–2890. doi: 10.1182/blood-2010-08-301515. [DOI] [PubMed] [Google Scholar]
- 36.Savasan S, Warrier I, Buck S, Kaplan J, Ravindranath Y. Increased lymphocyte Fas expression and high incidence of common variable immunodeficiency disorder in childhood Evans’ syndrome. Clin Immunol. 2007;125:224–229. doi: 10.1016/j.clim.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Evans RS, Takahashi K, Duane RT, Payne R, Liu C. Primary thrombocytopenic purpura and acquired hemolytic anemia; evidence for a common etiology. AMA Arch Intern Med. 1951;87:48–65. doi: 10.1001/archinte.1951.03810010058005. [DOI] [PubMed] [Google Scholar]
- 38.Aladjidi N, Leverger G, Leblanc T, Picat MQ, Michel G, Bertrand Y, Bader-Meunier B, Robert A, Nelken B, Gandemer V, et al. New insights into childhood autoimmune hemolytic anemia: a French national observational study of 265 children. Haematologica. 96:655–663. doi: 10.3324/haematol.2010.036053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bleesing JJ, Straus SE, Fleisher TA. Autoimmune lymphoproliferative syndrome. A human disorder of abnormal lymphocyte survival. Pediatr Clin North Am. 2000;47:1291–1310. doi: 10.1016/s0031-3955(05)70272-8. [DOI] [PubMed] [Google Scholar]
- 40.Rao VK, Price S, Perkins K, Aldridge P, Tretler J, Davis J, Dale JK, Gill F, Hartman KR, Stork LC, et al. Use of rituximab for refractory cytopenias associated with autoimmune lymphoproliferative syndrome (ALPS) Pediatr Blood Cancer. 2009;52:847–852. doi: 10.1002/pbc.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper N, Davies EG, Thrasher AJ. Repeated courses of rituximab for autoimmune cytopenias may precipitate profound hypogammaglobulinaemia requiring replacement intravenous immunoglobulin. Br J Haematol. 2009;146:120–122. doi: 10.1111/j.1365-2141.2009.07715.x. [DOI] [PubMed] [Google Scholar]
- 42*.Rao VK, Oliveira JB. How I treat autoimmune lymphoproliferative syndrome. Blood. 2011 doi: 10.1182/blood-2011-07-325217. Epub ahead of print. This article establishes the risk of post-splenectomy sepsis in ALPS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Neven B, Magerus-Chatinet A, Florkin B, Gobert D, Lambotte O, De Somer L, Lanzarotti N, Stolzenberg MC, Bader-Meunier B, Aladjidi N, et al. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood. 2011 doi: 10.1182/blood-2011-04-347641. Epub ahead of print. This article establishes the risk of post-splenectomy sepsis in ALPS. [DOI] [PubMed] [Google Scholar]
- 44.Rao VK, Dugan F, Dale JK, Davis J, Tretler J, Hurley JK, Fleisher T, Puck J, Straus SE. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. 2005;129:534–538. doi: 10.1111/j.1365-2141.2005.05496.x. [DOI] [PubMed] [Google Scholar]
- 45.Izeradjene K, Quemeneur L, Michallet MC, Bonnefoy-Berard N, Revillard JP. Mycophenolate mofetil interferes with interferon gamma production in T-cell activation models. Transplant Proc. 2001;33:2110–2111. doi: 10.1016/s0041-1345(01)01965-0. [DOI] [PubMed] [Google Scholar]
- 46.Rao VK, Price S, Perkins K, Aldridge P, Tretler J, Davis J, Dowdell K, Niemela JE, Brown M, Fleisher T. Use of mycophenolate mofetil in children with chronic, refractory immune cytopenias associated with autoimmune lymphoproliferative syndrome (ALPS) Pediatr Blood Cancer. 2009;52:697. doi: 10.1002/pbc.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kossiva L, Theodoridou M, Mostrou G, Vrachnou E, Le Deist F, Rieux-Laucat F, Kanariou MG. Mycophenolate mofetil as an alternate immunosuppressor for autoimmune lymphoproliferative syndrome. J Pediatr Hematol Oncol. 2006;28:824–826. doi: 10.1097/MPH.0b013e31802d7503. [DOI] [PubMed] [Google Scholar]
- 48.Teachey DT, Obzut DA, Axsom K, Choi JK, Goldsmith KC, Hall J, Hulitt J, Manno CS, Maris JM, Rhodin N, et al. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS) Blood. 2006;108:1965–1971. doi: 10.1182/blood-2006-01-010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teachey DT, Greiner R, Seif A, Attiyeh E, Bleesing J, Choi J, Manno C, Rappaport E, Schwabe D, Sheen C, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145:101–106. doi: 10.1111/j.1365-2141.2009.07595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teachey DT. Autoimmune lymphoproliferative syndrome: new approaches to diagnosis and management. Clin Adv Hematol Oncol. 9:233–235. [PubMed] [Google Scholar]
- 51.Janic MD, Brasanac CD, Jankovic JS, Dokmanovic BL, Krstovski RN, Kraguljac Kurtovic JN. Rapid regression of lymphadenopathy upon rapamycin treatment in a child with autoimmune lymphoproliferative syndrome. Pediatr Blood Cancer. 2009;53:1117–1119. doi: 10.1002/pbc.22151. [DOI] [PubMed] [Google Scholar]
- 52.Tommasini A, Valencic E, Piscianz E, Rabusin M. Immunomodulatory drugs in autoimmune lymphoproliferative syndrome (ALPS) Pediatr Blood Cancer. 2011 doi: 10.1002/pbc.23205. [DOI] [PubMed] [Google Scholar]
- 53.Hartford CM, Ratain MJ. Rapamycin: something old, something new, sometimes borrowed and now renewed. Clin Pharmacol Ther. 2007;82:381–388. doi: 10.1038/sj.clpt.6100317. [DOI] [PubMed] [Google Scholar]
- 54.Pizon M, Rampanarivo H, Tauzin SB, Chaigne-Delalande B, Daburon S, Castroviejo M, Moreau P, Moreau JF, Legembre P. PI3K/AKT inhibition elicits actin-independent and CD95-dependent apoptotic signalling through localisation of CD95 into lipid rafts. Eur J Immunol [Google Scholar]
- 55.Mittal SK, Sharma RK, Gupta A, Naik S. Increased interleukin-10 production without expansion of CD4+CD25+ T-regulatory cells in early stable renal transplant patients on calcineurin inhibitors. Transplantation. 2009;88:435–441. doi: 10.1097/TP.0b013e3181af20fd. [DOI] [PubMed] [Google Scholar]
- 56.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 57.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baker AK, Wang R, Mackman N, Luyendyk JP. Rapamycin enhances LPS induction of tissue factor and tumor necrosis factor-alpha expression in macrophages by reducing IL-10 expression. Mol Immunol. 2009;46:2249–2255. doi: 10.1016/j.molimm.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weichhart T, Saemann MD. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann Rheum Dis. 2008;67(Suppl 3):iii70–74. doi: 10.1136/ard.2008.098459. [DOI] [PubMed] [Google Scholar]