

Abstract
The use of biomarkers is becoming increasingly intrinsic to the practice of medicine and holds great promise for transforming the practice of rheumatology. Biomarkers have the potential to aid clinical diagnosis when symptoms are present or to provide a means of detecting early signs of disease when they are not. Some biomarkers can serve as early surrogates of eventual clinical outcomes or guide therapeutic decision making by enabling identification of individuals likely to respond to a specific therapy. Using biomarkers might reduce the costs of drug development by enabling individuals most likely to respond to be enrolled in clinical trials, thereby minimizing the number of participants required. In this Review, we discuss the current use and the potential of biomarkers in rheumatology and in select fields at the forefront of biomarker research. We emphasize the value of different types of biomarkers, addressing the concept of ‘actionable’ biomarkers, which can be used to guide clinical decision making, and ‘mechanistic’ biomarkers, a subtype of actionable biomarker that is embedded in disease pathogenesis and, therefore, represents a superior biomarker. We provide examples of actionable and mechanistic biomarkers currently available, and discuss how development of such biomarkers could revolutionize clinical practice and drug development.
Introduction
A biomarker is a characteristic that can be objectively measured as an indicator of normal or pathologic biological processes, or as an indicator of response to therapy.1 Although commonly used to describe a biochemical variable, such as the concentration of a circulating protein or other biomolecule, this broad definition can apply to many types of biological data. In fact, many biomarker studies focus on anatomical and structural features visualized by conventional radiography, ultrasonography, CT scanning (for example, positron emission tomography) or MRI, including functional MRI scans that can provide information about the neuronal activity in certain regions of the brain.2 Other variables considered biomarkers are cellular immune responses, genetic traits, histologic characteristics of diseased tissue and proteins or RNA expressed in tissues.
For many diseases, a single biomarker can be informative on a population level but not at the level of the individual patient. This inadequacy has shifted attention to the use of multiple biomarkers and, in parallel, to the development of technologies for the multiplex measurement of multiple variables.3 A panel of multiple biomarkers could comprise different entities of the same type of variable, for example, a number of distinct circulating proteins or expressed genes representing a specific molecular pathway. Alternatively, the panel could comprise a combination of disparate types of feature, such as a collection of radiographic, histologic, cellular, proteomic, and genetic variables.
Herein we review the field of biomarkers in rheumatology, and the concept of the ‘actionable’ biomarker. We discuss the superiority of biomarkers that are rooted in the pathogenesis of disease, how these ‘mechanistic’ biomarkers could be most effectively used in clinical practice and in drug development, and how close we are to having such tools for the management of rheumatic diseases.
Actionable biomarkers and their uses
The concept of an actionable biomarker is based on the expectation that results of biomarker testing can be used to guide clinical management of disease. Actionable biomarkers can inform clinical practice at many different stages of a disease (Figure 1).
Figure 1.

Possible clinical uses of actionable biomarkers at different stages of the development of RA. Screening the asymptomatic, at-risk population for biomarkers of RA-associated asymptomatic autoimmunity could identify individuals who will go on to develop RA, before they develop symptomatic disease. Profiling RA-associated biomarkers in individuals with undifferentiated arthritis who present with synovitis could enable the early diagnosis of RA, before the ACR criteria for a diagnosis of this disease are met and before cartilage and bone erosion has begun. Prognostic biomarkers could also enable the severity of disease course to be predicted in individuals with undifferentiated arthritis. Once cartilage and bone has begun to erode and the ACR criteria for diagnosis of RA are met, biomarker profiling could guide the selection of appropriate therapy by predicting disease activity and progression and aid in assessing the response to therapy by providing pharmacodynamic information. At each of these stages, biomarker profiling can inform physicians on how best to manage their patients to slow or even stop the progression of disease. Abbreviations: ACR, American College of Rheumatology; RA, rheumatoid arthritis.
Diagnosis of symptomatic disease
The most basic use of an actionable biomarker is in making a clinical diagnosis of symptomatic disease. For example, detection of antibodies directed against specific pathogens indicates the presence of infectious diseases (such as HIV or hepatitis virus), whereas detection of specific genetic aberrations is used in the diagnosis of certain cancers (including myelodysplastic syndrome and chronic myelocytic leukemia). In rheumatic diseases, diagnostic biomarkers are also central to clinical practice: the presence of rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA) aid diagnosis of rheumatoid arthritis (RA); and the presence and specificities of antinuclear antibodies (ANA) facilitate diagnosis of systemic lupus erythematosus (SLE). However, unlike in cancer, genetic traits do not seem to be generally useful as diagnostic biomarkers for rheumatic diseases at present. Although specific genetic mutations or polymorphisms are associated with certain autoinflammatory conditions, such as familial Mediterranean fever and Muckle-Wells syndrome,4,5 and minor subtypes of certain autoimmune diseases, SLE for example,6 most autoimmune and inflammatory diseases are polygenic, with individual gene polymorphisms conferring only a modest increase in disease risk.
Diagnosis of asymptomatic disease
Beyond their use in diagnosing symptomatic disease, actionable biomarkers can prove informative in the diagnosis of early, asymptomatic disease (Figure 1). Diagnosing a disease at the asymptomatic stage could enable early therapeutic intervention, with the ultimate goal of preventing the development of symptomatic disease or at least limiting the pathologic sequelae of the disease.7–9 For example, we have identified a profile of serum autoantibodies and cytokines that can be used to identify asymptomatic individuals who will develop RA within 2 years after testing.10 Prophylactic treatment has the potential to reduce the incidence or severity of RA in this group of individuals, given that current treatments result in impressive clinical and radiological improvements when used to treat recent-onset, symptomatic RA.11 Likewise, specific autoantibodies can be detected in the blood of individuals who will go on to develop symptomatic SLE,7 which might prove useful in guiding prophylactic treatment of this disease.
Assessment and prediction of disease activity
Biomarkers that aid assessment of disease activity are useful because they can reveal the presence or progression of disease despite the remission of symptoms. For example, such biomarkers are especially needed for osteoarthritis (OA), for which few, if any, disease-modifying therapies exist. In particular, structural and anatomical features assessed by MRI, such as articular cartilage integrity, bone marrow lesions, synovitis, and osteophytes, are being developed as biomarkers of disease activity in OA and have the potential to be used as surrogate endpoints in early-stage trials.12–14 Cartilage oligomeric matrix protein, a product of cartilage turnover, and metabolomic profiles are also being explored as potential biomarkers for assessment of disease activity in OA.15,16 In addition to providing potential surrogate endpoints, biomarkers could also facilitate enrollment of patient subgroups more likely to response to a candidate therapeutic.
Prognostic biomarkers predict severity of disease and, therefore, can guide the selection of an appropriate therapeutic regimen by providing information as to whether the disease is likely to be self-limiting or to develop into a severe form. Although several prognostic biomarkers, such as erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), complement proteins C3 and C4, and anti-DNA antibodies are currently used in the management of rheumatic diseases, the sensitivity and specificity of these biomarkers in predicting the course of disease are suboptimal.17 Profiles of inflammatory molecules are being developed as prognostic biomarkers for RA;18 although such multi-biomarker panels have been shown to correlate closely with disease activity,18 their specificity for RA (rather than other inflammatory and autoimmune diseases) remains to be fully assessed.
Dynamic biomarkers
Assessment of disease progression and response to therapy
Another actionable use of biomarkers is monitoring the progression of disease or response to therapy. Such ‘dynamic’ biomarkers can facilitate prediction of the ultimate clinical outcome by reporting early changes in disease-associated biological processes. Results of dynamic-biomarker profiling could prompt the clinician to initiate or intensify therapy in the setting of highly active disease or, conversely, to withdraw a specific treatment in the setting of an insufficient therapeutic response. For example, in nonrheumatic diseases, serum levels of prostate-specific antigen and α-fetoprotein can be informative in assessments of response to therapy in prostate cancer—for cases with abnormally high levels of this protein at the time of diagnosis—and hepatocellular cancer, respectively.19 Likewise, serial assessment of tumour size by semiquantitative imaging is a standard of care in the treatment of many solid cancers. Examples of dynamic biomarkers for rheumatic diseases include ESR in polymyalgia rheumatic and a profile of inflammatory mediators that reflects disease activity in RA.18 In addition, levels of proteinuria and composition of urine sediment are both used in assessing response to therapy in SLE.20 As routine imaging in rheumatology moves beyond conventional radiography, which predominantly reveals irreversible structural changes, to technologies such as MRI21 or ultrasound22 that enable serial assessment of synovitis, the results of interval imaging will probably become a commonly accepted dynamic biomarker of disease activity.
Beyond clinical practice, pharmacodynamic biomarkers that serve as surrogate endpoints—described by the FDA as biomarkers that can substitute for a clinical endpoint that “reflects how a patient feels, functions, or survives”98—can be useful in drug development. Such biomarkers can provide a ’yes’ or ‘no’ answer to the question of whether a therapy will be effective early in the development process; thus, time and resources spent on investigational drugs that will ultimately prove ineffective are minimized, enabling resources to be refocused on the development of a wider range of alternative potential therapies. Indeed, the identification of pharmacodynamic biomarkers that can provide early proof of concept is one of the research areas prioritized by the FDA’s Critical Path Initiative.23 Pharmacodynamic biomarkers have, in fact, been incorporated into the study of drugs for rheumatic diseases for decades. Even in early studies, identification of a decrease in serum levels of IL-1 in response to methotrexate24 and a reduction in levels of CRP in after infliximab treatment provided proof-of-concept supporting what are now well-established therapies for RA. More recently, a decrease in number of macrophages in the sublining of synovial tissue obtained by needle biopsy has been proposed as a sensitive and early biomarker of therapeutic efficacy in RA.25 In addition, profiling the expression of genes associated with the type I interferon (IFN) pathway in blood and skin samples has yielded proof-of-concept data in trials of anti-IFN antibody therapy in SLE26 and might do the same in future therapeutic trials in scleroderma.27 Nonetheless, pharmacodynamic biomarkers are used primarily to guide drug development, with clinical endpoints still forming the basis of regulatory approval.98
Assessment of drug toxicity
The use of pharmacodynamic biomarkers in drug development can also enable early detection of drug-related toxicity. The failure rate of novel investigational drugs in phase II and III clinical trials now approaches 85% and 50%, respectively, and, at each phase, adverse events and safety concerns account for approximately 20% of these failures.28,29 Examples of dynamic biomarkers of toxicity include traditional laboratory variables, such as biochemical indicators of liver and kidney dysfunction, but also surrogates of cardiac toxicity, such as increased blood pressure, an increase in levels of serum lipids and prolongation of the QT interval on an electrocardiogram. Going forward, it will be important to identify ‘next-generation’ biomarkers that can serve as early indicators of toxicity or other adverse events in clinical trials.
Predicting responsiveness to therapy
Just as useful as the pharmacodynamic biomarkers, which reflect the response to therapy, are biomarkers that predict responsiveness to therapy before therapy is initiated. Matching the right drug with each individual is important because a particular treatment will benefit only the subset of patients in whom the mechanism the drug targets is active. Thus, the ability to identify the individuals most likely to respond to a particular therapeutic would greatly benefit patients, preventing those who would not benefit from experiencing drug-related adverse events and incurring the costs of a treatment that is ineffective. The benefits of a targeted approach to therapy would also have considerable societal implications: improved drug development, achieved through the testing of candidate therapeutics only in patients likely to benefit,30,31 would optimize use of limited health-care resources, and a decrease in adverse effects of treatment would improve patient outcomes.
Biomarkers predictive of responsiveness to therapy have already proven invaluable in the treatment of several types of cancer. For example, expression of the estrogen receptor is indicative of responsiveness to hormonal therapy in breast cancer,32 the presence of the BCR–ABL1 translocation indicates responsiveness to the tyrosine kinase inhibitor imatinib in chronic myelocytic leukemia,33 and overexpression of the receptor tyrosine-protein kinase erbB-2 (also known as HER2) indicates responsiveness to specific anti-erbB-2 monoclonal antibodies (trastuzumab, pertuzumab)) and to tyrosine kinase inhibitors (lapatinib) in breast cancer.34 Other biomarkers that enable targeted therapy are specific mutations in the KRAS gene, which in lung and colorectal cancers indicate a lack of responsiveness to tyrosine kinase inhibitors targeting the epidermal growth factor receptor.35 Although most rheumatic diseases are more molecularly heterogeneous than the malignancies discussed, evidence suggests that ACPA could serve as biomarkers of responsiveness to B-cell-depletion therapy in patients with RA; however, the association is not as strong as for the described genetic mutations in cancer.36,37
In addition to their use in clinical practice, biomarkers predictive of responsiveness to therapy have great potential in improving the drug development process. As we have alluded to previously, predictive biomarkers can reduce the size of the cohorts needed in clinical trials by enabling selective recruitment of participants who are likely to benefit from the intervention being tested, thereby substantially reducing costs, streamlining clinical development, and, importantly, reducing exposure of individuals unlikely to respond. However, considerable costs are associated with the development of a ‘companion diagnostic’ biomarker for clinical trials that accurately identifies patients who are likely to respond to treatment; if a clear mechanistic biomarker is not available, the added costs and logistical and regulatory complexity can, in certain cases, outweigh the potential benefits. Nevertheless, the FDA has undertaken initiatives to facilitate the collaborative development of biomarkers and thereby lessen the resource burden on any individual, organization or company working towards qualification of a biomarker for use in drug development.98 Thus, as the cost of drug development soars,38 we believe that integrated use of predictive and pharmacodynamic biomarkers will probably be an important strategy for controlling costs and expediting clinical development programs.
Descriptive and mechanistic biomarkers
Biomarkers do not need to be directly involved in disease pathogenesis to be useful, though a biomarker is likely to be more informative if it has some mechanistic involvement. For example, ESR and CRP are components of the disease activity score (DAS),39 which is used in assessing disease activity in RA. Nevertheless, ESR and CRP are not specific to RA: they are markers of inflammation that are also associated with many other rheumatic and nonrheumatic inflammatory disorders, including infection, malignancy and even coronary artery disease. Such biomarkers are referred to as descriptive biomarkers because they describe the state of a disease but are not involved in the pathogenesis of the disease. Many descriptive biomarkers are associated with a disease because they are products of the disease process or of disease-induced damage; such factors are byproducts rather than intrinsic players in disease pathogenesis. Therefore, only limited pharmacodynamic, diagnostic or prognostic information can be derived from descriptive biomarkers,17 restricting their usefulness.
The most informative actionable biomarkers are rooted in the mechanism underlying the disease (Figure 2). Mechanistic biomarkers are superior to descriptive biomarkers for a number of reasons (Figure 3). First, a biomarker directly involved in the pathogenesis of the disease is more likely to be specific to that disease, compared with a descriptive biomarker that is a byproduct of the disease process, and therefore performs better in the differential diagnosis of disease. Second, mechanistic biomarkers often enable differentiation of distinct subtypes of the same disease and can, therefore, be used to stratify disease and target treatment. Third, a pharmacodynamic biomarker that is mechanistic can reliably reveal whether a therapy is efficaciously targeting the cause of a disease, rather than simply improving the symptoms of a disease, and thus represents the most useful type of biomarker for informing the development and expediting the assessment of rationally designed, mechanism-based therapies.
Figure 2.

Mechanistic biomarkers of autoimmune diseases. Mechanistic biomarkers can take the form of several different molecules or cell types that have distinct roles in the pathogenesis of autoimmune diseases. They can be cytokines secreted from many types of immune cells at different stages in the disease process that induce pathogenic cell signalling and the recruitment of additional immune cells. The immune cells themselves can serve as mechanistic biomarkers; dendritic cells activate T cells, which can be pathogenic by inducing cytotoxicity or by helping B cells to produce antibodies. T cells and macrophages, in particular, are major sources of pathogenic, inflammatory cytokines. Further along in the development of autoimmunity, autoantibodies produced by activated B cells can contribute to disease pathogenesis, thus representing mechanistic biomarkers, by triggering the complement cascade or by forming antigen-containing immune complexes that induce inflammatory cell signalling. Cytokines, autoantibodies, cell-surface receptors, and other stimuli activate distinct cell signalling pathways and molecules, which can also serve as mechanistic biomarkers. Cell signalling turns on or shuts down specific genes and, therefore, profiles of gene expression can likewise serve as mechanistic biomarkers.
Figure 3.
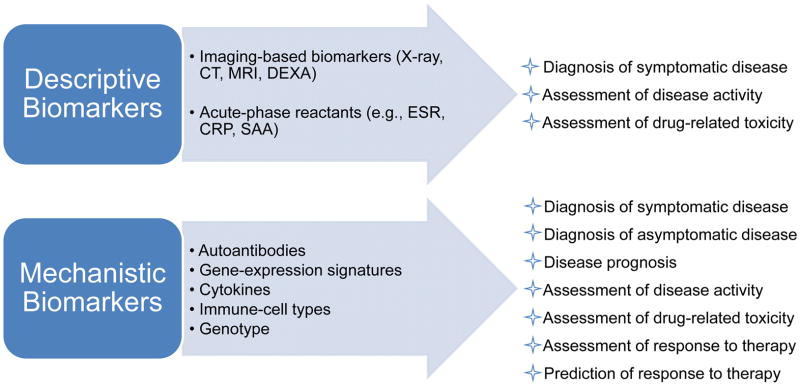
Types and uses of descriptive and mechanistic biomarkers for autoimmune rheumatic diseases. Mechanistic biomarkers are embedded in the pathogenesis of the disease and, thus, the biomarkers are generally more informative and more accurately reflect the disease state compared with descriptive biomarkers, which are byproducts of the disease process. The predictive or dynamic nature of mechanistic biomarkers offers clear advantages for disease diagnosis, prognosis and management, as is clear from the list of their uses. Genetic traits could serve as mechanistic biomarkers that are useful for diagnosis, prognosis or prediction of responsiveness, but not for assessment of disease progression or response to therapy; that is, they cannot serve as dynamic biomarkers. Abbreviations: DEXA, dual-energy X-ray absorptiometry; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; SAA, serum amyloid A.
In recognition of the importance of mechanistic biomarkers in drug development, increasing effort is put into integration of molecular diagnostics with therapeutics technologies.40 Researchers have even begun performing pathway-based biomarker discovery. Whereas the traditional paradigm of biomarker discovery involves seeking variables associated with a disease or clinical outcome and then evaluating the biological plausibility of candidates identified, in the pathway-based approach a signature of a pathway or process thought to be involved in the disease is tested for association with the disease or clinical outcome.41 For example, a pathway-based approach was used in identifying a gene-expression biomarker that can predict survival of individual women with breast cancer;42 in this case, wound healing was the mechanism assumed to be important (Table 1).42 A gene-expression signature of wound healing was experimentally derived in vitro, and a correlation score was developed for assessing how closely the gene-expression profile of a tumor specimen matched the wound-healing signature.42 This correlation score was able to accurately predict which women with breast cancer did not need adjuvant chemotherapy.41,42 Another example of pathway-based biomarker discovery is the derivation of a gene-expression signature in skin biopsies that identifies cases of systemic sclerosis that are driven by the tyrosine kinases PDGF and Abl and are, therefore, likely to respond to treatment with the tyrosine kinase inhibitor imatinib.43
Table 1.
Examples and uses of mechanistic biomarkers for immune-related diseases and cancer
| Type of Biomarker | Biomarker | Use of Biomarker | Development Stage |
|---|---|---|---|
| Cytokine | IL-7 | Prediction of responsiveness to IFN-β in MS | Retrospective testing in samples from clinical trials45 |
| Chemokine | CXCL13 | Assessment of disease activity in RA Prediction of disease progression in RA Assessment of response to TNF blockade Prediction of the rate of B-cell repopulation after rituximab therapy |
All uses are undergoing retrospective testing in samples from clinical trials55,56,57,58 |
| Cell type | TH1 cells, TH17 cells | Prediction of responsiveness to IFN-β in MS | Retrospective testing in samples from clinical trials44 |
| Autoantibodies | ACPA | Diagnosis of symptomatic RA Diagnosis of asymptomatic RA |
Clinical practice Retrospective testing in samples from clinical studies10 |
| ANA | Diagnosis of symptomatic SLE | Clinical practice | |
| Signalling molecule | ErbB2 | Prediction of responsiveness to anti-ErbB2 monoclonal antibodies and to tyrosine kinase inhibitors in breast cancer | Clinical practice |
| Gene expression | Wound-healing signature | Identification of women with breast cancer who do not need adjuvant chemotherapy | Retrospective testing in samples from clinical studies42 |
| IFN-α/β-inducible gene signature | Assessment of the efficacy of anti-IFNα monoclonal antibody therapy in neutralizing IFN-α and downstream signalling in SLE | Prospective testing in clinical trials26 | |
| Imatinib-responsive gene signature | Identification of systemic sclerosis patients most likely to respond to treatment with the tyrosine kinase inhibitor imatinib | Retrospective testing in samples from clinical trials43 |
Abbreviations: ACPA, anti-citrullinated protein antibodies; ANA, anti-nuclear antibodies; CXCL13, C-X-C motif chemokine 13; ErbB2, Receptor tyrosine-protein kinase erbB-2; MS, multiple sclerosis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; TH1, type 1 T helper (cell); TH17, type 17 T helper (cell).
Clear examples of mechanistic biomarkers include those we have discussed: the BCR–ABL1 translocation in chronic myelocytic leukemia, and the overexpression of the erbB-2 in breast cancer. Rooted in the disease mechanism, these biomarkers not only stratify disease but also provide a basis for selecting mechanism-based therapies. Allergy skin testing is another example of the use of mechanistic biomarkers: known allergens are injected subcutaneously, and development of an immune response to one of the allergens indicates that the person being tested is allergic to that particular allergen. Thus, the controlled immune response serves as a mechanistic biomarker of a specific allergy and provides information that can guide the development of antigen-specific tolerizing immunotherapy. The current status of mechanistic biomarkers for rheumatic diseases is discussed in the following sections.
Mechanistic biomarkers in rheumatology
Cytokines and chemokines
A clinical disease category (RA or SLE, for example) often comprises several distinct disease subtypes that can differ subtly in clinical presentation but markedly in molecular phenotype. Understanding the molecular pathogenesis of disease is essential for development of mechanistic biomarkers, a concept illustrated by findings of research in multiple sclerosis (MS). Although a common treatment for MS, IFN-β is not efficacious in 30–50% of cases. A recent study showed that a form of mouse MS driven by type 1 T helper (TH1) cells responded to IFN-β treatment, whereas a form of the disease driven by type 17 T helper (TH17) cells did not and was, in fact, exacerbated by the treatment.44 Moreover, a follow-on study showed that IL-7 promoted TH1-cell-driven, but not TH17-cell-driven, autoimmune demyelinating disease, and that high levels of IL-7 in the blood of individuals with relapsing remitting MS were predictive of responsiveness to IFN-β therapy.45 Thus, identifying the molecular mechanisms of disease revealed biomarkers directly involved in pathogenesis of the disease that might prove useful in guiding personalized clinical care.
In rheumatology too, efforts are increasingly being made to use our advancing knowledge of molecular pathogenesis to identify mechanistic biomarkers (Figure 2). This approach has been used to search for biomarkers for autoinflammatory diseases known to be driven by IL-1, such as familial Mediterranean fever, Muckle–Wells syndrome and the related cryopyrin-associated periodic fever syndromes, and systemic onset juvenile idiopathic arthritis.46 However, neither levels of IL-1 in the blood nor levels of IL-1 derived from peripheral blood mononuclear cells (PBMCs) activated ex vivo can accurately predict the response of these diseases to IL-1 blocking therapy;47 biomarkers downstream of IL-1, such as IL-1-induced transcriptional profiles,48 are now being assessed as potential biomarkers for these autoinflammatory diseases.
More advanced is the search for mechanistic biomarkers for SLE, which centres on type I IFNs. Type I IFNs have an important role in SLE pathogenesis,49 and individuals with SLE have abnormally high levels of type I IFNs in their blood,50 as well as a signature of type I IFN-associated gene expression in their circulating immune cells.51 Measuring type I IFN directly is challenging because many different IFN isoforms exist; therefore, levels of transcripts induced by type I IFNs are measured as surrogates for the levels of type I IFN. Increased levels of these transcripts are associated with SLE disease activity.52 Indeed, transcriptional profiles of genes induced by type I IFNs are already being used as surrogates of disease activity in early-phase clinical trials26 and could prove to be actionable, mechanistic biomarkers for SLE.
Another cytokine that could serve as a mechanistic biomarker is C-X-C motif chemokine 13 (CXCL13; also known as B lymphocyte chemoattractant; Table 1). CXCL13 has an essential role in organizing germinal centres, and high expression of CXCL13 mRNA in the inflamed RA synovium is a strong predictor of the presence of germinal centres in this tissue,53 suggesting that CXCL13 contributes to the autoimmune synovitis in RA. In addition to B cells, osteoblasts express the receptor for CXCL13, and activation of this receptor induces osteoblasts to release extracellular-matrix-degrading enzymes,54 suggesting that CXCL13 also contributes to bone remodeling in RA joints. Indeed, in silico modeling of an RA joint, taking into account synovitis, cartilage destruction and bone erosion, identified CXCL13 as a candidate prognostic biomarker of erosiveness,55 further implicating CXCL13 in the pathogenesis of RA. That CXCL13 might be a useful mechanistic biomarker has so far been borne out by the findings of studies in patients with RA. The findings suggest that high levels of serum CXCL13, which positively correlate with levels of synovial CXCL13 expression,56 could serve not only as a biomarker of active disease but also as a biomarker predictive of severe RA.55,57,58 Moreover, assessment of the usefulness of CXCL13 as a pharmacodynamic biomarker showed that serum levels of CXCL13 decreased after TNF blockade, correlating positively with changes in DAS28 (disease activity score using 28 joint counts).58 Finally, high levels of serum CXCL13 were predictive of a faster rate of B-cell repopulation after rituximab therapy in patients with RA.56 Whether CXCL13 will prove useful in the clinic or in drug development depends on whether levels of this chemokine can be reliably detected in blood samples and on its sensitivity and specificity as a biomarker.
Autoantibodies
Autoantibodies are emerging as useful, possibly mechanistic, biomarkers for autoimmune rheumatic diseases. Autoantibodies that bind to and form immune complexes with DNA, RNA or chromatin autoantigens implicated in SLE augment type I IFN production in plasmacytoid dendritic cells by providing a second stimulatory signal that synergizes with the signal delivered by the autoantigen. The autoantibodies in the immune complexes activate Fcγ receptors on the cell surface, after which the autoantigens are internalized and directly activate Toll-like receptor (TLR) 9 (in the case of a DNA autoantigen) or TLR7 (in the case of an RNA autoantigen).59–64 Indeed, autoantibodies that target RNA-binding proteins (such as Ro and La antigens, and U1 small nuclear ribonucleoprotein A) or DNA are associated with increased serum IFN-α activity—determined using an in vitro reporter-cell-based assay of IFN-induced gene expression)—in patients with SLE.65 Autoantibodies in immune complexes can likewise augment the ability of the corresponding autoantigen to activate autoreactive B cells by triggering B-cell receptor signaling, which synergizes with TLR9 or TLR7 signalling in inducing the production of autoantibodies.62
In RA, ACPA autoantibodies target a wide variety of citrullinated antigens, including citrullinated fibrinogen. Although citrullinated fibrinogen alone can induce the production of the inflammatory cytokine TNF by activating TLR4, citrullinated fibrinogen bound to autoantibodies induces macrophage TNF production more effectively through the synergistic activation of both TLR4 and Fcγ receptors.66 Together, these findings suggest that autoantibodies targeting immunologically active autoantigens (such as DNA, RNA, chromatin, citrullinated fibrinogen) could be pathogenic by augmenting the activation of specific molecular pathways that underlie disease pathogenesis. Thus, such autoantibodies could themselves be considered mechanistic biomarkers.
If such autoantibodies are found to contribute to disease pathogenesis, autoantibody profiling in at-risk individuals could be useful for diagnosis of disease before the onset of symptoms (Table 1). Indeed, autoantibodies are present in the blood long before the clinical onset of many autoimmune diseases, including SLE,7 MS,8 type 1 diabetes mellitus,67–69 and RA.70,71 Likewise, increases in the levels of cytokines72,73 and acute-phase reactants74 in the blood occur before the onset of clinical disease in RA. Moreover, accumulating evidence indicates that the range of different autoantibodies present and levels of specific autoantibodies increase as the onset of clinical disease approaches.8,10,68
Besides aiding in the prediction of clinical disease onset, profiling pathogenic autoantibodies can be used to guide therapy. Autoantibody profiling can identify the critical antigens targeted by the disease-associated immune response and thereby guide the development of tolerizing therapies, an approach that has been used for MS.75,76 Autoantibody profiling could also guide early or preventive intervention; in RA, for example, methotrexate treatment markedly decreased the incidence of progression from undifferentiated arthritis to clinical RA in a randomized, placebo-controlled trial, but this effect was only in the ACPA+ population.77 Thus, profiling the specificity of ACPA represents an example of an actionable, mechanistic biomarker that could guide early or even preventive intervention in RA. Furthermore, identifying the specific ACPA present during the asymptomatic phase10,72,73 could not only improve the identification of those at risk of developing clinical RA but also pinpoint the time closest to the onset of clinical RA—a time at which the disease might be most amenable to immunomodulatory intervention. Finally, one can envision performing diagnostic tests for autoantibodies targeting immunologically active autoantigens and then using this information to select a therapy that targets the downstream mediators of the specific pathways activated in an individual patient (for example, therapies targeting IFN-α, TNF, IL-6, IL-1, IL-17, or IL-12p40).
Not only autoantibodies but also anti-drug antibodies (ADA) could serve as mechanistic biomarkers. Certain individuals develop ADA against biologic therapeutics;78 depending on their specificity, these ADA can neutralize or otherwise interfere with the activity of the biologic agent, affect the rate at which the drug is cleared from the body, or trigger serious adverse effects by cross-reacting with self proteins. Biologically active ADA could thus serve as mechanistic, pharmacodynamic biomarkers that are an early sign of a lack of response or of an adverse response to a biologic therapeutic.
MicroRNAs
An emerging concept is the potential of microRNAs (miRNAs) to serve as biomarkers for rheumatic diseases.79,80 A single miRNA can regulate the translation of multiple genes and thus have far-reaching biological effects. Of particular interest in rheumatology is miR-146a, which has important roles in the control of inflammation and immunity. Expression of miR-146a is induced during activation of T cells, in which it suppresses apoptosis and IL-2 production.81 A reduction in apoptosis of inflammatory cells, including T cells, is a feature of RA,82 and miR-146a expression is, in fact, upregulated in IL-17-expressing T cells, macrophages and B cells in the RA synovium.83,84 Moreover, miR-146a expression is upregulated in PBMCs of individuals with RA,84,85 and miR-146a expression was found to be increased in the synovium of individuals with greater disease activity in a study that enrolled a small number of patients with RA (n = 6).84 miR-146a is also overexpressed in PBMCs from individuals with Sjögren’s syndrome.86 Conversely, miR-146a expression is downregulated in PBMCs from patients with SLE, and the level of this microRNA in these cells correlates inversely with disease activity and with the expression of IFN-inducible genes implicated in SLE pathogenesis.87
Another miRNA that shows promise as a biomarker is miR-155. This miRNA promotes the development of inflammatory TH1 cells and TH17 cells and T-cell-dependent tissue inflammation,88 key processes in the pathogenesis of RA. Indeed, miR-155 knockout mice were resistant to development of collagen-induced arthritis, producing markedly fewer autoreactive B cells and T cells in response to immunization with collagen, and were partially protected from bone erosion in the K/BxN serum-transfer model of RA due to a decrease in osteoclast formation.89 Furthermore, miR-155 expression is upregulated in PBMCs from patients with RA.83,84 Together, these findings suggest that miRNAs might prove to be mechanistic biomarkers of rheumatic diseases, and increasing efforts in this area are uncovering further miRNAs that could fulfill such a function.80 Nevertheless, the clinical utility provided by miRNA biomarkers will depend on their predictive value and whether they can be reliably detected in blood samples.
Future of biomarkers in rheumatology
Despite the current paucity of definitive mechanistic biomarkers for rheumatic diseases, a deeper understanding of disease pathogenesis is starting to uncover putative mechanistic biomarkers (Table 1), as well as identifying mechanistic roles for certain known biomarkers. In the future, biomarkers might help to establish a molecular taxonomy of diseases that are currently classified according to phenotype alone. Most rheumatic diseases are currently diagnosed on the basis of medical history, clinical findings, and basic laboratory tests, all of which reflect the phenotype but not the underlying molecular pathology. Given the wide heterogeneity in disease course, disease-related damage and response to therapy, defining the molecular features of rheumatic diseases, and thus establishing a molecular taxonomy of the diseases and their subtypes, is essential. Among rheumatic diseases, molecular characterization is perhaps most advanced in RA, which can already be divided into ACPA− and ACPA+ subtypes, with the latter generally representing the more severe form of disease.90 As a means of identifying further molecular markers capable of classifying and stratifying rheumatic diseases, the field has begun to uncover the primary inflammatory drivers of such conditions and their subtypes. For example, IL-1 is the main driver of inflammasome-mediated diseases, including several autoinflammatory diseases, gout and possibly pseudogout, and these conditions are responsive to IL-1 antagonism.91 By contrast, RA is only minimally responsive to IL-1 antagonism and less than half of patients with RA respond to TNF antagonism.
In addition to inflammatory cytokines, different immune cell types can also distinguish different subtypes of the same clinical disease. Focusing on cell types as stratifying biomarkers is a relatively new area of biomarker research, best exemplified by the classification of MS according to whether disease is driven by TH1 cells or by TH17 cells.44 Although IFN-β treatment (which is effective in TH1-cell-driven but not TH17-cell-driven MS)44 improved disease outcomes in TH1-cell-driven mouse models of RA, this therapeutic was, however, not efficacious in clinical trials of patients with RA,92 suggesting that other molecular pathways are driving pathogenesis in this disease. SLE might, likewise, comprise different subtypes distinguished by the involvement of distinct immune cell types. As the main producers of type I IFN, plasmacytoid dendritic cells make an important contribution to the pathogenesis of SLE; however, T cells, B cells and neutrophils are also involved, such that the type I IFN signature might serve as an actionable biomarker only in a subset of patients with SLE.93
A growing area of biomarker research in rheumatology, and particularly in RA, is the search for biomarkers that can predict successful drug-free remission. An increasing number of patients with RA achieve long-term clinical remission whilst being treated with DMARDs in clinical trials, and focus is increasingly placed on determining whether such patients can remain in remission once they stop taking these therapeutics—that is, whether they can achieve ‘true’ drug-free remission. Findings suggest that drug-free remission is achieved in 17–29% of patients with RA and that most patients who have to restart medication are able to once again achieve medicated remission.94 Nonetheless, because some patients do not achieve remission upon restarting medication, predictors of successful drug-free remission are needed. At present, ACPA negativity, shared-epitope negativity and short duration of symptoms before initiation of treatment have been identified as predictors of drug-free remission in RA, but longer follow-up after treatment cessation is needed and could unearth additional and better predictive biomarkers.94
Stratifying diseases classified according to phenotype is not the only way that biomarkers can be used to forge a molecular taxonomy of disease: they can do so also by breaking down the boundaries of current classifications. That is, biomarkers can be used to uncover molecular similarities between diseases thought to be distinct. For instance, the type I IFN signature is associated not only with SLE, but also with dermatomyositis and polymyositis,95 Sjögren’s syndrome,96 and some cases of scleroderma.97 Thus, this signature might serve as an actionable biomarker for multiple autoimmune pathologies.
Conclusions
For each of the rheumatic diseases, great opportunity exists for identifying actionable biomarkers, whether based on imaging, profiling of autoantibodies, measuring levels of inflammatory mediators or other molecular analyses. The shortcomings of many biomarkers currently used in the diagnosis and management of the rheumatic diseases, at least relative to those available for certain cancers discussed in this Review, is that they are not related to the underlying disease mechanism. Thus, stratifying current clinical classifications and identifying molecular pathways that mediate the pathogenesis of disease represents an important first step towards defining a new molecular taxonomy of disease and the subsequent identification of diagnostic, predictive and prognostic mechanistic biomarkers. We anticipate that involvement of certain molecular pathways will be shared across subsets of multiple different rheumatic diseases, whereas other pathways will be disease-specific. Molecular classification of disease could enable the identification of disease subtypes that are responsive to specific therapeutics and eventually the use of patient-derived biomarkers for guiding targeted therapy.
As a field, rheumatology has long been at the forefront of biomarker discovery; some of the biomarkers identified will prove to be descriptive whereas others could prove to be mechanistic. Nevertheless, as our understanding of the molecular immunology of rheumatic disease progresses, we envision a future with biomarker-based molecular subtyping of disease that can guide clinical decision making.
Key points.
Biomarkers can aid in the management of disease by helping to diagnose and stratify disease, as well as assess or predict disease severity or response to therapy.
Biomarkers can aid in drug development by enabling selective recruitment of individuals likely to benefit from the intervention being tested and/or rapid assessment of response to a candidate therapeutic.
Biomarkers rooted in the mechanism underlying the disease (mechanistic biomarkers) are likely to be more useful than those that are byproducts of the disease process (descriptive biomarkers).
Compared to descriptive biomarkers, mechanistic biomarkers are more likely to perform better in differential diagnosis of disease, disease stratification and targeting of treatment, and as surrogate endpoints in clinical trials.
Mechanistic biomarkers for rheumatic diseases may include cytokines, chemokines, autoantibodies, microRNAs, gene-expression profiles, and immune cell types.
Mechanistic biomarkers might help to establish a molecular taxonomy of diseases.
Acknowledgments
The work W. H. Robinson is supported by N01-HV-00242/HV/NHLBI NIH HSS/United States, RC1 AR058713/AR/NIAMS NIH HHS/United States, R01 AR-054822/AR/NIAMS NIH HHS/United States, and U01 U01AI101981/NIAID NIH HSS/United States grants, and Veterans Affairs Health Care System funding. J. Sokolove receives salary support from an American College of Rheumatology Research and Education Foundation Physician Scientist Development Award and a Veterans Affairs Career Development 2 Award.
Biographies
WILLIAM H. ROBINSON
Dr William H. Robinson received his MD and PhD from Stanford University, Stanford, CA, USA and completed his residency training in internal medicine at the University of California San Francisco, San Francisco, CA, USA. His laboratory pioneered the development and application of protein arrays, lipid arrays, and, most recently, high-throughput sequencing approaches. He holds a joint appointment at the Department of Veterans Affairs Palo Alto Health Care System, Palo Alto, CA, USA.
TAMSIN M. LINDSTROM
Dr Tamsin M. Lindstrom completed a BSc in Biology at the University of Nottingham (Nottingham, UK) and a PhD in Reproductive Sciences at Imperial College London (London, UK). She spent one year at GlaxoSmithKline (Stevenage, UK) in the Departments of Virology and Respiratory Disease. Dr Lindstrom is currently a Research Associate working with Professor William Robinson’s laboratory at Stanford University School of Medicine (Stanford, CA, USA).
REGINA K. CHEUNG
Regina K. Cheung is a PhD student in the Immunology Program at Stanford University School of Medicine. Ms Cheung is a member of P. J. Utz’s and Garry Nolan’s laboratories, where she has expertise in autoimmunity, lymphocyte signaling, and bioengineering. Ms Cheung’s thesis work aims to discover and characterize plasmacytoid dendritic cell subsets.
JEREMY SOKOLOVE
Dr. Sokolove received his undergraduate degree from the University of New Hampshire and his medical degree from Boston University. He completed rheumatology fellowship at Stanford University, and is now an Instructor in Medicine at Stanford University and a Staff Physician and the Department of Veterans Affairs Palo Alto Health Care System. His laboratory group is investigating the role of citrullination in inducing inflammatory and autoimmune responses.
Footnotes
Author contributions
All authors contributed to the preparation of this manuscript for publication.
Competing interests
In print:
W. H. Robinson has declared associations with the following companies: Bio-Rad Laboratories; Crescendo Biosciences; Roche Diagnostics; Genentech. See the article online for full details of the relationships. All other authors declare no competing interests.
Online:
W. H. Robinson is an inventor on patents owned by Stanford University that have been licensed to, or for which option agreements to license have been taken by, Bio-Rad Laboratories, Crescendo Biosciences and Roche Diagnostics, and has received research support from Genentech and Roche Diagnostics. All other authors declare no competing interests
Contributor Information
William H. Robinson, VA Palo Alto Health Care System, 3801 Miranda Ave, Palo Alto, CA 94304 USA
Tamsin M. Lindstrom, Division of Immunology and Rheumatology, Stanford University School of Medicine, CCSR 4135, 269 Campus Dr, Stanford, CA USA
Regina K. Cheung, Division of Immunology and Rheumatology, Stanford University School of Medicine, CCSR 4135, 269 Campus Dr, Stanford, CA USA
Jeremy Sokolove, VA Palo Alto Health Care System, 3801 Miranda Ave, Palo Alto, CA 94304 USA.
References
- 1.Atkinson AJ, et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 2.Logothetis NK. What we can do and what we cannot do with fMRI. Nature. 2008;453:869–878. doi: 10.1038/nature06976. [DOI] [PubMed] [Google Scholar]
- 3.Fu Q, Schoenhoff FS, Savage WJ, Zhang P, Van Eyk JE. Multiplex assays for biomarker research and clinical application: translational science coming of age. Proteomics Clin Appl. 2010;4:271–284. doi: 10.1002/prca.200900217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011;365:1612–1623. doi: 10.1056/NEJMra1100030. [DOI] [PubMed] [Google Scholar]
- 5.Walsh GM. Canakinumab for the treatment of cryopyrin-associated periodic syndromes. Drugs Today (Barc) 2009;45:731–735. doi: 10.1358/dot.2009.45.10.1416583. [DOI] [PubMed] [Google Scholar]
- 6.Karlson EW, et al. Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis. 2010;69:54–60. doi: 10.1136/ard.2008.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arbuckle MR, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 8.Berger T, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. 2003;349:139–145. doi: 10.1056/NEJMoa022328. [DOI] [PubMed] [Google Scholar]
- 9.Soeldner JS, Tuttleman M, Srikanta S, Ganda OP, Eisenbarth GS. Insulin-dependent diabetes mellitus and autoimmunity: islet-cell autoantibodies, insulin autoantibodies, and beta-cell failure. N Engl J Med. 1985;313:893–894. doi: 10.1056/NEJM198510033131417. [DOI] [PubMed] [Google Scholar]
- 10.Sokolove J, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS ONE. 2012;7:e35296. doi: 10.1371/journal.pone.0035296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klarenbeek NB, et al. The impact of four dynamic, goal-steered treatment strategies on the 5-year outcomes of rheumatoid arthritis patients in the BeSt study. Ann Rheum Dis. 2011;70:1039–1046. doi: 10.1136/ard.2010.141234. [DOI] [PubMed] [Google Scholar]
- 12.Guermazi A, et al. Assessment of synovitis with contrast-enhanced MRI using a whole-joint semiquantitative scoring system in people with, or at high risk of, knee osteoarthritis: the MOST study. Ann Rheum Dis. 2011;70:805–811. doi: 10.1136/ard.2010.139618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunter DJ, et al. The reliability of a new scoring system for knee osteoarthritis MRI and the validity of bone marrow lesion assessment: BLOKS (Boston Leeds Osteoarthritis Knee Score) Ann Rheum Dis. 2008;67:206–211. doi: 10.1136/ard.2006.066183. [DOI] [PubMed] [Google Scholar]
- 14.Peterfy CG, et al. Whole-Organ Magnetic Resonance Imaging Score (WORMS) of the knee in osteoarthritis. Osteoarthritis Cartilage. 2004;12:177–190. doi: 10.1016/j.joca.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Blanco FJ, Ruiz-Romero C. Osteoarthritis: metabolomic characterization of metabolic phenotypes in OA. Nat Rev Rheumatol. 2012;8:130–132. doi: 10.1038/nrrheum.2012.11. [DOI] [PubMed] [Google Scholar]
- 16.Hoch JM, Mattacola CG, Medina McKeon JM, Howard JS, Lattermann C. Serum cartilage oligomeric matrix protein (sCOMP) is elevated in patients with knee osteoarthritis: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2011;19:1396–1404. doi: 10.1016/j.joca.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Leeuwen MA, et al. The acute-phase response in relation to radiographic progression in early rheumatoid arthritis: a prospective study during the first three years of the disease. Br J Rheumatol. 1993;32 (Suppl 3):9–13. doi: 10.1093/rheumatology/32.suppl_3.9. [DOI] [PubMed] [Google Scholar]
- 18.Eastman PS, et al. Characterization of a multiplex, 12-biomarker test for rheumatoid arthritis. J Pharm Biomed Anal. 2012;70:415–424. doi: 10.1016/j.jpba.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Ludwig JA, Weinstein JN. Biomarkers in cancer staging, prognosis and treatment selection. Nat Rev Cancer. 2005;5:845–856. doi: 10.1038/nrc1739. [DOI] [PubMed] [Google Scholar]
- 20.Kasitanon N, Petri M, Haas M, Magder LS, Fine DM. Mycophenolate mofetil as the primary treatment of membranous lupus nephritis with and without concurrent proliferative disease: a retrospective study of 29 cases. Lupus. 2008;17:40–45. doi: 10.1177/0961203307085114. [DOI] [PubMed] [Google Scholar]
- 21.Syversen SW, et al. Biomarkers in early rheumatoid arthritis: longitudinal associations with inflammation and joint destruction measured by magnetic resonance imaging and conventional radiographs. Ann Rheum Dis. 2010;69:845–850. doi: 10.1136/ard.2009.122325. [DOI] [PubMed] [Google Scholar]
- 22.Hama M, et al. Power Doppler ultrasonography is useful for assessing disease activity and predicting joint destruction in rheumatoid arthritis patients receiving tocilizumab—preliminary data. Rheumatol Int. 2012;32:1327–1333. doi: 10.1007/s00296-011-1802-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.US Department of Health and Human Services. Draft guidance for industry on qualification process for drug development tools. 2010 [online], http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM230597.pdf.
- 24.Woodcock J, Woosley R. The FDA critical path initiative and its influence on new drug development. Annu Rev Med. 2008;59:1–12. doi: 10.1146/annurev.med.59.090506.155819. [DOI] [PubMed] [Google Scholar]
- 25.Chang DM, Weinblatt ME, Schur PH. The effects of methotrexate on interleukin 1 in patients with rheumatoid arthritis. J Rheumatol. 1992;19:1678–1682. [PubMed] [Google Scholar]
- 26.Haringman JJ, et al. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:834–838. doi: 10.1136/ard.2004.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao Y, et al. Neutralization of interferon-α/β-inducible genes and downstream effect in a phase I trial of an anti-interferon-α monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1785–1796. doi: 10.1002/art.24557. [DOI] [PubMed] [Google Scholar]
- 28.Farina G, Lafyatis D, Lemaire R, Lafyatis R. A four-gene biomarker predicts skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2010;62:580–588. doi: 10.1002/art.27220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arrowsmith J. Trial watch: phase II failures: 2008–2010. Nat Rev Drug Discov. 2011;10:328–329. doi: 10.1038/nrd3439. [DOI] [PubMed] [Google Scholar]
- 30.Arrowsmith J. Trial watch: phase III and submission failures: 2007–2010. Nat Rev Drug Discov. 2011;10:87. doi: 10.1038/nrd3375. [DOI] [PubMed] [Google Scholar]
- 31.Finkielman JD, et al. ANCA are detectable in nearly all patients with active severe Wegener’s granulomatosis. Am J Med. 2007;120:643.e9–e14. doi: 10.1016/j.amjmed.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 32.Hueber W, et al. Blood autoantibody and cytokine profiles predict response to anti-tumor necrosis factor therapy in rheumatoid arthritis. Arthritis Res Ther. 2009;11:R76. doi: 10.1186/ar2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higgins MJ, Baselga J. Targeted therapies for breast cancer. J Clin Invest. 2011;121:3797–3803. doi: 10.1172/JCI57152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ernst T, La Rosee P, Muller MC, Hochhaus A. BCR-ABL mutations in chronic myeloid leukemia. Hematol Oncol Clin North Am. 2011;25:997–1008. v–vi. doi: 10.1016/j.hoc.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 35.Sachdev JC, Jahanzeb M. Blockade of the HER family of receptors in the treatment of HER2-positive metastatic breast cancer. Clin Breast Cancer. 2012;12:19–29. doi: 10.1016/j.clbc.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Pao W, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen SB, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–2806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 38.Emery P, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–400. doi: 10.1002/art.21778. [DOI] [PubMed] [Google Scholar]
- 39.Dickson M, Gagnon JP. Key factors in the rising cost of new drug discovery and development. Nat Rev Drug Discov. 2004;3:417–429. doi: 10.1038/nrd1382. [DOI] [PubMed] [Google Scholar]
- 40.Fransen J, van Riel PL. The Disease Activity Score and the EULAR response criteria. Clin Exp Rheumatol. 2005;23:S93–S99. [PubMed] [Google Scholar]
- 41.Park JW, et al. Rationale for biomarkers and surrogate end points in mechanism-driven oncology drug development. Clin Cancer Res. 2004;10:3885–3896. doi: 10.1158/1078-0432.CCR-03-0785. [DOI] [PubMed] [Google Scholar]
- 42.Liu ET. Mechanism-derived gene expression signatures and predictive biomarkers in clinical oncology. Proc Natl Acad Sci USA. 2005;102:3531–3532. doi: 10.1073/pnas.0500244102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang HY, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung L, et al. Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheum. 2009;60:584–591. doi: 10.1002/art.24221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Axtell RC, et al. T helper type 1 and 17 cells determine efficacy of interferon-β in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee LF, et al. IL-7 promotes T(H)1 development and serum IL-7 predicts clinical response to interferon-β in multiple sclerosis. Sci Transl Med. 2011;3:93ra68. doi: 10.1126/scitranslmed.3002400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agostini L, et al. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 48.Gattorno M, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1505–1515. doi: 10.1002/art.23437. [DOI] [PubMed] [Google Scholar]
- 49.Allantaz F, et al. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med. 2007;204:2131–2144. doi: 10.1084/jem.20070070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niewold TB. Interferon alpha as a primary pathogenic factor in human lupus. J Interferon Cytokine Res. 2011;31:887–892. doi: 10.1089/jir.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hooks JJ, et al. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301:5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 52.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 53.Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006;54:1906–1916. doi: 10.1002/art.21890. [DOI] [PubMed] [Google Scholar]
- 54.Takemura S, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 55.Lisignoli G, et al. Human osteoblasts express functional CXC chemokine receptors 3 and 5: activation by their ligands, CXCL10 and CXCL13, significantly induces alkaline phosphatase and β-N-acetylhexosaminidase release. J Cell Physiol. 2003;194:71–79. doi: 10.1002/jcp.10188. [DOI] [PubMed] [Google Scholar]
- 56.Meeuwisse CM, et al. Identification of CXCL13 as a marker for rheumatoid arthritis outcome using an in silico model of the rheumatic joint. Arthritis Rheum. 2011;63:1265–1273. doi: 10.1002/art.30273. [DOI] [PubMed] [Google Scholar]
- 57.Rosengren S, Wei N, Kalunian KC, Kavanaugh A, Boyle DL. CXCL13: a novel biomarker of B-cell return following rituximab treatment and synovitis in patients with rheumatoid arthritis. Rheumatology (Oxford) 2011;50:603–610. doi: 10.1093/rheumatology/keq337. [DOI] [PubMed] [Google Scholar]
- 58.Bugatti S, et al. Serum levels of CXCL13 are associated with ultrasonographic synovitis and predict power Doppler persistence in early rheumatoid arthritis treated with non-biological disease-modifying anti-rheumatic drugs. Arthritis Res Ther. 2012;14:R34. doi: 10.1186/ar3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rioja I, et al. Potential novel biomarkers of disease activity in rheumatoid arthritis patients: CXCL13, CCL23, transforming growth factor alpha, tumor necrosis factor receptor superfamily member 9, and macrophage colony-stimulating factor. Arthritis Rheum. 2008;58:2257–2267. doi: 10.1002/art.23667. [DOI] [PubMed] [Google Scholar]
- 60.Boule MW, et al. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med. 2004;199:1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kelly KM, et al. “Endogenous adjuvant” activity of the RNA components of lupus autoantigens Sm/RNP and Ro 60. Arthritis Rheum. 2006;54:1557–1567. doi: 10.1002/art.21819. [DOI] [PubMed] [Google Scholar]
- 62.Lovgren T, et al. Induction of interferon-α by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjogren’s syndrome autoantigen-associated RNA. Arthritis Rheum. 2006;54:1917–1927. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 63.Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 64.Means TK, et al. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savarese E, et al. U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood. 2006;107:3229–3234. doi: 10.1182/blood-2005-07-2650. [DOI] [PubMed] [Google Scholar]
- 66.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 2011;63:53–62. doi: 10.1002/art.30081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van de Sande MG, et al. Different stages of rheumatoid arthritis: features of the synovium in the preclinical phase. Ann Rheum Dis. 2011;70:772–777. doi: 10.1136/ard.2010.139527. [DOI] [PubMed] [Google Scholar]
- 69.Verge CF, et al. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996;45:926–933. doi: 10.2337/diab.45.7.926. [DOI] [PubMed] [Google Scholar]
- 70.Ziegler AG, Hummel M, Schenker M, Bonifacio E. Autoantibody appearance and risk for development of childhood diabetes in offspring of parents with type 1 diabetes: the 2-year analysis of the German BABYDIAB Study. Diabetes. 1999;48:460–468. doi: 10.2337/diabetes.48.3.460. [DOI] [PubMed] [Google Scholar]
- 71.Nielen MM, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 72.Rantapaa-Dahlqvist S, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 73.Deane KD, et al. The number of elevated cytokines/chemokines in pre-clinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum. 2010;62:3161–3172. doi: 10.1002/art.27638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kokkonen H, et al. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010;62:383–391. doi: 10.1002/art.27186. [DOI] [PubMed] [Google Scholar]
- 75.Nielen MM, et al. Increased levels of C-reactive protein in serum from blood donors before the onset of rheumatoid arthritis. Arthritis Rheum. 2004;50:2423–2427. doi: 10.1002/art.20431. [DOI] [PubMed] [Google Scholar]
- 76.Robinson WH, et al. Protein microarrays guide tolerizing DNA vaccine treatment of autoimmune encephalomyelitis. Nat Biotechnol. 2003;21:1033–1039. doi: 10.1038/nbt859. [DOI] [PubMed] [Google Scholar]
- 77.Garren H, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol. 2008;63:611–620. doi: 10.1002/ana.21370. [DOI] [PubMed] [Google Scholar]
- 78.van Dongen H, et al. Efficacy of methotrexate treatment in patients with probable rheumatoid arthritis: a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2007;56:1424–1432. doi: 10.1002/art.22525. [DOI] [PubMed] [Google Scholar]
- 79.De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol. 2007;28:482–490. doi: 10.1016/j.it.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 80.Alevizos I, Illei GG. MicroRNAs as biomarkers in rheumatic diseases. Nat Rev Rheumatol. 2010;6:391–398. doi: 10.1038/nrrheum.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ceribelli A, et al. MicroRNAs in systemic rheumatic diseases. Arthritis Res Ther. 2011;13:229. doi: 10.1186/ar3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Curtale G, et al. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood. 2010;115:265–273. doi: 10.1182/blood-2009-06-225987. [DOI] [PubMed] [Google Scholar]
- 83.Pope RM. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nat Rev Immunol. 2002;2:527–535. doi: 10.1038/nri846. [DOI] [PubMed] [Google Scholar]
- 84.Nakasa T, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2008;58:1284–1292. doi: 10.1002/art.23429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Niimoto T, et al. MicroRNA-146a expresses in interleukin-17 producing T cells in rheumatoid arthritis patients. BMC Musculoskelet Disord. 2010;11:209. doi: 10.1186/1471-2474-11-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pauley KM, et al. Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther. 2008;10:R101. doi: 10.1186/ar2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pauley KM, et al. Altered miR-146a expression in Sjögren’s syndrome and its functional role in innate immunity. Eur J Immunol. 2011;41:2029–2039. doi: 10.1002/eji.201040757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tang Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60:1065–1075. doi: 10.1002/art.24436. [DOI] [PubMed] [Google Scholar]
- 89.O’Connell RM, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bluml S, et al. Essential role of microRNA-155 in the pathogenesis of autoimmune arthritis in mice. Arthritis Rheum. 2011;63:1281–1288. doi: 10.1002/art.30281. [DOI] [PubMed] [Google Scholar]
- 91.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 92.Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232–241. doi: 10.1038/nrrheum.2010.4. [DOI] [PubMed] [Google Scholar]
- 93.van Holten J, et al. A multicentre, randomised, double blind, placebo controlled phase II study of subcutaneous interferon beta-1a in the treatment of patients with active rheumatoid arthritis. Ann Rheum Dis. 2005;64:64–69. doi: 10.1136/ard.2003.020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 95.van den Broek M, Huizinga TW, Dijkmans BA, Allaart CF. Drug-free remission: is it already possible? Curr Opin Rheumatol. 2011;23:266–272. doi: 10.1097/BOR.0b013e32834563e3. [DOI] [PubMed] [Google Scholar]
- 96.Walsh RJ, et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56:3784–3792. doi: 10.1002/art.22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bave U, et al. Activation of the type I interferon system in primary Sjögren’s syndrome: a possible etiopathogenic mechanism. Arthritis Rheum. 2005;52:1185–1195. doi: 10.1002/art.20998. [DOI] [PubMed] [Google Scholar]
- 98.York MR, et al. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum. 2007;56:1010–1020. doi: 10.1002/art.22382. [DOI] [PubMed] [Google Scholar]