

Abstract
Much of current cancer research is aimed at exploiting cancers’ molecular addictions with targeted therapeutics, with notable successes documented in clinical trials. By their nature, these agents have different side effect profiles than conventional chemotherapeutics. While very few targeted agents have attained regulatory approval for use in children, pediatric oncologists are gaining experience with these drugs, which may have unique effects, both short- and long-term, on the developing child, unrecognized in adults. This Review summarizes the rationale for targeted therapy, challenges in pediatric drug development, unique side effect profiles of targeted agents, limited data from children treated with targeted agents, as well as implications of the current knowledge and gaps thereof. The demonstrated and potential impacts of targeted therapies on normal tissue development and function are discussed. We conclude that future clinical trial design should include carefully considered assessment of developmental effects of targeted therapy, as well as informed supportive care recommendations.
Keywords: targeted therapy, oncogene addiction, non-oncogene addiction, children, pediatric oncology, adverse effects, side effects
INTRODUCTION
Recent major advances in cancer therapy have come through the identification of molecular drivers of oncogenesis, and the subsequent suppression of their activity to inhibit the malignant phenotype of cancer cells. Such advancement has also provided critical new insight into normal cellular functions. Importantly, it is now clear that due to numerous interdependent pathways, the overlap between normal and abnormal gene activity may have clinical consequences. While inhibiting oncogenic activity is critical for effective targeted cancer therapy, if normal pathways are also affected, the potential for unanticipated or undesirable side effects also exists.
Targeting Cancers’ Addictions
A central hypothesis driving current basic and clinical oncology research is that of oncogene addiction (1), positing that single mutated oncogenes are necessary to both initiate and maintain the malignant behavior of cancer cells (Figure 1A). The corollary is that inhibition of the resultant altered protein will restrict the malignant behavior of cancerous cells, ideally resulting in cancer cell death. This has been most successfully proven with chronic myeloid leukemia (CML), a once uniformly fatal disease driven by the oncogenic fusion protein Bcr-Abl1. Now, almost 70% of patients remain in sustained remission, taking imatinib mesylate or other tyrosine kinase inhibitors (TKI) orally with relatively few side effects (2). Patients generally must remain on therapy long-term, though, as discontinuation typically leads to relapse. Nonetheless, this success fostered great hope that targeting a cancer’s addiction to a single oncogene could render a potentially fatal disease into a chronic disease with simpler management. This success ushered in the era of targeted therapy, inspiring the search for other oncogene addictions and agents to target them. There are several putative oncogene addictions in pediatric tumors, including Bcr-Abl1, and the addition of imatinib to conventional chemotherapy substantially improves survival rates for children with BCR-ABL1+ ALL (3).
Figure 1. Cancers’ Addictions.

A. Oncogene addiction. Bcr-abl1 leads to aberrant proliferation as well as impaired differentiation and apoptosis. When inhibited, the malignant phenotype is abrogated. B. Non-oncogene addiction. Ews-Fli1 enhances PARP1 expression and results in DNA damage, leading to dependence upon PARP1. C. Escape pathway addiction. Cytarabine results in activation of DNA damage checkpoints, including WEE1, providing chemoresistance.
Further laboratory research indicates that oncogenes can be quite toxic to cells, and that oncogene activation renders cancer cells dependent upon the activation of other genes and pathways that are not mutated to survive the oncogenic stress (4). Different oncogenes likely activate different non-oncogenic pathways that differ from cancer to cancer. Determination of which non-oncogenes a given tumor is addicted to may lead to novel therapeutic approaches for that tumor. For example, due to deficiency in the homologous recombination DNA damage repair mechanism, tumors with mutated BRCA2 rely on PARP1, a key molecule in the base-excision repair DNA damage repair pathway (5). Thus, interruption of PARP1 activity is “synthetic lethal” with BRCA2 mutation and is being explored clinically with targeted inhibitors of PARP1. Relevant in pediatrics, EWS-FL1, the translocation responsible for Ewing’s sarcoma family of tumors (ESFT), causes DNA damage, enhanced expression of, and dependence on PARP1 (Figure 1B) (6).
As non-oncogene addiction is context-dependent (i.e., on the oncogene), cancers can similarly become dependent on particular pathways in the context of specific therapies, which we refer to as escape pathway addiction (Figure 1C). Using genome-wide RNA-interference screens, we and others have identified several escape pathway addictions (7–10). With agents targeting specific components of pathways that cancer cells engage in order to escape the toxic effects of targeted and conventional anti-cancer therapeutics, we have validated their importance in pre-clinical models of leukemia and lung cancer, and have translated one set of discoveries into a clinical trial (NCT01456988).
For each putative cancer addiction that is discovered, depending on the nature of the molecule, several targeted inhibitors may be developed, each with a side effect profile that may be class- or molecule-specific. Some of the genes that cancers depend on may be important in the normal development of a child, raising the possibility of more adverse effects in younger patients as compared to adults (Figure 2). In this review, we will address some of the unique adverse effects associated with targeted therapies, as they relate to the development of novel pediatric therapies. For reasons discussed, there are a paucity of data from pediatric trials compared to those in adults; thus, much of the review will focus on the nature and adverse effects of drugs with which pediatric oncologists have the most experience. Unfortunately this is at the exclusion of many recently approved and experimental agents now available.. Reasonable extrapolation from preclinical and adult clinical data is made by necessity. We conclude with the implications of current knowledge, calling for carefully considered inclusion of specific adverse effects in clinical trials of targeted agents in children.
Figure 2. Targeting developmental pathways in children.

Some genes that drive cancer are normally active only in specific stages of development (blue triangle). Thus targeting one of these developmental oncogenes in young children (red) may also impair a developmental pathway, in this example linear growth. However, if targeted later in life (black), this adverse effect will not occur.
TARGETED ANTI-CANCER AGENTS IN CHILDREN
Although current conventional therapeutic strategies cure the majority of children with cancer, most pediatric oncologists have some experience with targeted agents (11), through prescription in clinical trial regimens or “off-study,” presumably for a biologically plausible reason. However, with notable exceptions, most targeted agents have not been tested in large numbers of children, and thus lack clear demonstration of efficacy and toxicity. This fact that should raise caution in pediatric oncologists considering their use as very few targeted agents are approved for use in children by regulatory authorities. For example, among the targeted agents discussed in this article, the United States Food and Drug Administration (FDA) has approved only imatinib for children with chronic-phase CML, an extremely rare diagnosis in children, and the mTOR inhibitors, sirolimus and everolimus, for children over 13 years for prevention of organ graft rejection and specific tuberous sclerosis associated astrocytomas, respectively.
Braod success with targeted agents in children has been elusive for several reasons. One of the biggest limitations is the lack of development of drugs specifically for pediatric cancers, a consequence of the relative rarity of childhood cancer and the market implications thereof. With the exception of clofarabine in 2004, no new drug has been approved specifically for childhood cancer by the FDA since 1990. Instead, agents developed for adult cancers are retrofitted for pediatric use, where pre-clinical data suggests potential benefit. Second, the sequence of drug testing in children is similar to that in adults, beginning in heavily pre-treated patients with relapsed or refractory disease. As the incidence of childhood cancer is quite low and the therapeutic successes generally quite high, agents must be prioritized with respect to which trials will be opened to which populations. Undoubtedly, this leads to potentially effective agents not being adequately tested in children. Finally, the bar is quite high for statistically improving upon the relatively good success rates of front-line therapy for many tumor types.
Nonetheless, as many of these agents are approved in adults, they are available for use in children, most appropriately in the setting of carefully designed clinical trials. The pediatric oncologist must be aware of not only the known on-target, class effects, and the more specific off-target effects of individual targeted agents, but also the potential adverse effects specific to the developing child. As new molecular entities are being tested and used more commonly in children, the developmental effects of targeted agents are only now coming to light.
ADVERSE EFFECTS OF TARGETED AGENTS IN CHILDREN
Essential to the comprehensive care of children with any medical illness is an understanding of normal human growth and development, including the molecular mechanisms of these processes. Cancer cells often hijack key nodes of the molecular networks that control normal development (12), thus predictably, targeted anti-cancer agents will have unique adverse effect profiles in children. For example, homeobox (Hox) genes, which are critical regulators of patterning during development (with repressed expression in adults), are re-expressed in cancers, contributing to malignancy (13). In fact, Hox genes are frequently deregulated in leukemias and suggested as therapeutic targets, but their roles in normal hematopoiesis and tissue development need to be carefully considered, particularly in children (14). The hedgehog (HH) pathway is another developmental program that is dysregulated in some tumors, most notably in about 25% of medulloblastoma (15, 16), and can be selectively inhibited by vismodegib (17). The developmental importance of HH signaling is highlighted by permanent bone defects in young mice treated with another HH antagonist (18). Similarly, the receptor tyrosine kinase anaplastic lymphoma kinase (ALK) plays a role in normal neuronal and visceral muscle development, as well as oncogenesis in several cancer types, including neuroblastoma and anaplastic large cell lymphoma (19–21). While Hox modulators have not yet been tested, and the ALK inhibitor crizotinib has recently entered pediatric clinical trials in children, experience with other agents highlight the importance of carefully considering the risks associated with targeted drugs.
Unique side effect profiles of targeted agents
Some side effects of cancer therapies are broad and involve normal cellular processes that are not susceptible specifically in cancer cells. This is most prevalent with the use of conventional cytotoxic agents. These agents are effective because they generally target cells that are rapidly dividing and/or lack appropriate DNA damage checkpoints, including those that make up the bulk of a tumor. Unfortunately, many normal cells and their precursors, including the cells that line the gastrointestinal tract, the hair follicles, and the bone marrow, are also rapidly dividing and are essential for homeostasis and human growth. The undesirable by-product of general cytotoxic therapy is that while killing the malignant cells, healthy cells are also affected. As such, alopecia, GI toxicity, and myelosuppression are frequent and often dose limiting side effects of these important agents. In children, the undesirable general adverse effects are manifest long-term in the form of short stature, impaired fertility, cardiac dysfunction, and neurocognitive deficits (22).
For a number of newer targeted agents such as TKIs, monoclonal antibodies, and anti-sense molecules, side effect profiles are distinct and hoped to be less severe than those associated with conventional agents, although they still cause side effects that can be quite significant (23). These unique profiles may be based on the intended target (on-target effects), such that agents directed against the same target may share a similar side effect profile. For example, patients treated with epidermal growth factor receptor (EGFR) inhibitors frequently develop a characteristic rash, likely due to the inhibition of EGFR in the basal layer of the skin & hair follicles, and subsequent immune cell infiltration (24). Another example of an on-target effect is the well-described side effect of hypertension associated with the use of agents targeting vascular endothelial growth factor and its receptors (collectively termed VEGF inhibitors). Interestingly, the rash and hypertension associated with EGFR and VEGF inhibitors respectively have been correlated with positive outcomes (25, 26), but correlations between adverse effects and outcomes may be limited to specific classes (23). Thus, clinical oncologists must be aware of on-target, class effects, their implications related to outcomes, as well as the best ways manage them. Moreover, it is incumbent upon clinical scientists to anticipate and recognize class effects, and prospectively collect data on their incidence and severity within early and late phase trials.
In addition to inhibiting the putative addiction, targeted agents typically inhibit known and unknown targets (off-target effects) that may have unexpected consequences. Tyrosine kinase inhibitors are inherently “dirty” and inhibit several kinases in addition to the target for which they are developed. By definition, these effects are harder to categorize, as they are more specific to the various kinases inhibited by a particular molecule. For example, the greater hematopoietic toxicity associated with small molecule inhibitors sorafenib and sunitinib, as compared to the anti-VEGF antibody, bevacizumab, implicates off-target inhibition of other targeted kinases such as c-Kit or Flt3 (27). Monoclonal antibodies may have off-target effects as well. Type 1 insulin-like growth factor receptor (IGF1R) antibodies, in development for the treatment of solid tumors, cause hyperglycemia, possibly due to indirect effects on the insulin receptor (28). Other off-target effects may be associated with the origin, structure or size of the molecule, as are the infusion reactions associated with monoclonal antibody administration.
Impaired Linear Growth & Endocrine Function
Unfortunately, the extent of overlap between normal developmental networks and cancers’ addictions is not necessarily obvious before testing in large numbers of children. One unanticipated observation is that of growth suppression or failure in children treated with TKIs for BCR-ABL1+ leukemias. This effect is most pronounced in children treated before the onset of puberty (29, 30). One proposed mechanism modeled in rats, is through the off-target direct inhibition of PDGFR of chondrocytes, impairing proliferation, and consequently, linear bone growth (31). Indirectly, treatment with TKIs has been shown to cause growth hormone deficiency (32), although the relevant off-target molecules remain undefined. More directly, inhibition of the growth hormone axis by IGF1R inhibitors may also impair linear growth in children (28), although this has yet to be proven.
As the number of adult survivors of childhood cancer increased due to the optimization of conventional cytotoxic therapy, the consequence of therapy on fertility became clear (33). With the introduction of targeted therapy, the hope is that this adverse effect will not be as prevalent, but there are hypothetical reasons and case reports suggesting that infertility may emerge in this context, too. Normal spermatogenesis is dependent upon PDGFR and c-Kit, protein kinases that are inhibited by many TKIs. Experiments in neonatal rats reveal permanent deleterious effects on the maturation of testes after short-term exposure to imatinib, although fertility was preserved, leading to speculation that pre-pubertal exposure to TKIs may have more adverse effects on fertility than later exposure (34). Interestingly, the two case reports of oligospermia after imatinib exposure are from patients started on therapy during childhood (35, 36) and azoospermia was observed in a post-pubertal patient treated only with dasatinib for Ph+ leukemia (Personal Communication, Dr. C. M. Zwann). While there are reports of successful pregnancy in men and women treated with imatinib, its use is not recommended during pregnancy due to potential teratogenicity, and the extent and reversibility of impaired fertility due to tyrosine kinase inhibition in children with cancer remain to be defined.
Other endocrine abnormalities have been reported in adults treated with tyrosine kinase inhibitors, including hypothyroidism and testosterone deficiency (37, 38). The testosterone deficiency associated with crizotinib should be of particular concern for pediatric oncologists, given its apparent potential and current active studies (20). While not formally reported, hypothyroidism has also been observed in children enrolled in clinical trials and treated with sunitinib and sorafenib (personal observation, LG). As with adults, these toxicities appear to be cumulative and related to the duration of treatment with the particular agent (38). Unrecognized, this deficiency can have devastating effects on the developing child.
The developing immune system
A critical aspect of normal growth and development is the maturation of the adaptive immune system. This highly regulated phenomenon is most active in infancy and early childhood, not coincidentally when the incidence of acute lymphoblastic leukemia is highest (39). Notably, several conventional chemotherapeutics are highly immunosuppressive due to their lympholytic properties. The effects of conventional chemotherapy on the immune system can persist long after therapy is discontinued. This phenomenon has clinical significance as even after re-immunization, not all patients demonstrate normal antibody production and serious infections have been reported (40).
The potential immune suppressive effects of targeted agents should not be disregarded, as their mechanisms of action may not be toxic only to the cancer cell. Rather, the possibility of off- and on-target effects on the immune system could be substantial. This is perhaps most commonly observed with rituximab, which is known to lead to substantial suppression of B-cell immunity, and with inhibitors of mTOR, such as rapamycin and its analogs (“rapalogs”). Rapamycin was approved as an immunosuppressive agent in the 1990s, and is still used in combination with other agents to prevent the rejection of solid organ transplants. The mTOR pathway has more recently emerged as a target for anti-cancer therapy, and rapalogs have been tested in early phase clinical trials as single agents and in combination with conventional chemotherapeutics in children and adults. Everolimus is approved for use in five different cancers, including subependymal giant-cell astrocytomas associated with tuberous sclerosis (41). These agents are associated with a two-fold increased risk of infection in adults, although these are generally mild (42). Similar adverse effects of everolimus were reported in children (41). While this degree of immunosuppression is generally manageable, for example, with altered dosing schedules, the extent to which persistent immunosuppression and the promotion of tolerance contributes to treatment failure and even potential for secondary malignancies is in question, and may limit the utility of these drugs, especially for chronic use.
The immune suppressive effects of targeted agents are not limited to small molecule inhibitors. Rituximab is a monoclonal antibody directed against CD20, expressed on normal mature and malignant B-cells. It has improved outcomes in non-Hodgkin’s lymphoma (NHL) in adults (43), and is being studied for efficacy and safety in clinical trials for children with high-risk NHL (NCT01595048). The addition of rituximab to chemotherapy regimens does not appear to increase the rate of infections in children or adults with lymphoma (43, 44), although some patients have hypogammaglobulinemia due to the elimination of immunoglobulin-producing mature B-cells (45). This laboratory abnormality can be quite severe and prolonged (46, 47). The clinical significance of this finding in patients with lymphomas has not been well studied, but treating physicians may prescribe infusions of intravenous immune globulin preparations as prophylaxis or in response to fever. An unexpected, off-target effect of rituximab is the development of late-onset neutropenia (48). This clinical laboratory finding is observed weeks after therapy (49), is variable in severity and duration, but is typically not associated with severe infections (48).
In addition to BCR-ABL1, dasatinib inhibits several members of the Src family of kinases (SFK). In fact, dasatinib was initially developed as an SFK inhibitor. Lck is a member of this family that is a critical component of T-cell receptor signaling and resultant activation and proliferation (50). Indeed, dasatinib inhibits T-cell activation and proliferation ex vivo (51), and has been associated with atypical infections in patients treated at higher doses (52). When combined with other immunomodulators, the effect is more complex, as the inhibitory effect is enhanced when dasatinib is combined with cyclosporine A or rapamycin in some experimental conditions (51), but is antagonistic in others (53). Data such as these prompted the inclusion of correlative laboratory studies in our clinical trial (NCT1456988) combining dasatinib and cyclosporine for patients with BCR-ABL1+ leukemia.
The tissue landscape
One of the most devastating side effect of conventional chemotherapeutics is the development of secondary malignancies, occurring in 2–15% of patients treated for cancer (54). These may arise due to direct DNA damage from the anti-cancer agents, but emerging evidence indicates that non-cell autonomous factors play a major role. Cancer development, from initiation to metastasis is an evolutionary process that is highly influenced by the tissue microenvironment (55, 56). We have previously proposed that a key tumor suppressive mechanism evolved by complex multicellular species is the maintenance of normal tissue structure and cellular fitness, which should impede somatic cellular evolution (55, 57). A normal tissue microenvironment and high cellular fitness should favor the status quo, as trait-altering mutations should mostly be disadvantageous. On the other hand, alterations in the microenvironment and reductions in progenitor cell fitness, such as during aging or following stress-inducing insults (e.g., radiation exposure), will promote selection for adaptive oncogenic mutations. These concepts should also be relevant for oncologic therapies, as they not only impinge upon cancer cells but will also impact the tissue landscape and normal stem and progenitor cell fitness. Impacts on non-target tissues could contribute to secondary cancers, not only by directly increasing the frequency of oncogenic mutations but also by promoting selection for particular oncogenic mutations adaptive to the altered landscape. While secondary cancers are expected to occur less frequently with the use of targeted agents, current data from patients treated with several small molecule inhibitors confirm the possibility.
In the context of conventional chemotherapy, secondary AML (sAML) is a significant risk for survivors occurring in 1–10% of treated patients depending on the treatment regimen and accounting for up to 30% of AML cases (58). While anti-topoisomerase-II treatment is involved in the initiation of sAML due to the DNA damage it causes (59), subsequent myelosuppressive therapies alter the tissue landscape and impair the fitness of competing stem and progenitor cells, promoting selection for adaptive oncogenic mutations. Indeed, we have shown that treatment of mice with the 6-thioguanine (6TG) can promote selection for early hematopoietic progenitors expressing the MLL-AF9 translocation product, accelerating the development of AML (60). Importantly, by transplanting bone marrow cells from hprt mutant mice whose cells are resistant to 6TG, the ability of 6TG to promote selection for MLL-AF9 and promote AML is reversed. Thus, the ability of 6TG to promote MLL-AF9 initiated leukemogenesis requires 6TG mediated impairment of hematopoiesis, which engenders an altered adaptive landscape.
The consequences of altered landscape are not limited to conventional cytotoxic agents like topo-II-inhibitors and 6TG. In fact, while imatinib and dasatinib restore polyclonal hematopoiesis in the majority of patients that achieve cytogenetic remission, clonal chromosomal abnormalities are detected in Philadelphia chromosome negative (Phneg) cells in 3–15% of these patients (61–63). Some of these chromosomal changes are associated with myelodysplasia and leukemia (e.g., t(3;21) and monosomy 7), and a few patients have been reported to manifest myelodysplastic syndrome and AML (61, 64). This phenomenon raises the question as to whether the emergence of Phneg clonal hematopoiesis represents an underlying predisposition to the development of CML, or whether TKIs are responsible for clonal hematopoiesis. The absence of these chromosomal abnormalities in the Ph+ clone in some of these patients suggests that the former is less likely. The latter hypothesis from a cell autonomous point of view, would suggest that TKI treatment results in DNA damage in normal hematopoietic progenitors, for example, due to an aberrant DNA damage response resulting from Abl inhibition (61). Alternatively, TKI may cause mild or moderate myelosuppression on the Phneg population which promotes the emergence of aberrant clones, either pre-existing due to previous DNA damage, or that evolve in the altered landscape. This possibility does not implicate TKI specifically, but any agent that alters hematopoiesis. Indeed, Phneg clonal cytogenetic abnormalities were observed in the context of interferon treatment (65). Most importantly, though, the emergence of Phneg clonal cytogenetic abnormalities appears to be rarely associated with the development of MDS or AML, and generally does not negatively influence prognosis for chronic phase CML, with the exception of monosomy 7, the emergence of which may necessitate a change in therapy (63). To our knowledge, Phneg clonal hematopoiesis has not been reported in children, but if this phenomenon is a consequence of the therapy, presumably the longer-term use in children may increase the risk of its development.
THERAPEUTIC IMPLICATIONS IN CHILDREN
More Isn’t Necessarily Better
The “if some is good, more must be better” principle of dosing chemotherapy has held some value since the inception of chemotherapy as a therapeutic strategy, and most pediatric regimens employ higher doses and greater dose intensity than are used in treatment of adults. In general, higher doses of cytotoxics have usually demonstrated greater tumor kill than lower doses, although myelosuppression often limits further dose escalation (Figure 3). In fact, hematopoietic stem cell transplantation emerged as a therapeutic strategy as a means to allow for myeloablative doses of anti-cancer therapy, in an effort to induce more tumor kill.
Figure 3. Dose Response Effects on Normal and Malignant Tissue.
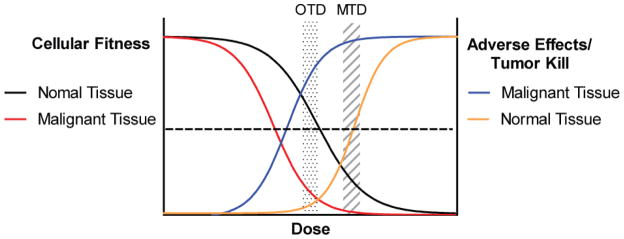
Targeted agents should have a wide therapeutic window, but likely impair fitness of normal cells at higher doses leading to adverse effects. Here, lower doses affect the fitness of cancer more than normal tissue, with high tumor kill and low adverse effects. This is the OTD. More tumor kill may be achieved at higher doses, but more adverse effects occur.
However, in the era of targeted therapy, in which molecules are designed to be highly potent and specific inhibitors of cancers’ addictions, dose escalation to the MTD is not biologically rational, as the dose limiting side effects at higher doses are likely to be unrelated to inhibition of the desired target. This concept has led to consideration of defining the optimal biologic dose (OBD) as an endpoint in early phase trials, rather than the MTD. The OBD is that dose at which there is maximal inhibition of the identified cancer’s addiction (66). This is, of course, predicated in the deep understanding of the target and the ability to measure its modulation, both of which can be elusive (67). While the OBD is expected to be lower than the MTD, as normal cells should be less dependent upon the targeted protein, it is possible that the OBD is higher than the MTD, which would diminish the utility of that agent. We suggest that an optimal therapeutic dose (OTD) to be defined by experimentally determined, pre-set measure(s) of target engagement, as well as a tolerable side effect profile. Ideally, the extent of target engagement and outcomes will be correlated in early phase trials, further refining the OTD (68). This necessitates a strong understanding of the cancer’s addiction, a reliable biomarker to measure target inhibition, and access to tumor samples after the initiation of therapy. The latter is particularly troublesome in pediatric trial settings, as invasive procedures may be necessary. The problem is less significant in leukemias, the most common childhood cancer, with readily available tumor cells in blood or bone marrow.
Vigilance for on-target and off-target adverse effects
Pediatric oncologists are well-trained to recognize the common side effects associated with conventional chemotherapeutics. The consequences of therapy are so frequent that many practices have clinical care guidelines to address acute problems such as fever and neutropenia, and staff multi-disciplinary clinics for management of late effects. As cancer therapy evolves to include targeted agents, pediatric oncologists will need to screen for more obscure adverse effects that may have considerable consequences. Fortunately, many common side effects can be effectively prevented or treated: growth hormone replacement for growth failure, levothyroxine for hypothyroidism, and antibiotic prophylaxis to prevent Pneuomocystis pneumonia. Nonetheless, the effects must be considered prior to clinical presentation to avoid long term consequences. Thus, pediatric oncologists must be aware of emerging side effect profiles of drug classes, as well as those related to specific drugs.
FUTURE IMPLICATIONS
With the notable exception of CML, single targeted agents have yet to improve long-term survival in most patients with cancer. It is clear from clinical experience and laboratory studies, that targeting a single cancer addiction will likely have only short-lived effects, as cancer cells engage other mechanisms to manifest the malignant phenotype (69). Thus, most targeted agents will only be successful when combined with other agents, resulting in elimination of minimal residual disease. This will, of course, complicate the assessment and separation of adverse effects from that of other medications included in a given regimen, a problem addressed recently by an international consensus conference (70). Unfortunately, as new agents are first tested in patients who have a relatively poor prognosis, and thus a likely shortened life expectancy, long-term effects cannot be assessed, further complicating drug development strategies. Nonetheless, as targeted agents progress to later stage trials, plans for monitoring for late effects should be included, perhaps through the Childhood Cancer Survivor Study or similar efforts, with international collaborations having increasing importance.
As cancer therapy evolves, the fundamentals of how we study novel therapeutic strategies need to be reassessed, as well. Novel approaches to early phase trials of targeted agents should be considered, including adaptive protocols and randomization in phase 2 (71, 72). Phase 1 trials should include biomarkers of target engagement when possible (67, 71), as well as intra-patient dose escalation to achieve the OTD (73). The inclusion of predictive companion biomarkers, based on robust pre-clinical modeling and validation in clinical trials, to predict populations most likely to respond to therapy should be sought, as well (74). This should enhance success rates in drug development and reduce exposures for those not likely to benefit from a particular therapy. For the pediatric oncologist involved in early phase trials, consideration of on- and off- target effects on the developing child is imperative, and specific hypotheses need to be developed and tested in these trials. Effective conduct of Phase 1 and 2 trials have the potential to mitigate adverse effects in larger numbers of patients in Phase 3 trials, with the collection of developmental data and inclusion of informed supportive care guidelines. For example, protocol directed collection of growth data before therapy and during therapy may alert treating physicians to unexpected growth impairment. Recommendations for periodic thyroid function testing for patients taking TKI (38) or skin care measures for those taking EGFR inhibitors could also be included (75).
SUMMARY AND CONCLUSIONS
The understanding of cancers’ addictions and the development of targeted agents has already, and will most certainly continue to revolutionize cancer therapy. As these agents are used more effectively, though, it is perhaps likely that some cancers will be modulated to a chronic disease state requiring long-term therapy, as is the case with CML. The prospect of lifelong therapy and its inherent risks is daunting, particularly for the developing child. Nonetheless, this non-monetary cost for high-quality, productive life-years is most certainly worthwhile. While data from children are currently scarce, as cancer therapy evolves, pediatric oncologists must be aware of the side-effect profiles that emerge. Those involved in early and late phase clinical trials need to consider and include predictive biomarkers and developmental endpoints in pediatric trials, so that the patients most likely to benefit are included and so proper supportive care guidelines can be developed in real-time with the novel therapeutic strategies. The era of molecular oncology in pediatrics is exciting, but is clearly accompanied by considerable challenge, as well.
SEARCH STRATEGY AND SELECTION CRITERIA
Published data for this review were identified by searches of PubMed with the term “children” and “side effects” or “developmental” in combination with terms such as “targeted therapy,” “imatinib,” “dasatinib,” “rituximab,” “hedgehog,” “IGF1R” and “mTOR.” Relevant references within identified publications were also identified and reviewed. Only articles published in English and relevant to the Review were included. No date range was specified.
Table 1.
Examples of targeted agents and their side effects.
| Drug | intended target | Approved in children | Other notable targets | On target side effect | Off target side effect |
|---|---|---|---|---|---|
| bevacizumab | Vegf | no | ND | hypertension, delayed healing, bleeding | infusion reactions |
| sunitinib | Vegfr1-3 | no | Axl, Pdgfr, Flt3, Ckit, Ret | hypertension | hypothyroidism, myelosuppression |
| sorafenib | Vegfr1-3 | no | Pdgfr, Flt3, C-Kit, Ret, B-Raf, | hypertension | hypothyroidism, myelosuppression |
| cetuximab | Egfr | no | ND | rash | infusion reactions |
| erlotinib | Egfr | no | Erbb4, Blk, Flt3, Gak, Stk10, Slk | rash | ND |
| sirolimus | Mtor | yes** | ND | immunosuppression | ND |
| everolimus | Mtor | yes | ND | immunosuppression | ND |
| imatinib | Bcr-Abl1, c-Kit | yes | Abl, Pdgfr, Arg | azoospermia | delayed growth |
| dasatinib | Bcr-abl1 | no | SFK†, Pdgfr, Blk, Btk, Csk, Ddr1, Epha1-5,8, Fgr | altered hematopoietic landscape | delayed growth, immunosuppression |
| rituximab | CD20 | no | ND | hypogammaglobulinemia, b-cell lineage immunosuppression | anaphylaxis/infusion reaction |
| crizotinib | Alk | no | Met | altered neural development? | hypogonadism |
| vismodegib | Smo | no | ND | abnormal bone growth, alopecia | muscle spasms |
| cixutumumab* | Igf1r | no | insulin receptor (indirect) | delayed growth? | hyperglycemia |
| olaparib* | Parp1 | no | ND | cytopenia | ND |
ND – Not defined
Not currently approved for clinical use.
For the prevention of graft rejection.
Src family kinases (Src, Lck, Yes, Fyn, Hck)
Speculative adverse associations
Acknowledgments
The authors thank Dr. Frank Haluska for his critical review of the manuscript. The authors are supported by The Leukemia & Lymphoma Society (JD, CP), the National Cancer Institute (LG, JD, CP), the Morgan Adams and McCormick Foundations (LG). Dr. DeGregori receives research funding from AstraZeneca.
Footnotes
Conflict of interest statement: The authors have no conflict of interest to disclose.
Contributors: LG, JD and CCP did the literature search, designed the figures and wrote and revised the manuscript.
References
- 1.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002 Jul 5;297(5578):63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian H, O’Brien S, Garcia-Manero G, Faderl S, Ravandi F, Jabbour E, et al. Very long-term follow-up results of imatinib mesylate therapy in chronic phase chronic myeloid leukemia after failure of interferon alpha therapy. Cancer. 2012 Jun 15;118(12):3116–22. doi: 10.1002/cncr.26568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol. 2009 Nov 1;27(31):5175–81. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009 Mar 6;136(5):823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005 Apr 14;434(7035):913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 6.Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou-Maroun LM, et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012 Apr 1;72(7):1608–13. doi: 10.1158/0008-5472.CAN-11-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casa-Selves M, Kim J, Zhang Y, Helfrich BA, Gao D, Porter CC, Bunn PA, Chan DC, Tan AC, DeGregori J. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer Research. doi: 10.1158/0008-5472.CAN-11-2848. epub Aug 6. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D, et al. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia. 2012 Jun;26(6):1266–76. doi: 10.1038/leu.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O’Hare T, et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010 Jul 13;18(1):74–87. doi: 10.1016/j.ccr.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007 Apr 12;446(7137):815–9. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 11.Grunewald TG, Greulich N, Kontny U, Fruhwald M, Rutkowski S, Kordes U, et al. Targeted therapeutics in treatment of children and young adults with solid tumors: an expert survey and review of the literature. Klin Padiatr. 2012 Apr;224(3):124–31. doi: 10.1055/s-0032-1301930. [DOI] [PubMed] [Google Scholar]
- 12.Moore SW. Developmental genes and cancer in children. Pediatr Blood Cancer. 2009 Jul;52(7):755–60. doi: 10.1002/pbc.21831. [DOI] [PubMed] [Google Scholar]
- 13.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010 May;10(5):361–71. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- 14.McGonigle GJ, Lappin TR, Thompson A. Grappling with the HOX network in hematopoiesis and leukemia. Front Biosci. 2008;13:4297–308. doi: 10.2741/3006. [DOI] [PubMed] [Google Scholar]
- 15.Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997 Mar 1;57(5):842–5. [PubMed] [Google Scholar]
- 16.Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002 Jul;31(3):306–10. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 17.Low JA, de Sauvage FJ. Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol. 2010 Dec 20;28(36):5321–6. doi: 10.1200/JCO.2010.27.9943. [DOI] [PubMed] [Google Scholar]
- 18.Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008 Mar;13(3):249–60. doi: 10.1016/j.ccr.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 19.Pulford K, Lamant L, Espinos E, Jiang Q, Xue L, Turturro F, et al. The emerging normal and disease-related roles of anaplastic lymphoma kinase. Cell Mol Life Sci. 2004 Dec;61(23):2939–53. doi: 10.1007/s00018-004-4275-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carpenter EL, Mosse YP. Targeting ALK in neuroblastoma-preclinical and clinical advancements. Nat Rev Clin Oncol. 2012;9(7):391–9. doi: 10.1038/nrclinonc.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008 Jan;8(1):11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 22.Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006 Oct 12;355(15):1572–82. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 23.Niraula S, Seruga B, Ocana A, Shao T, Goldstein R, Tannock IF, et al. The price we pay for progress: a meta-analysis of harms of newly approved anticancer drugs. J Clin Oncol. 2012 Aug 20;30(24):3012–9. doi: 10.1200/JCO.2011.40.3824. [DOI] [PubMed] [Google Scholar]
- 24.Li T, Perez-Soler R. Skin toxicities associated with epidermal growth factor receptor inhibitors. Target Oncol. 2009 Apr;4(2):107–19. doi: 10.1007/s11523-009-0114-0. [DOI] [PubMed] [Google Scholar]
- 25.Schuster C, Eikesdal HP, Puntervoll H, Geisler J, Geisler S, Heinrich D, et al. Clinical efficacy and safety of bevacizumab monotherapy in patients with metastatic melanoma: predictive importance of induced early hypertension. PLoS One. 2012;7(6):e38364. doi: 10.1371/journal.pone.0038364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wacker B, Nagrani T, Weinberg J, Witt K, Clark G, Cagnoni PJ. Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studies. Clin Cancer Res. 2007 Jul 1;13(13):3913–21. doi: 10.1158/1078-0432.CCR-06-2610. [DOI] [PubMed] [Google Scholar]
- 27.Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009 Aug;6(8):465–77. doi: 10.1038/nrclinonc.2009.94. [DOI] [PubMed] [Google Scholar]
- 28.Yuen JS, Macaulay VM. Targeting the type 1 insulin-like growth factor receptor as a treatment for cancer. Expert Opin Ther Targets. 2008 May;12(5):589–603. doi: 10.1517/14728222.12.5.589. [DOI] [PubMed] [Google Scholar]
- 29.Shima H, Tokuyama M, Tanizawa A, Tono C, Hamamoto K, Muramatsu H, et al. Distinct impact of imatinib on growth at prepubertal and pubertal ages of children with chronic myeloid leukemia. J Pediatr. 2011 Oct;159(4):676–81. doi: 10.1016/j.jpeds.2011.03.046. [DOI] [PubMed] [Google Scholar]
- 30.Bansal D, Shava U, Varma N, Trehan A, Marwaha RK. Imatinib has adverse effect on growth in children with chronic myeloid leukemia. Pediatr Blood Cancer. 2011 Nov 2; doi: 10.1002/pbc.23389. [DOI] [PubMed] [Google Scholar]
- 31.Vandyke K, Dewar AL, Fitter S, Menicanin D, To LB, Hughes TP, et al. Imatinib mesylate causes growth plate closure in vivo. Leukemia. 2009 Nov;23(11):2155–9. doi: 10.1038/leu.2009.150. [DOI] [PubMed] [Google Scholar]
- 32.Hobernicht SL, Schweiger B, Zeitler P, Wang M, Hunger SP. Acquired growth hormone deficiency in a girl with chronic myelogenous leukemia treated with tyrosine kinase inhibitor therapy. Pediatr Blood Cancer. 2011 Apr;56(4):671–3. doi: 10.1002/pbc.22945. [DOI] [PubMed] [Google Scholar]
- 33.Byrne J, Mulvihill JJ, Myers MH, Connelly RR, Naughton MD, Krauss MR, et al. Effects of treatment on fertility in long-term survivors of childhood or adolescent cancer. N Engl J Med. 1987 Nov 19;317(21):1315–21. doi: 10.1056/NEJM198711193172104. [DOI] [PubMed] [Google Scholar]
- 34.Apperley J. CML in pregnancy and childhood. Best Pract Res Clin Haematol. 2009 Sep;22(3):455–74. doi: 10.1016/j.beha.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Seshadri T, Seymour JF, McArthur GA. Oligospermia in a patient receiving imatinib therapy for the hypereosinophilic syndrome. N Engl J Med. 2004 Nov 11;351(20):2134–5. doi: 10.1056/NEJM200411113512024. [DOI] [PubMed] [Google Scholar]
- 36.Mariani S, Basciani S, Fabbri A, Agati L, Ulisse S, Lubrano C, et al. Severe oligozoospermia in a young man with chronic myeloid leukemia on long-term treatment with imatinib started before puberty. Fertil Steril. 2011 Mar 1;95(3):1120 e15–7. doi: 10.1016/j.fertnstert.2010.08.060. [DOI] [PubMed] [Google Scholar]
- 37.Weickhardt AJ, Rothman MS, Salian-Mehta S, Kiseljak-Vassiliades K, Oton AB, Doebele RC, et al. Rapid-onset hypogonadism secondary to crizotinib use in men with metastatic nonsmall cell lung cancer. Cancer. 2012 Apr 4; doi: 10.1002/cncr.27450. [DOI] [PubMed] [Google Scholar]
- 38.Torino F, Corsello SM, Longo R, Barnabei A, Gasparini G. Hypothyroidism related to tyrosine kinase inhibitors: an emerging toxic effect of targeted therapy. Nat Rev Clin Oncol. 2009 Apr;6(4):219–28. doi: 10.1038/nrclinonc.2009.4. [DOI] [PubMed] [Google Scholar]
- 39.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006 Mar;6(3):193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 40.Smith S, Schiffman G, Karayalcin G, Bonagura V. Immunodeficiency in long-term survivors of acute lymphoblastic leukemia treated with Berlin-Frankfurt-Munster therapy. J Pediatr. 1995 Jul;127(1):68–75. doi: 10.1016/s0022-3476(95)70259-8. [DOI] [PubMed] [Google Scholar]
- 41.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010 Nov 4;363(19):1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 42.Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010 Apr;7(4):209–19. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- 43.Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002 Jan 24;346(4):235–42. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 44.Meinhardt A, Burkhardt B, Zimmermann M, Borkhardt A, Kontny U, Klingebiel T, et al. Phase II window study on rituximab in newly diagnosed pediatric mature B-cell non-Hodgkin’s lymphoma and Burkitt leukemia. J Clin Oncol. 2010 Jul 1;28(19):3115–21. doi: 10.1200/JCO.2009.26.6791. [DOI] [PubMed] [Google Scholar]
- 45.Brusamolino E, Rusconi C, Montalbetti L, Gargantini L, Uziel L, Pinotti G, et al. Dose-dense R-CHOP-14 supported by pegfilgrastim in patients with diffuse large B-cell lymphoma: a phase II study of feasibility and toxicity. Haematologica. 2006 Apr;91(4):496–502. [PubMed] [Google Scholar]
- 46.Irie E, Shirota Y, Suzuki C, Tajima Y, Ishizawa K, Kameoka J, et al. Severe hypogammaglobulinemia persisting for 6 years after treatment with rituximab combined chemotherapy due to arrest of B lymphocyte differentiation together with alteration of T lymphocyte homeostasis. Int J Hematol. 2010 Apr;91(3):501–8. doi: 10.1007/s12185-010-0528-6. [DOI] [PubMed] [Google Scholar]
- 47.Walker AR, Kleiner A, Rich L, Conners C, Fisher RI, Anolik J, et al. Profound hypogammaglobulinemia 7 years after treatment for indolent lymphoma. Cancer Invest. 2008 May;26(4):431–3. doi: 10.1080/07357900701809068. [DOI] [PubMed] [Google Scholar]
- 48.Wolach O, Bairey O, Lahav M. Late-onset neutropenia after rituximab treatment: case series and comprehensive review of the literature. Medicine (Baltimore) 2010 Sep;89(5):308–18. doi: 10.1097/MD.0b013e3181f2caef. [DOI] [PubMed] [Google Scholar]
- 49.Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997 Sep 15;90(6):2188–95. [PubMed] [Google Scholar]
- 50.Molina TJ, Kishihara K, Siderovski DP, van Ewijk W, Narendran A, Timms E, et al. Profound block in thymocyte development in mice lacking p56lck. Nature. 1992 May 14;357(6374):161–4. doi: 10.1038/357161a0. [DOI] [PubMed] [Google Scholar]
- 51.Schade AE, Schieven GL, Townsend R, Jankowska AM, Susulic V, Zhang R, et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood. 2008 Feb 1;111(3):1366–77. doi: 10.1182/blood-2007-04-084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sillaber C, Herrmann H, Bennett K, Rix U, Baumgartner C, Bohm A, et al. Immunosuppression and atypical infections in CML patients treated with dasatinib at 140 mg daily. Eur J Clin Invest. 2009 Dec;39(12):1098–109. doi: 10.1111/j.1365-2362.2009.02206.x. [DOI] [PubMed] [Google Scholar]
- 53.Blake SJ, Hughes TP, Lyons AB. Drug-interaction studies evaluating T-cell proliferation reveal distinct activity of dasatinib and imatinib in combination with cyclosporine A. Exp Hematol. 2012 Apr 19; doi: 10.1016/j.exphem.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 54.Super HJ, McCabe NR, Thirman MJ, Larson RA, Le Beau MM, Pedersen-Bjergaard J, et al. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood. 1993 Dec 15;82(12):3705–11. [PubMed] [Google Scholar]
- 55.DeGregori J. Evolved tumor suppression: why are we so good at not getting cancer? Cancer Res. 2011 Jun 1;71(11):3739–44. doi: 10.1158/0008-5472.CAN-11-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008 Jan;8(1):56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 57.Degregori J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene. 2012 Jul 2; doi: 10.1038/onc.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pui CH, Relling MV. Topoisomerase II inhibitor-related acute myeloid leukaemia. Br J Haematol. 2000 Apr;109(1):13–23. doi: 10.1046/j.1365-2141.2000.01843.x. [DOI] [PubMed] [Google Scholar]
- 59.Libura J, Slater DJ, Felix CA, Richardson C. Therapy-related acute myeloid leukemia-like MLL rearrangements are induced by etoposide in primary human CD34+ cells and remain stable after clonal expansion. Blood. 2005 Mar 1;105(5):2124–31. doi: 10.1182/blood-2004-07-2683. [DOI] [PubMed] [Google Scholar]
- 60.Porter CC, Baturin D, Choudhary R, Degregori J. Relative fitness of hematopoietic progenitors influences leukemia progression. Leukemia. 2011 Feb 18; doi: 10.1038/leu.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bumm T, Muller C, Al-Ali HK, Krohn K, Shepherd P, Schmidt E, et al. Emergence of clonal cytogenetic abnormalities in Ph- cells in some CML patients in cytogenetic remission to imatinib but restoration of polyclonal hematopoiesis in the majority. Blood. 2003 Mar 1;101(5):1941–9. doi: 10.1182/blood-2002-07-2053. [DOI] [PubMed] [Google Scholar]
- 62.De Melo VA, Milojkovic D, Khorashad JS, Marin D, Goldman JM, Apperley JF, et al. Philadelphia-negative clonal hematopoiesis is a significant feature of dasatinib therapy for chronic myeloid leukemia. Blood. 2007 Oct 15;110(8):3086–7. doi: 10.1182/blood-2007-05-092437. [DOI] [PubMed] [Google Scholar]
- 63.Deininger MW, Cortes J, Paquette R, Park B, Hochhaus A, Baccarani M, et al. The prognosis for patients with chronic myeloid leukemia who have clonal cytogenetic abnormalities in philadelphia chromosome-negative cells. Cancer. 2007 Oct 1;110(7):1509–19. doi: 10.1002/cncr.22936. [DOI] [PubMed] [Google Scholar]
- 64.Kovitz C, Kantarjian H, Garcia-Manero G, Abruzzo LV, Cortes J. Myelodysplastic syndromes and acute leukemia developing after imatinib mesylate therapy for chronic myeloid leukemia. Blood. 2006 Oct 15;108(8):2811–3. doi: 10.1182/blood-2006-04-017400. [DOI] [PubMed] [Google Scholar]
- 65.Fayad L, Kantarjian H, O’Brien S, Seong D, Albitar M, Keating M, et al. Emergence of new clonal abnormalities following interferon-alpha induced complete cytogenetic response in patients with chronic myeloid leukemia: report of three cases. Leukemia. 1997 May;11(5):767–71. doi: 10.1038/sj.leu.2400642. [DOI] [PubMed] [Google Scholar]
- 66.Adjei AA. What is the right dose? The elusive optimal biologic dose in phase I clinical trials. J Clin Oncol. 2006 Sep 1;24(25):4054–5. doi: 10.1200/JCO.2006.07.4658. [DOI] [PubMed] [Google Scholar]
- 67.Ratain MJ, Glassman RH. Biomarkers in phase I oncology trials: signal, noise, or expensive distraction? Clin Cancer Res. 2007 Nov 15;13(22 Pt 1):6545–8. doi: 10.1158/1078-0432.CCR-07-2133. [DOI] [PubMed] [Google Scholar]
- 68.Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005 Apr 10;23(11):2445–59. doi: 10.1200/JCO.2005.11.890. [DOI] [PubMed] [Google Scholar]
- 69.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12(7):487–93. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horton TM, Sposto R, Brown P, Reynolds CP, Hunger SP, Winick NJ, et al. Toxicity assessment of molecularly targeted drugs incorporated into multiagent chemotherapy regimens for pediatric acute lymphocytic leukemia (ALL): review from an international consensus conference. Pediatr Blood Cancer. 2010 Jul 1;54(7):872–8. doi: 10.1002/pbc.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rubin EH, Gilliland DG. Drug development and clinical trials--the path to an approved cancer drug. Nat Rev Clin Oncol. 2012 Apr;9(4):215–22. doi: 10.1038/nrclinonc.2012.22. [DOI] [PubMed] [Google Scholar]
- 72.Sharma MR, Stadler WM, Ratain MJ. Randomized phase II trials: a long-term investment with promising returns. J Natl Cancer Inst. 2011 Jul 20;103(14):1093–100. doi: 10.1093/jnci/djr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC. Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst. 1997 Aug 6;89(15):1138–47. doi: 10.1093/jnci/89.15.1138. [DOI] [PubMed] [Google Scholar]
- 74.Rodon J, Saura C, Dienstmann R, Vivancos A, Ramon y Cajal S, Baselga J, et al. Molecular prescreening to select patient population in early clinical trials. Nat Rev Clin Oncol. 2012 Jun;9(6):359–66. doi: 10.1038/nrclinonc.2012.48. [DOI] [PubMed] [Google Scholar]
- 75.Lacouture ME, Anadkat MJ, Bensadoun RJ, Bryce J, Chan A, Epstein JB, et al. Clinical practice guidelines for the prevention and treatment of EGFR inhibitor-associated dermatologic toxicities. Support Care Cancer. 2011 Aug;19(8):1079–95. doi: 10.1007/s00520-011-1197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]