

Abstract
We have previously identified a putative tumor suppressor gene, NISCH, whose promoter is methylated in lung tumor tissue as well as in plasma obtained from lung cancer patients. NISCH was observed to be more frequently methylated in smoker lung cancer patients than in non-smoker lung cancer patients. Here, we investigated the effect of tobacco smoke exposure on methylation of the NISCH gene. We tested methylation of NISCH after oral keratinocytes were exposed to mainstream and side stream cigarette smoke extract in culture. Methylation of the promoter region of the NISCH gene was also evaluated in plasma obtained from lifetime non-smokers and light smokers (< 20 pack/year), with and without lung tumors, and heavy smokers (20+ pack/year) without disease. Promoter methylation of NISCH was tested by quantitative fluorogenic real-time PCR in all samples. Promoter methylation of NISCH occurred after exposure to mainstream tobacco smoke as well as to side stream tobacco smoke in normal oral keratinocyte cell lines. NISCH methylation was also detected in 68% of high-risk, heavy smokers without detectable tumors. Interestingly, in light smokers, NISCH methylation was present in 69% of patients with lung cancer and absent in those without disease. Our pilot study indicates that tobacco smoke induces methylation changes in the NISCH gene promoter before any detectable cancer. Methylation of the NISCH gene was also found in lung cancer patients’ plasma samples. After confirming these findings in longitudinally collected plasma samples from high-risk populations (such as heavy smokers), examining patients for hypermethylation of the NISCH gene may aid in identifying those who should undergo additional screening for lung cancer.
Keywords: lung cancer, Nisch, methylation, smoking, tobacco
Introduction
Lung cancer is the leading cause of cancer death in both men and women.1 The majority (80–90%) of lung cancer cases are due to cigarette smoking. Tobacco use is involved in the development of human cancers by inducing DNA methylation changes.2-5
Epigenetic changes such as hypermethylation of tumor suppressor genes (TSGs) are implicated in malignant transformation.6 DNA hypermethylation refers to the addition of a new methyl group to the cytosine ring of those cytosines that precede a guanosine (called CpG dinucleotides) to form methyl-cytosine (5-methyl-cytosine). CpG dinucleotides are found at increased frequency in the promoter region of many genes. Hypermethylation of CpG islands of tumor suppressors leads to loss of gene expression.7 These epigenetic changes are early events in tumorigenesis and have been found in precursor lesions of the lung,8 thyroid9 and colon.10
In 2000, Alahari et al. discovered a novel protein, Nischarin, which inhibits cell migration.11 Nischarin reverses the effects of the Rac oncogene on lamellipodia formation. Further investigation of Nischarin by Alarhai in 2003 demonstrated that Nischarin impairs Rac-induced cell migration and invasion in breast and colon carcinomas.12 Nischarin was also found to inhibit Rac-1 stimulated transformation of NIH3T3 cells.13 Tumor suppressive activity of NISCH (the gene that codes for Nischarin) is described by Baranwal et al. 2011.14 The authors showed that normal human breast tissue had higher expression of Nischarin mRNA than tumor tissue. They also demonstrated that overexpression of NISCH in the breast cancer cell line MDA-MB-231 reduced tumor growth and metastasis.14 The NISCH gene is located on chromosome 3p21, an area known to be lost in lung cancers. Together, these data suggest that Nischarin acts as a tumor suppressor. We have found the NISCH gene to be hypermethylated and inactivated in lung cancer tissue15 as well as in circulating DNA in the plasma of 86% of lung cancer patients (with and without smoking history),16 suggesting that NISCH hypermethylation could serve as a tumor marker for the early detection/screening of lung cancer. One observation made in our plasma study16 was that methylation of the NISCH gene was also found in plasma DNA in 66% of controls before a statistical cutoff was made. This control group was primarily made up of high-risk, heavy smokers (20+ pack/year). This interesting observation prompted us to determine whether there is any relationship between exposures of cigarette smoke extract (CSE) and NISCH promoter methylation.
In this study we aimed to determine whether NISCH methylation is related to cigarette smoke exposure. To this end, we tested methylation of NISCH after oral keratinocytes were exposed to mainstream and side stream CSE in culture. The effect of mainstream smoke (MSE) or smoke inhaled directly by the smokers was tested and compared with side stream smoke (SSE), smoke from the burning end of the cigarette. To determine human relevance, we also examined NISCH methylation in plasma from a cohort of lifetime non-smokers and light smokers (< 20 pack/year), with and without lung tumors, and heavy smokers without disease.
Results
To determine whether cigarette smoking has a direct effect on methylation of the NISCH gene promoter, we treated normal human oral keratinocytes (HOK) with CSE. Cells were treated at passage 3 and with 0.1% MSE and 0.1% SSE in separate flasks. After one week of treatment (round 1), cells were passaged and DNA was extracted for quantitative methylation-specific PCR (QMSP). No morphological changes were noticeable. Cells at passage 4 then underwent a second round (round 2) of treatment with 0.1% MSE and 0.1% SSE. After the second round, the cells were again harvested and DNA was extracted for QMSP. All treated cells were found to harbor promoter methylation of the NISCH gene, while untreated cells were unmethylated (Fig. 1). As expected, quantitative methylation values were higher in MSE than SSE. We also noticed that methylation already occurred during round 1 of treatment with no further increase in methylation after the second round. These results suggest that methylation of the NISCH promoter is directly induced by cigarette smoke.
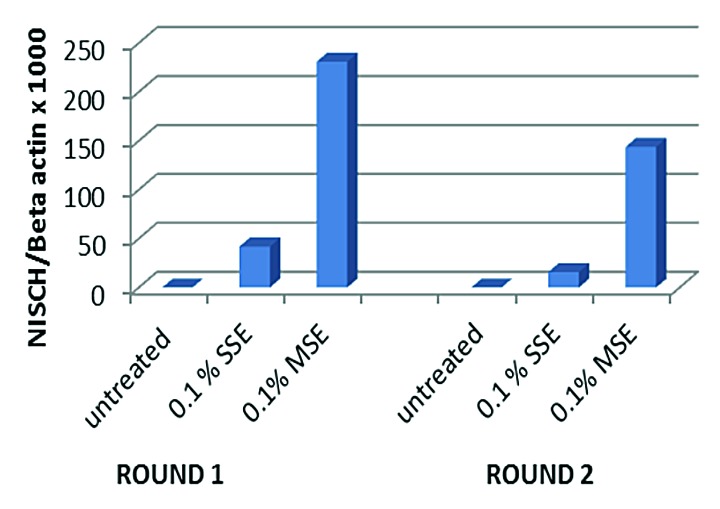
Figure 1. Bar graph of quantitative fluorogenic real-time PCR analysis of the NISCH gene promoter. Normal human oral keratinocytes (HOK) cells were treated with 0.1% side stream smoke extract (SSE) and 0.1% mainstream smoke extract (MSE). Cells exposed to smoke showed hypermethylation of the NISCH gene promoter while untreated cells did not.
To determine the effect of smoking in human subjects, hypermethylation of the NISCH gene was analyzed by QMSP in the plasma DNA of healthy controls. The controls were made up of lifetime non-smokers, light smokers (< 20 pack/year), 20–35 pack/year smokers, and 35+ pack/year smokers (Table 1). The smokers were either current smokers or former smokers. Promoter methylation of NISCH was not seen in lifetime non-smokers or light smokers (0/20) (Fig. 2). A significant difference in methylation was seen in healthy individuals who smoked over 20 pack/year (p < 0.0001) (Fig. 2). Methylation of NISCH was detected in 65% (24/37) of 20–35 pack/year smokers and 71% (34/38) of those who smoked more than 35 pack/year (Fig. 2). Methylation differences were not seen between the groups that smoke above 20 pack/year (p = 0.64) (Fig. 2). Promoter hypermethylation of NISCH was detected in current as well as in former smokers (Table 2). However, no significant differences in NISCH methylation were observed among former and current smokers (Table 3). These findings suggested that NISCH promoter methylation is an indicator of tobacco smoke exposure.
Table 1. Demographics of samples tested.
| I. Controls | |
|---|---|
| i. Lifetime non-smokers and < 20 pack/year smokers (n = 20) | |
|
Mean age (range) |
52.8 (34–78) |
|
Gender | |
| Male |
5 |
| Female |
15 |
|
Smoking History | |
| Former |
6 |
| Current |
2 |
| Non-Smoker |
12 |
|
ii. 20–35 pack/year smokers (n = 37) | |
|
Mean age (range) |
50 (34–83) |
|
Gender | |
| Male |
19 |
| Female |
18 |
|
Smoking History | |
| Former |
17 |
| Current |
20 |
|
iii. 35+ pack/year smokers (n = 48) | |
|
Mean age (range) |
58 (43–83) |
|
Gender | |
| Male |
23 |
| Female |
25 |
|
Smoking History | |
| Former |
25 |
| Current |
23 |
|
II. Cancer Cases | |
|
Lifetime non-smokers and < 20 pack/year smokers (n = 13) | |
|
Mean age (range) |
59 (31–85) |
|
Gender | |
| Male |
7 |
| Female |
6 |
|
Smoking History | |
| Former |
7 |
| Current |
3 |
| Non-Smoker | 3 |
Patients underwent chest CT scan and were divided into two groups based on CT findings. Controls (n = 105) had no nodules identified on CT scan. Cancer cases (n = 13) had confirmed solid cancerous tumors. The control group was further divided by pack/year smoking history: Lifetime non-smokers and < 20 pack/year smokers (n = 20), 20–35 pack/year smokers (n = 38) and 35+ pack/year smokers (n = 47).

Figure 2. Scatter plot of quantitative fluorogenic real-time PCR analysis of the NISCH gene promoter in the plasma DNA of the control group. Hypermethylation was detected in controls who smoked 20+ pack/year. *LNS, life time non-smokers; **py, pack/year.
Table 2. Methylation of NISCH was seen in plasma from controls in both former smokers and current smokers who smoked 20+ pack/year.
| Controls | Former | Current |
|---|---|---|
| 20–35 py |
11/17 (64%) |
13/20 (65%) |
| 35+ py |
15/25 (60%) |
19/23 (83%) |
| Combined (20+ py) | 26/42 (62%) | 32/43 (75%) |
Py, pack/year.
Table 3. Statistical associations for methylation of NISCH in controls and cancer cases.
| p values |
|
||
|---|---|---|---|
| pack/year in controls | Controls |
Cancer (LNS and < 20 py) | |
| (LNS and < 20 py) | 35+ py | ||
| LNS and < 20 py |
- |
< 0.0001 |
< 0.0001 |
| ≥ 20–35 py |
< 0.0001 |
0.64 |
- |
| Combined (20+ py) | < 0.0001 | - | - |
LNS, lifetime non-smokers; py, pack/year.
We also examined methylation of NISCH in the free circulating plasma DNA of lifetime non-smokers or light smokers diagnosed with lung cancer. Methylation of NISCH in the plasma DNA was seen in 69% (9/13) of lung cancer patients with a light smoking history, yet completely absent in non-smoking controls (p < 0.0001) (Fig. 3). Thus, NISCH promoter methylation is absent in non-smokers and light smokers without disease, but present in heavier smokers and those with lung cancer, even if the latter were lighter smokers. Consistent with our previous report,16 these findings demonstrated that NISCH promoter methylation could be a marker of presence of disease, although additional studies are needed to confirm this observation.
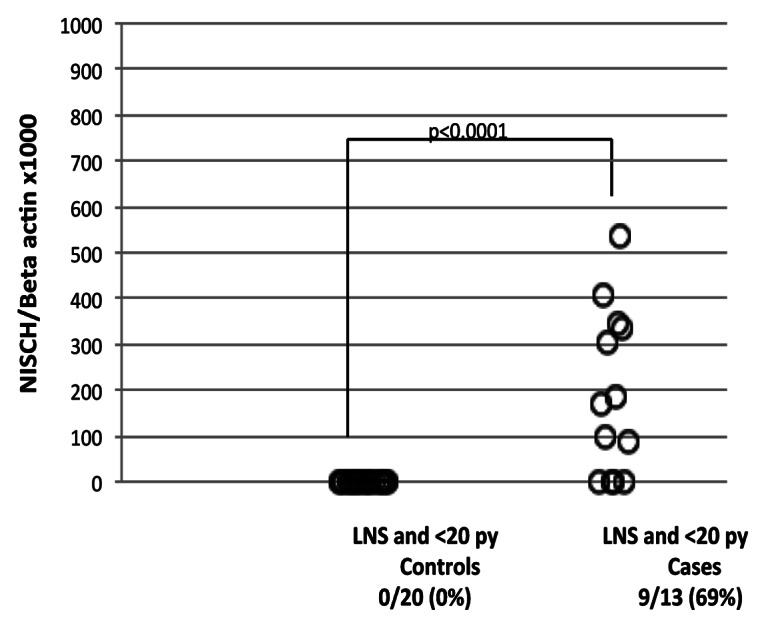
Figure 3. Scatter plot of quantitative fluorogenic real-time PCR analysis of the NISCH gene promoter in plasma DNA from lifetime non-smokers and light smokers: control group vs. cancer cases. *LNS, life time non-smokers; **py, pack/year
Discussion
Identifying those patients at increased risk for lung cancer is important in developing effective early detection strategies. While low-dose CT scanning is a promising screening test for detecting lung cancer, the scans cannot distinguish between benign nodules and cancerous tumors.17 Early detection screening would be enhanced by combining CT scan with accurate biomarker tests, thus determining which patients with nodules are likely to actually harbor cancer17. Our current works describes a tumor suppressor gene, NISCH, which is methylated in cells directly exposed to CSE, in patients with lung cancer, and in patients who are at high risk (20+ pack/year—current and former smokers) for the development of this disease. Therefore, NISCH promoter methylation appears to be a marker of prolonged cigarette exposure and displays an interesting “cliff pattern” in the plasma, present only after approximately 20+ pack/year of smoking. What needs to be determined is whether detection of NISCH methylation in plasma DNA can help “rule in” patients with nodules for more invasive diagnostic biopsies of a screened nodule. In our previous study,16 NISCH methylation was detected in 86% of high-risk 20+ pack/year smokers with cancer as well as in 68% of high-risk smokers without disease (p = 0.03) detected by CT scan. Follow up on these high-risk patients without disease but with methylated NISCH will elucidate the usefulness of NISCH as an early biomarker of disease. Alternatively, NISCH methylation may play a “rule out” role for patients with nodules because it can accurately identify those patients with much less cumulative cigarette exposure as a biologic “dosimeter” of exposure. Therefore, those with a negative NISCH methylation test may not need further screening. Again, before clinical use, this findings need to be confirmed in a longitudinally follow up cohort.
NISCH hypermethylation is seen in healthy controls that are heavy smokers; hence, NISCH methylation alone is clearly not enough to induce the development of cancer. It is widely accepted that lung cancer development is a multistep process that involves genetic mutations and epigenetic modifications.18,19 Epigenetic alterations have been previously described in non-malignant tissues and plasma of cancer-free individuals.5,20 Promoter methylation in these individuals may be a marker of disease, the result of exposure to a carcinogen, or both.20 We do not know how CSE or active smoking leads to methylation of the NISCH promoter and why this promoter is so susceptible to this methylation. It is possible that NISCH promoter methylation is simply a consequence of selection whereby cells that harbor this methylation survive the toxic effects of cigarette smoke carcinogens. If so, the percentage of cells at baseline with this methylation must be very low, because QMSP has a sensitivity of at least 1 in 1,000 alleles and did not identify NISCH methylation in untreated keratinocytes.
Tobacco smoke is the major contributor to the development of lung cancer. Smoking cessation has helped reduce the number of current smokers but almost 50% of all lung cancer cases occur in former smokers.19 Therefore, a major effort is focused on identifying high-risk former smokers. As all the smokers do not develop lung cancer, it is important to identify markers of early-stage disease that predict the molecular risk factors for developing lung cancer. The NISCH gene remains methylated in the plasma DNA of former smokers even after quitting. Sixty two percent of former smokers (20+ pack/year) still showed methylation of the NISCH gene (Table 2), even though they were not found to harbor cancerous nodules. Because patients were grouped into pack/year categories we could not directly compare the exact level of NISCH methylation in plasma DNA with exact number of years of smoking. There could be an undefined quantitative relationship in the first years of smoking as patients reach and then pass 20 pack/year that could be useful in defining cancer risk. Prospective studies with serial sampling of plasma DNA are needed to further elucidate the role of NISCH methylation in early detection efforts.
We have shown that NISCH methylation is induced by mainstream smoke and, to a lesser degree, in side stream smoke in cell culture. Exposure to second-hand smoke is associated with an increased risk of lung cancer among lifetime non-smokers.21 Approximately 10% of all lung cancer cases occur in lifetime non-smokers.22 In this study, we detected NISCH methylation in 69% of lifetime non-smokers and light smokers with lung cancer. We do not have information from these patients in regards to second-hand smoke (SHS) exposure. The development of a risk-associated biomarker for lung cancer is especially important for those who were exposed to SHS but who are non-smokers themselves. Promoter methylation of TSGs was reported previously in SHS-exposed lung cancer patients.23 It would be interesting to test patients exposed to SHS for the presence of NISCH promoter methylation. Several epidemiological studies have suggested that exposure to SHS in childhood increases the risk of lung cancer in adulthood.24-27 A study by Asomaning et al. (2008) demonstrated an increased risk for lung cancer in people who were exposed to SHS before age 25.24 Data from a 2009 NCI-MD lung cancer study demonstrated that childhood SHS exposure correlates with increased risk for lung cancer in adults who were lifetime non-smokers.26 In lifetime non-smokers who were exposed to SHS, diagnosis of lung cancer occurred 15 y earlier than in those not exposed.26 Although legislation is decreasing SHS in public areas, many smokers still smoke in the home and around their children.28 NISCH may be useful as a marker of those who have been exposed to SHS and who may be at increased risk for lung cancer.
Materials and Methods
Collection of samples
Individuals were selected from study populations recruited by the Specialized Program of Research Excellence in Lung Cancer at the University of Pittsburgh and the New
York University (NYU) Lung Cancer Biomarker Center. Plasma was collected from individuals with no history of smoking or < 20 pack/year of smoking history (20 disease-free individuals and 13 individuals with histologically confirmed cases of primary lung cancer). Also plasma was collected from 85 disease-free individuals who were heavy smokers (> 20 pack/year smoking history). A complete smoking history was collected for each individual.16 Those with a positive smoking history were current smokers, and ex-smokers (Table 1).
DNA extraction
Plasma DNA was extracted by digestion with 50 μg/mL proteinase K (Boehringer Mannheim) in the presence of 1% SDS at 48°C overnight followed by phenol/chloroform extraction and ethanol precipitation.
Bisulfite treatment
DNA extracted from 1 mL of blood plasma was subjected to bisulfite treatment, using the EpiTect Bisulfite kit from Qiagen according to the manufacturer's conditions, www.Qiagen.com. Bisulfite-treated DNA was eluted in 30 μL of elution buffer and stored at −80°C.
Methylation analysis by Quantitative Methylation Specific PCR (QMSP)
Bisulfite-modified DNA was used as template for fluorescence-based real-time PCR, as previously described.29 Amplification reactions were performed in duplicate in a volume of 20 μL that contained 2 μL bisulfite modified DNA; 600 nmol/L forward and reverse primers; 200 nmol/L probe; 5 units of platinum Taq polymerase (Invitrogen); 200 μmol/L each of dATP, dCTP, and dGTP; 200 μmol/L dTTP; and 5.5 mmol/L MgCl2. Primers and probes were designed to specifically amplify the promoter of the NISCH gene and the promoter of a reference gene, ACTB as described.15,16 Amplifications were performed using the following profile: one step at 95°C for 3 min, 50 cycles at 95°C for 15 sec, and 58 C for 1 min. Amplification reactions were performed in 384-well plates in a 7900 Sequence detector (Perkin-Elmer Applied Biosystems) and were analyzed by SDS 2.2.1 Sequence Detector System (Applied Biosystems). Each plate included patient or cell line DNA samples, positive (in vitro methylated leukocyte DNA) and negative (normal leukocyte DNA or DNA from a known unmethylated cell line) controls, and multiple water blanks. In vitro methylated leukocyte DNA was purchased from Zymo laboratories, and serial dilutions (90–0.009 ng) of this DNA were used to construct a calibration curve for each plate. All samples were within the assay's range of sensitivity and reproducibility based on amplification of internal reference standard (CT value for ACTB of 40 or less). The relative level of methylated DNA for each gene in each sample was determined as a ratio of methylation-specific PCR-amplified gene to ACTB (reference gene) and then multiplied by 1,000 for easier tabulation (average value of duplicates of gene of interest/ average value of duplicates of ACTB × 1,000).
Cell culture
Normal Human Oral Kerationcytes (HOK) were purchased from Sciencell Research Laboratories and grown in keratinocytes medium with supplements as specified by the supplier.
Preparation of cigarette smoke extract
Mainstream (MSE) and side stream smoke extracts (SSE) were made from research-grade cigarettes (2R4F, from Tobacco Health Research, University of Kentucky, contains nicotine: 0.85 mg/cigarette and tar: 9.70 mg/cigarette). Side stream smoke was collected from the burning end of the cigarettes without puffing at a rate of 200 ml/min and mainstream smoke with 35 ml/min puff per 2 sec from the opposite end using two smoking machines (MasterFlex Pump Systems, Cole-Parmer Instrument) as described.30 Briefly, the smoke of 20 cigarettes for MSE and 40 cigarettes for SSE was bubbled into each flask containing 20 ml of pre-warmed PBS. The aqueous smoke extract was filtered through 0.22-lm pore syringe filter to remove large particles. The smoke bubbled into MSE flask was acidic (yellow in color) and that into SSE flask basic (pink), so the pH of each solution was adjusted to 7.4. The solution was aliquoted and kept frozen at -80 C until use.
Treatment of HOK cells with CSE
Cells at passage 3 were treated with CSE as follows. Passage 3 cells were divided into 3 T-25 flasks. These flasks were treated with 0.1% MSE, 0.1% SSE, and one was untreated as a control. Cells were treated every other day until confluent (~8 d). The flasks were then passaged and DNA was extracted (Round 1). Cells at passage 4 continued treatment until confluent (Round 2). The cells were then harvested and DNA was extracted for QMSP analysis.
Statistical analysis
Statistical analysis of the clinical and biological characteristics of study subjects was performed using Fisher’s exact test. p < 0.05 was considered to be statistically significant.
Glossary
Abbreviations:
- QMSP
quantitative methylation specific PCR
- MSE
mainstream smoke
- SSE
side stream smoke
- HOK
human oral keratinocytes
- NS
non-smoker
- LNS
lifetime non-smoker
- PY
pack/year
Disclosure of Potential Conflicts of Interest
Early Detection Research Network (EDRN) grant U01-CA084986 from the National Cancer Institute (NCI), MDHealthDX, SA., NCI grant EDRN U01 CA086137 NYU Lung Cancer Biomarker Center; and FAMRI Young Clinical Scientist Award, International Association for the Study of Lung Cancer. M.O.H. is supported by FAMRI Clinical Innovative Award#103015_CIA. The funding agency had no role in the design of the study, data collection or analysis, in the interpretation of the results, in the preparation of the manuscript or the decision to submit the manuscript for publication. Under a licensing agreement between MDHealthDX, SA and the Johns Hopkins University, D.S. is entitled to a share of royalty received by the University upon sales of diagnostic products described in this article. D.S. owns MDHealthDX, SA stock, which is subject to certain restrictions under University policy. D.S. is a paid consultant to MDHealthDX, SA and is a paid member of the company's Scientific Advisory Board. The Johns Hopkins University in accordance with its conflict of interest policies is managing the terms of this agreement.
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/24195
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Belinsky SA, Palmisano WA, Gilliland FD, Crooks LA, Divine KK, Winters SA, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002;62:2370–7. [PubMed] [Google Scholar]
- 3.Chang HW, Ling GS, Wei WI, Yuen AP. Smoking and drinking can induce p15 methylation in the upper aerodigestive tract of healthy individuals and patients with head and neck squamous cell carcinoma. Cancer. 2004;101:125–32. doi: 10.1002/cncr.20323. [DOI] [PubMed] [Google Scholar]
- 4.Soria JC, Rodriguez M, Liu DD, Lee JJ, Hong WK, Mao L. Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res. 2002;62:351–5. [PubMed] [Google Scholar]
- 5.Zöchbauer-Müller S, Lam S, Toyooka S, Virmani AK, Toyooka KO, Seidl S, et al. Aberrant methylation of multiple genes in the upper aerodigestive tract epithelium of heavy smokers. Int J Cancer. 2003;107:612–6. doi: 10.1002/ijc.11458. [DOI] [PubMed] [Google Scholar]
- 6.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–92. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 7.Leonhardt H, Rahn HP, Cardoso MC. Functional links between nuclear structure, gene expression, DNA replication, and methylation. Crit Rev Eukaryot Gene Expr. 1999;9:345–51. doi: 10.1615/CritRevEukarGeneExpr.v9.i3-4.190. [DOI] [PubMed] [Google Scholar]
- 8.Belinsky SA, Liechty KC, Gentry FD, Wolf HJ, Rogers J, Vu K, et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006;66:3338–44. doi: 10.1158/0008-5472.CAN-05-3408. [DOI] [PubMed] [Google Scholar]
- 9.Hoque MO, Rosenbaum E, Westra WH, Xing M, Ladenson P, Zeiger MA, et al. Quantitative assessment of promoter methylation profiles in thyroid neoplasms. J Clin Endocrinol Metab. 2005;90:4011–8. doi: 10.1210/jc.2005-0313. [DOI] [PubMed] [Google Scholar]
- 10.Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000;60:4366–71. [PubMed] [Google Scholar]
- 11.Alahari SK, Lee JW, Juliano RL. Nischarin, a novel protein that interacts with the integrin alpha5 subunit and inhibits cell migration. J Cell Biol. 2000;151:1141–54. doi: 10.1083/jcb.151.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alahari SK. Nischarin inhibits Rac induced migration and invasion of epithelial cells by affecting signaling cascades involving PAK. Exp Cell Res. 2003;288:415–24. doi: 10.1016/S0014-4827(03)00233-7. [DOI] [PubMed] [Google Scholar]
- 13.Reddig PJ, Xu D, Juliano RL. Regulation of p21-activated kinase-independent Rac1 signal transduction by nischarin. J Biol Chem. 2005;280:30994–1002. doi: 10.1074/jbc.M502546200. [DOI] [PubMed] [Google Scholar]
- 14.Baranwal S, Wang Y, Rathinam R, Lee J, Jin L, McGoey R, et al. Molecular characterization of the tumor-suppressive function of nischarin in breast cancer. J Natl Cancer Inst. 2011;103:1513–28. doi: 10.1093/jnci/djr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoque MO, Kim MS, Ostrow KL, Liu J, Wisman GB, Park HL, et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008;68:2661–70. doi: 10.1158/0008-5472.CAN-07-5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ostrow KL, Hoque MO, Loyo M, Brait M, Greenberg A, Siegfried JM, et al. Molecular analysis of plasma DNA for the early detection of lung cancer by quantitative methylation-specific PCR. Clin Cancer Res. 2010;16:3463–72. doi: 10.1158/1078-0432.CCR-09-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider J. Early detection of lung cancers - Comparison of computed tomography, cytology and fuzzy-based tumor markers panels. Cancer Biomark. 2010;6:149–62. doi: 10.3233/CBM-2009-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osada H, Takahashi T. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene. 2002;21:7421–34. doi: 10.1038/sj.onc.1205802. [DOI] [PubMed] [Google Scholar]
- 19.Sato M, Shames DS, Gazdar AF, Minna JD. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol. 2007;2:327–43. doi: 10.1097/01.JTO.0000263718.69320.4c. [DOI] [PubMed] [Google Scholar]
- 20.Brait M, Ford JG, Papaiahgari S, Garza MA, Lee JI, Loyo M, et al. Association between lifestyle factors and CpG island methylation in a cancer-free population. Cancer Epidemiol Biomarkers Prev. 2009;18:2984–91. doi: 10.1158/1055-9965.EPI-08-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Husgafvel-Pursiainen K. Genotoxicity of environmental tobacco smoke: a review. Mutat Res. 2004;567:427–45. doi: 10.1016/j.mrrev.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Levitz JS, Bradley TP, Golden AL. Overview of smoking and all cancers. Med Clin North Am. 2004;88:1655–75, xiii. doi: 10.1016/j.mcna.2004.07.005. [xiii.] [DOI] [PubMed] [Google Scholar]
- 23.Scesnaite A, Jarmalaite S, Mutanen P, Anttila S, Nyberg F, Benhamou S, et al. Similar DNA methylation pattern in lung tumours from smokers and never-smokers with second-hand tobacco smoke exposure. Mutagenesis. 2012;27:423–9. doi: 10.1093/mutage/ger092. [DOI] [PubMed] [Google Scholar]
- 24.Asomaning K, Miller DP, Liu G, Wain JC, Lynch TJ, Su L, et al. Second hand smoke, age of exposure and lung cancer risk. Lung Cancer. 2008;61:13–20. doi: 10.1016/j.lungcan.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janerich DT, Thompson WD, Varela LR, Greenwald P, Chorost S, Tucci C, et al. Lung cancer and exposure to tobacco smoke in the household. N Engl J Med. 1990;323:632–6. doi: 10.1056/NEJM199009063231003. [DOI] [PubMed] [Google Scholar]
- 26.Olivo-Marston SE, Yang P, Mechanic LE, Bowman ED, Pine SR, Loffredo CA, et al. Childhood exposure to secondhand smoke and functional mannose binding lectin polymorphisms are associated with increased lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18:3375–83. doi: 10.1158/1055-9965.EPI-09-0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang FL, Love EJ, Liu N, Dai XD. Childhood and adolescent passive smoking and the risk of female lung cancer. Int J Epidemiol. 1994;23:223–30. doi: 10.1093/ije/23.2.223. [DOI] [PubMed] [Google Scholar]
- 28.StatBite: reported secondhand smoke exposure in the home among Americans. J Natl Cancer Inst. 2008;100:1278. doi: 10.1093/jnci/djn328. [DOI] [PubMed] [Google Scholar]
- 29.Harden SV, Tokumaru Y, Westra WH, Goodman S, Ahrendt SA, Yang SC, et al. Gene promoter hypermethylation in tumors and lymph nodes of stage I lung cancer patients. Clin Cancer Res. 2003;9:1370–5. [PubMed] [Google Scholar]
- 30.Kim MS, Huang Y, Lee J, Zhong X, Jiang WW, Ratovitski EA, et al. Cellular transformation by cigarette smoke extract involves alteration of glycolysis and mitochondrial function in esophageal epithelial cells. Int J Cancer. 2010;127:269–81. doi: 10.1002/ijc.25057. [DOI] [PMC free article] [PubMed] [Google Scholar]