

Abstract
Polo-like kinase 1 has been established as one of the most attractive targets for molecular cancer therapy. In fact, multiple small-molecule inhibitors targeting this kinase have been developed and intensively investigated. Recently, it has been reported that the cytotoxicity induced by Plk1 inhibition is elevated in cancer cells with inactive p53, leading to the hypothesis that inactive p53 is a predictive marker for the response of Plk1 inhibition. In our previous study based on different cancer cell lines, we showed that cancer cells with wild type p53 were more sensitive to Plk1 inhibition by inducing more apoptosis, compared with cancer cells depleted of p53. In the present work, we further demonstrate that in the presence of mitotic stress induced by different agents, Plk1 inhibitors strongly induced apoptosis in HCT116 p53+/+ cells, whereas HCT116 p53−/− cells arrested in mitosis with less apoptosis. Depletion of p53 in HCT116 p53+/+ or U2OS cells reduced the induction of apoptosis. Moreover, the surviving HCT116 p53−/− cells showed DNA damage and a strong capability of colony formation. Plk1 inhibition in combination with other anti-mitotic agents inhibited proliferation of tumor cells more strongly than Plk1 inhibition alone. Taken together, the data underscore that functional p53 strengthens the efficacy of Plk1 inhibition alone or in combination by strongly activating cell death signaling pathways. Further studies are required to investigate if the long-term outcomes of losing p53, such as low differential grade of tumor cells or defective DNA damage checkpoint, are responsible for the cytotoxicity of Plk1 inhibition.
Keywords: p53, BI 2536, BI 6727, Poloxin, monastrol
Introduction
Polo-like kinase 1 (Plk1), the best-characterized member of the Plk family, is crucial for the cell cycle and exerts multiple functions throughout mitosis.1-3 Overexpression of Plk1 enables cells to override control checkpoints and to promote transformation of mammalian cells.1,4 In line with these observations, numerous studies reveal that elevated Plk1 in tumor tissues is well correlated with a poor prognosis of tumor patients.4,5 Moreover, Plk1 has been identified as the only kinase selectively required for the viability of Ras cancer cells in a genome-wide RNA interference screening.6 Thus, Plk1 has been widely considered as one of the most promising targets for molecular intervention. In fact, multiple small-molecule inhibitors targeting the enzymatic kinase domain and the regulatory Polo-box binding domain (PBD) have been recently developed and characterized.1,7-19 In particular, BI 2536 and BI 6727 are the most intensively investigated Plk1 inhibitors.20-25 Poloxin, the first reported non-peptidic inhibitor of the PBD of Plk1, shows its specificity and anti-proliferative activity in vitro as well as in vivo.15-17 While the preclinical data of Plk1 inhibitors are encouraging, the clinical results are rather less inspiring, showing limited anticancer activity.20,23,26,27 It is of importance to identify the molecules and mechanisms responsible for the sensitivity of Plk1 inhibitors.
It has been reported that the cytotoxicity resulting from Plk1 inhibition is elevated in cancer cells with defective p53,28-30 leading to the hypothesis that the inactive p53 might be a predictive marker for sensitive response of Plk1 inhibition. However, in our previous work based on various cancer cell lines with or without functional p53, we demonstrated that inactive p53 is clearly not a predictor for the sensitive response to Plk1 inhibition.15 In contrast, cancer cells with wild type p53 responded more strongly in apoptosis induction than cancer cells without p53.15 We could not exclude the possibility that other circumstances, such as mitotic stress or DNA damage, could render cancer cells with inactive p53 more susceptible to Plk1 inhibitors. In the current work, we have systematically addressed whether mitotic stress, which is very often observed in cancer cells, could affect the efficiency of Plk1 inhibitors in cancer cells with or without functional p53.
Results
Plk1 inhibitors trigger more apoptosis in HCT116 p53+/+ cells than in HCT116 p53−/− cells under mitotic stress
To address the impact of mitotic stress on the efficiency of Plk1 inhibitors in context of the p53 status of cancer cells, we have chosen the isogenic HCT116 p53+/+ and HCT116 p53−/− cell lines, as they comprise comparable cellular context with the exception of the p53 status and are very well characterized.31 Microtubule destabilizer nocodazole and vincristine, microtubule stabilizer paclitaxel and the kinesin Eg5 inhibitor monastrol were chosen as mitotic stress inducers for pretreatment. As indicated in the figure legend, all mitotic stress inducers were used in a low dose after performing dose-kinetics, so that they induce mitotic stress but not yet apoptosis during pretreatment. BI 2536 and BI 6727,12,32,33 two of the most intensively studied kinase domain inhibitors, and Poloxin, the selective PBD inhibitor,16,17 were taken as representatives of Plk1 inhibitors.
As illustrated in Figure 1A, cells were treated with a mitotic stress inducer for 10 h and then further incubated with Plk1 inhibitor Poloxin, BI 2536 or BI 6727 for 28 h. Cells were harvested, and cellular lysates were prepared for western blot analysis. HCT116 p53+/+ cells, pretreated with monastrol then with Plk1 inhibitor, clearly displayed more apoptosis by showing cleaved poly(ADP-ribose) polymerase (PARP), an apoptosis marker (Fig. 1B, first row), increased pro-apoptotic protein Bax (Fig. 1B, fourth row) and enhanced caspase-3/7 activity (Fig. 1F). By contrast, monastrol-Plk1 inhibitor-treated HCT116 p53−/− cells exhibited mitotic arrest, as evidenced by increased mitotic proteins Plk1 and cyclin B1 (Fig. 1B, second and third rows) and less apoptosis (Fig. 1F) accompanied with increased anti-apoptotic proteins Mcl-1 and Bcl-2 (Fig. 1B, fifth and sixth rows). Mitotic arrest in HCT116 p53−/− cells was further underscored by displaying an increased G2/M peak via FACS analysis (data not shown). The same experiments were also performed using microtubule stabilizer paclitaxel, microtubule destabilizer nocodazole and vincristine. As shown in Figure 1C–E, similar to monastrol-Plk1 inhibitor-treated cells (Fig. 1B), HCT116 p53+/+ cells demonstrated a strong apoptosis induction (Fig. 1C–E, first row) with increased Bax (Fig. 1C–E, fourth rows), whereas HCT116 p53−/− cells showed a mitotic arrest (Fig. 1C–E, second and third rows) with increased pro-survival molecules Mcl-1 and Bcl-2 (Fig. 1C, fifth and sixth rows; Fig. 1D and E, fifth row). The activities of caspase-3/7 in HCT116 p53+/+ cells were much higher upon vincristine alone or combined treatment than in HCT116 p53−/− cells (Fig. 1G). Taken together, in the presence of mitotic stress, Plk1 inhibitors trigger a strong apoptosis in HCT116 cells with functional p53, whereas they arrest HCT116 cells without p53 in mitosis with less apoptosis.

Figure 1. Plk1 inhibitors trigger more apoptosis in HCT116 p53+/+ cells than in HCT116 p53−/− cells in the presence of mitotic stress. (A) Illustration of experimental schedule. Cells were pretreated with a low dose of mitotic stress inducers (15 µM monastrol, 7 nM paclitaxel, 1.2 nM vincristine or 50 ng/ml nocodazole) for 10 h, then Plk1 inhibitor (25 µM Poloxin, 25 nM BI 2536 or BI 6727) was added for further 28 h. (B) Western blot analysis for cells treated with monastrol and Plk1 inhibitors. HCT116 p53+/+ or HCT116 p53−/− cells were treated as described in (A) and harvested for western blot analysis using indicated antibodies. β-actin served as loading control. (C) Western blot analysis for cells treated with paclitaxel and Plk1 inhibitors. (D) Western blot analysis for cells treated with vincristine and Plk1 inhibitors. (E) Western blot analysis for cells treated with nocodazole and Plk1 inhibitors. (F) Quantification of relative activity of caspase-3/7 for cells treated as in (B). (G) Quantification of relative activity of caspase-3/7 for cells treated as in (D). The results are presented as mean ± SD; mona, monastrol; vincris, vincristine.
Depletion of p53 reduces apoptosis in HCT116 p53+/+ and U2OS cells
To rule out the genetic background responsible for the different response, HCT116 p53+/+ cells were depleted of p53 with siRNA and then treated as described (Fig. 2A). As shown in Figure 2B, depletion of p53 in HCT116 p53+/+ cells reduced the cleavage of PARP (Fig. 2B, first row), associated with reduced pro-apoptotic protein Bax (Fig. 2B, sixth row). Decreased induction of apoptosis was further corroborated by reduced activity of caspase-3/7 (Fig. 2C). U2OS cells, osteosarcoma cell line with wild type p53, were also treated with siRNA against p53, and comparable results were observed (Fig. 2D and E). The data indicate that induction of apoptosis is indeed dependent on functional p53 in cells upon combined Plk1 treatment.

Figure 2. Depletion of p53 reduces apoptosis in HCT116 p53+/+ and U2OS cells. (A) Experimental schedule. (B) HCT116 p53+/+ cells were treated with 20 nM control siRNA (siRNA con) or siRNA against p53 (siRNA p53) for 24 h and then further incubated as described in (A). Cellular lysates were prepared for western blot analysis with antibodies as indicated. β-actin served as loading control. (C) Cellular lysates were also used for measuring the activity of caspase-3/7. The results are presented as mean ± SD (D) U2OS cells were treated as described in (A). Cellular lysates were prepared for western blot analysis with antibodies as indicated. β-actin served as loading control. (E) Cellular lysates were also used for measuring the activity of caspase-3/7. The results are presented as mean ± SD.
Activation of the spindle assembly checkpoint in HCT116 p53−/− cells
Next, we took a close look into the happening of these cells after combined treatment. HCT116 cells were treated as illustrated in Figure 3A and harvested at indicated time points for western blot analysis. Treated HCT116 cells, regardless of the p53 status, arrested in mitosis at 6 h, showing increased Plk1 and cyclin B1 (Fig. 3B, first and second rows). While HCT116 p53−/− cells kept further in mitosis during 36 h (Fig. 3B, right panel, first and second rows), HCT116 p53+/+ cells were no longer in mitosis (Fig. 3B, left panel, second row). Further analysis by measuring the activity of caspase-3/7 showed that apoptosis occurred already at 6 h, and the activities increased throughout the 36 h duration with single (Fig. 3C, left and middle panels) or double treatment in HCT116 p53+/+ cells (Fig. 3C, right panel). In HCT116 p53−/− cells, apoptosis also took place, yet to a much lesser extent (Fig. 3C). In particular, more apoptosis was induced at 24 h upon combined treatment (Fig. 3C, right panel) than that with single treatment (Fig. 3C, left and middle panels), suggesting efficacy of combined strategy. To examine why HCT116 p53−/− cells still arrested in mitosis, the staining of BubR1, a marker for active spindle assembly checkpoint, was performed. We observed that 95.4% of arrested mitotic cells displayed a strong staining of BubR1 (Fig. 3D). The results indicate that upon combined treatment, HCT116 cells without p53 activate the spindle assembly checkpoint, consequently leading to mitotic arrest, but are not much capable of triggering apoptosis within cells.

Figure 3. Activation of the spindle assembly checkpoint in HCT116 p53−/− cells. (A) Experimental schedule. (B) HCT116 p53+/+ and HCT116 p53−/− cells were treated as described in (A) and harvested at 0, 6, 12, 18, 24 and 36 h for western blot analysis with indicated antibodies. (C) The same lysates were also used for measurement of the caspase-3/7 activity. The results are presented as mean ± SD (D) Treated cells with monastrol and BI 6727 were fixed and stained for BubR1, pericentrin and DNA. The representatives of confocal microscopy are shown. Scale: 5 µm
Phenotype of treated HCT116 cells with or without p53
To understand the mechanisms by which the treated cells activate the spindle assembly checkpoint and arrest in mitosis, indirect immunofluorescence staining was performed. Cells were treated as described in Figure 1A, fixed and stained for Plk1, centrosome (pericentrin), tubulin and DNA. Interestingly, while a low dose of monastrol combined with BI 2536 induced almost 100% monopolar spindle cells (Fig. 4A–C), monastrol together with BI 6727 triggered much less monopolar spindle cells (Fig. 4C). Instead, monastrol with BI 6727 induced rather multipolar spindle cells in both cell lines (Fig. 4A, B and D). It is obvious that the working mechanism of BI 2536 differs from that of BI 6727, although both are targeting the kinase domain of Plk1: while BI 2536 appears to majorly disrupt centrosome separation, showing monopolar spindle cells, BI 6727 impacts centrosome duplication and cytokinesis by displaying multipolar cells (Fig. 4 and our previous data15). We speculate that BI compounds interfere with possibly not only Plk1, but also other kinases responsible for these processes within tumor cells.

Figure 4. Phenotype of treated HCT116 cells with or without p53. Cells were treated with monastrol and Plk1 inhibitors as shown in Figure 1A and stained for Plk1, pericentrin, tubulin and DNA. (A and B) The representatives from CLSM are shown for HCT116 p53+/+ cells (A) and for HCT116 p53−/− cells (B). Scale: 5 µm. (C) Quantification of monopolar spindle. About 300 to 400 mitotic cells were evaluated from each cell line. The results are presented as mean ± SD; con, control; mona,: monastrol. (D) Quantification of multipolar spindle. About 300 to 400 mitotic cells were evaluated from each cell line. The results are presented as mean ± SD; con, control; mona,: monastrol.
HCT116 p53−/− cells show more DNA damage in mitosis than cells with p53 upon treatment
Missegregated chromosomes are frequently damaged during cytokinesis, triggering a DNA double-strand break response even in respective daughter cells.34 Since cells with monopolar or multipolar spindles were not capable of proper chromosome segregation, we were interested in seeing if DNA damage could happen in surviving HCT116 cells after combined treatment. To answer this question, cells were pre-treated with paclitaxel for 10 h and then treated with BI 6727 for 28 h. Treated cells were fixed and stained for γH2AX, a DNA damage marker, tubulin and DNA. As shown in Figure 5, surviving HCT116 p53−/− cells displayed more strong staining of γH2AX than the cells with p53 (Fig. 5A and B). Further analysis revealed that paclitaxel or combined treatment induced more positive staining of γH2AX in HCT116 cells without p53 than HCT116 cells with p53 (Fig. 5C). Even DMSO induced almost 10% cells showing this staining (Fig. 5C), indicating that cells without p53 are susceptible to any damage inducer.

Figure 5. Survived HCT116 p53−/− cells show more mitotic DNA damage than the cells with p53. (A and B) HCT116 p53+/+ cells (A) and HCT116 p53−/− cells (B) were pre-treated with paxlitaxel for 10 h and then treated with BI 6727 for 28 h. Cells were fixed and stained for DNA damage marker γH2AX, for centromere (ACA, anti-centromere antibody), for tubulin and DNA. The representatives are shown. Scale: 5 µm. (C) Quantification of γH2AX foci in about 100–150 mitotic nuclei. The results are presented as mean ± SD. PTX, paclitaxel.
Long-term kinetics of treated cells with or without p53
Next we studied the fate of these DNA-damaged cells. Cells were treated as described in Figure 1A and harvested on day 1, day 2 and day 3 for western blot analysis. Again, HCT116 cells without p53 remained in mitosis after combined treatment even on day 2 and day 3, as evidenced by increased Plk1 and cyclin B1 (Fig. 6A, second and third rows) and showed less apoptosis (Fig. 6A, first row) with enhanced anti-apoptotic protein Bcl-2 (Fig. 6A, fifth row). Interestingly, p21 was also induced in HCT116 p53−/− cells on day 2 and day 3 (Fig. 6A, seventh row) in a p53-independent manner (Fig. 6A, sixth row). By contrast, apoptosis was strongly induced in HCT116 p53+/+ cells (Fig. 6A, first row), accompanied by induction of p53 and p21 (Fig. 6A, sixth and seventh rows), and increased pro-apoptotic protein Bax (Fig. 6A, fourth row). These observations were further underlined by phospho-histone H3 (S10) staining, a mitotic marker (Fig. 6B and C), and active caspase-3 measurement (Fig. 6D and E). Intriguingly, almost 35% of HCT116 p53−/− cells were arrested in mitosis (Fig. 6C, day 1), whereas only 15% of HCT116 p53+/+ cells stayed in mitosis on day 1 upon combined treatment (Fig. 6B, day 1). On day 3, there were still 10% of HCT116 p53−/− cells kept in mitosis (Fig. 6C, day 3). Moreover, 60–80% of HCT116 p53+/+ cells were positively stained with active caspase-3, whereas 20–30% of HCT116 p53−/− cells exhibited the positive staining during these three days (Fig. 6D and E). The data suggest further that in the presence of p53, HCT116 cells are susceptible to combined mitotic agents by showing a strong induction of apoptosis.

Figure 6. Long-time kinetics. (A) Cells were treated with monastrol for 10 h and further incubated together with Plk1 inhibitor BI 6727 for 1 d, 2 d and 3 d. Cells were harvested and cellular lysates were prepared for western blot analysis with indicated antibodies. β-actin served as loading control. (B and C) Treated HCT116 p53+/+ cells (B) and HCT116 p53−/− cells (C) were also collected for phospho-histone H3 (S10) staining and measured by FACS. The results are presented as mean ± SD. (D and E) HCT116 p53+/+ cells (D) and HCT116 p53−/− cells (E) were treated with paclitaxel for 10 h and further incubated with Plk1 inhibitor BI 6727 for 1 d, 2 d and 3 d. Cells were then collected for active caspase-3 staining and measured by FACS. The results are presented as mean ± SD and statistically analyzed. * p < 0.05. ***p < 0.001.
More strongly inhibited proliferation in HCT116 p53+/+ cells upon treatment
Furthermore, we examined anti-proliferation effect of combined treatment on HCT116 cells with or without p53. Cells were treated, and cell titer assay was performed at indicated time points. Proliferation of HCT116 p53+/+ cells was more strongly inhibited than that in HCT116 p53−/− cells (Fig. 7A, left and right panels). Moreover, colony-formation assay was performed. Upon treatment, the colony numbers of HCT116 p53+/+ cells were much less than that of HCT116 p53−/− cells (Fig. 7B).
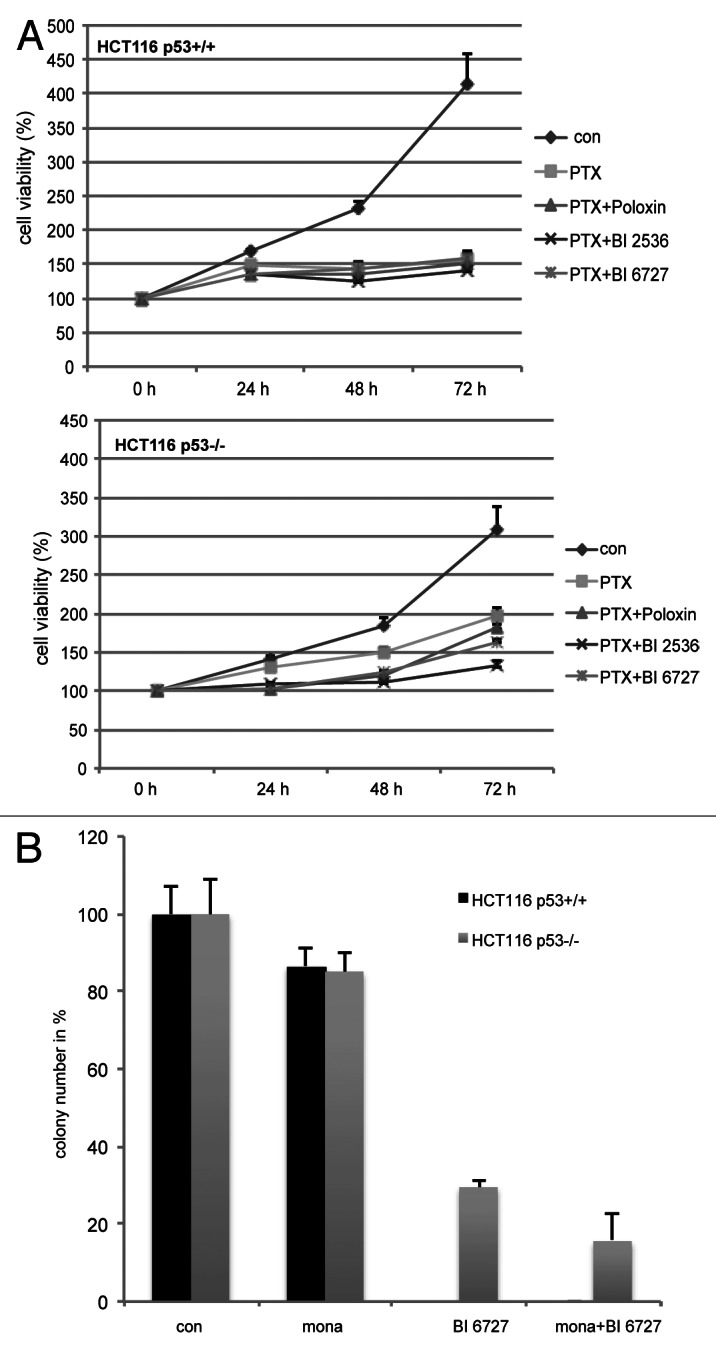
Figure 7. More HCT116 without p53 survived upon combined treatment. (A) Cells were seeded in 96-well plates, pre-treated with paclitaxel for 10 h then incubated with Plk1 inhibitors for indicated time periods and cell viability was measured. The results are presented as mean ± SD (B) Colony-formation assay. Treated cells were seeded in 6-well plates and cultured for 12–14 d. Cells were then stained, and the colony numbers were counted. The results are presented as mean ± SD.
Discussion
Multiple small-molecule inhibitors targeting Plk1 have been intensively investigated with encouraging preclinical data.1 The data from clinical trials are, however, less promising.20,26 It is of importance to explore predictive biomarkers for the response of Plk1 inhibitors to successfully select suitable patients for therapy. Plk1 associates with p53 in many aspects.35-37 In the search of biomarkers for Plk1 inhibitors, several reports suggest that Plk1 inhibition preferably affects p53-deficient cancer cells.29,30 However, we and others observed that Plk1 inhibitors affect both tumor cells with functional or deficient p53 as well as primary/normal non-transformed proliferating cells.16,19,32,38 In the previous work, based on different cancer cell lines, we have demonstrated that cancer cells with wild type p53 actually responded more sensitively to Plk1 inhibitors, as evidenced by a high degree of apoptosis induction.15 In the present work, we have focused on the question whether mitotic stress influences the effect of Plk1 inhibitors in cancer cells in context of the p53 status.
The tumor suppressor p53 serves as a critical signaling hub to determine cell fate in response to genotoxic and other cellular stress.39-43 The inactivation of functional p53 is the most common single mutation observed in a wide variety of cancers.44 As mutant p53 is often associated with malignancy of tumors and with therapy resistance,45,46 restoration of p53 function is widely regarded as an important strategy for combating cancer.47,48 p53 is activated in response to various mitotic stresses, such as aberrant spindle formation, abnormal centrosome separation and chromosome damage or mis-segregation.15,49,50 Our data show that in the presence of mitotic stress induced by various anti-mitotic agents and Plk1 inhibitors, p53 is increased in HCT116 p53+/+ cells, accompanied by elevated downstream effectors Bax and p21 (Fig. 1B–E). Moreover, HCT116 p53+/+ cells respond to combined Plk1 inhibition with a strong induction of apoptosis, whereas HCT116 p53−/− cells arrest in mitosis with less apoptosis (Fig. 1). Interestingly, reduced apoptosis in HCT116 p53−/− cells is associated not only with weak Bax expression, the consequence of the absence of p53, but also with increased anti-apoptotic proteins, such as Mcl-1 and Bcl-2 (Fig. 1B–E). Depletion of p53 reduces apoptosis in HCT116 p53+/+ and U2OS cells, indicating that apoptosis induction is directly associated with functional p53 (Fig. 2). In response to cellular stress, p53 determines cell fate, activating apoptosis or arresting the cell cycle. The latter is usually considered as a means for tumor cells to escape cytotoxicity of anticancer agents. Plk1 combined therapy induces severe mitotic defects, such as aberrant spindles (Fig. 4), which make p53 vote for inducing a strong apoptosis in HCT116 p53+/+ cells. By contrast, in the absence of p53, HCT116 cells treated with combined Plk1 inhibition are neither able to execute a strong apoptosis nor to exit from mitosis, as observed with other anti-microtubule agents.51,52 In addition, γH2AX staining, a DNA damage marker, was enhanced in HCT116 p53−/− cells after combined treatment (Fig. 5). These cells with DNA damage survived longer (Fig. 7A) with less caspase-3 activity (Fig. 6D and E), compared with HCT116 p53+/+ cells. Moreover, the ability of clonogenic survival of HCT116 p53−/− cells was much better than that of HCT116 cells with p53 after treatment (Fig. 7B). In this regard, Plk1 inhibition in cancer cells with inactive p53 could lead to accumulation of polyploidy/aneuploidy due to the lack of p53-mediated cell death signaling pathway. These surviving tumor cells could be more malignant. Collectively, the present data, in line with our previous results,15 demonstrate that wild type p53 makes tumor cells more susceptible to Plk1 inhibitors, alone or in combination with other anti-mitotic agents, than tumor cells without p53.
Furthermore, anti-microtubule agents remain the most reliable anticancer therapy. They interfere with microtubule dynamics, leading to many defects in mitosis and activation of spindle assembly checkpoint.53,54 Undesirable side effects and resistance challenge the use of microtubule-targeting agents.54,55 It is thus desired to develop novel drugs targeting key but nonstructural components of the mitotic machinery. In fact, various inhibitors targeting specific mitotic molecules, like mitotic kinases (Plk1) and mitotic motor proteins (Eg5), have been developed. These drugs are indeed more mitosis selective, yet the response in patients in clinical trials is still limited. Combinatorial strategy may harness the full potential of new anti-mitotic drugs. Interestingly, retinoids sensitize Plk1 inhibitor GSK461364.56 BI 2536 in combination with pemetrexed shows an encouraging antitumor activity.24 In this work, our results show that combined anti-mitotic agents are effective and support further investigations of Plk1 inhibitors in a combined manner.
Taken together, based on the data from our studies and others,10,15,16,19,38 we suggest that loss of functional p53 does not directly facilitates the cytotoxicity of Plk1 inhibition. Rather, by activating cell death signaling pathways, functional p53 strengths the efficacy of Plk1 inhibitors and prevents possibly genome instability caused by Plk1 inhibitors.38 Further studies are required to investigate whether the long-term outcomes of losing p53, such as low differential grade of tumor cells, defective DNA damage checkpoint or unbalanced metabolism in tumor cells, which makes possible the survival of cancer cells that are more dependent on Plk1 function, are responsible for the cytotoxicity of Plk1 inhibition.
Materials and Methods
Cell culture, cell cycle and phospho-histone H3 and active caspase-3 measurement
HCT116 p53+/+, HCT116 p53−/− and U2OS cells were cultured as instructed. BI 2536 and BI 6727 were purchased from Selleck Chemicals LLC. Monastrol, paclitaxel, nocodazole and vincristine were obtained from Sigma-Aldrich. All mitotic stress inducers were used in a low dose after performing dose kinetics: 15 µM monastrol, 7 nM paclitaxel, 50 ng/ml nocodazole and 1.2 nM vincristine. Cell cycle was analyzed using a FACSCalibur (BD Biosciences, Heidelberg), as described.57 The staining of phospho-histone H3 and active caspase-3 was also evaluated by FACS. Briefly, treated HCT116 cells were trypsinized, washed twice with pre-warmed PBS, fixed and permeabilized with 2% paraformaldehyde and 0.1% Triton X-100 for 15 min at 37°C. Cells were blocked with 2% BSA for 15 min at 37°C prior to be incubated with antibody against phospho-histone H3 (pS10, Millipore) or with antibody against active caspase-3 (Cell Signaling) for 1 h at 37°C and then washed twice. Cells were then incubated with secondary FITC-labeled polyclonal donkey anti-mouse antibody (DAKO) for 30 min at 37°C. Finally, the stained cells were evaluated by a FACSCalibur (BD Biosciences).
siRNA treatment, cellular extract preparation and western blot analysis
siRNA targeting p53 and control siRNA were obtained from Sigma-Aldrich and Qiagen, respectively. siRNA was transiently transfected as described.15 Cell lysis was performed with RIPA buffer [50 mM TRIS-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% Na-desoxycholate, 0.1% SDS, 1 mM NaF, 1 mM DTT, 0.4 mM PMSF, 0.1 mM Na3VO4, and protease inhibitor cocktail complete (Roche)]. Western blot analysis was performed, as previously described.57 Mouse monoclonal antibodies against cyclin B1, Plk1, Bax, Bcl-2 and p53 were obtained from Santa Cruz (Heidelberg). Mouse monoclonal antibody against p21 and rabbit polyclonal antibody against PARP were purchased from Cell Signaling. Mouse monoclonal antibody against Mcl-1 was obtained from PharMingen. Mouse monoclonal antibody against β-actin was from Sigma-Aldrich.
Indirect immunofluorescence staining
The staining was performed as described.58,59 In brief, control or treated cells were fixed for 15 min with 4% paraformaldehyde containing 0.2% Triton X-100 at room temperature. The following primary antibodies were used for staining: polyclonal rabbit antibodies against pericentrin (Abcam), polyclonal rabbit antibody against γH2AX (Upstate Biotechnology), monoclonal mouse antibody against BubR1 (BD Transduction), monoclonal mouse antibody against Plk1 (Santa Cruz), immune serum against centromere (anti-centromere antibody, ACA, Immuno Vision) and monoclonal rat antibodies against α-tubulin (Sigma-Aldrich). FITC goat anti-mouse, Cy3 goat anti-rabbit, Cy5 donkey anti-human and Cy3 donkey anti-rat antibodies (Jackson Immunoresearch, West Grove) were used as secondary antibodies. DNA was stained using DAPI (4’,6-diamidino-2-phenylindol-dihydrochlorid) (Roche). Slides were examined using an Axio Imager 7.1 microscope (Zeiss, Göttingen) and images were taken using an Axio Cam MRm camera (Zeiss). To quantify γH2AX foci formation 150–200 mitotic nuclei from each cell line were evaluated. The immunofluorescence stained slides were also examined by a confocal laser scanning microscope (CLSM) (Leica CTR 6500, Heidelberg, Germany). Images were processed using Photoshop.
Active caspase-3/7 measurement, cell proliferation assay and colony-formation assay
The activity of caspase-3/7 was analyzed with Caspase-Glo® 3/7 Assay (Promega, Mannheim), as instructed. Cell proliferation assays were performed by using Cell Titer-Blue® Cell Viability Assay on treated cells in 96-well plates, based on the reduction of the indicator dye Resazurin into Resorufin by viable cells (Promega). Twenty µl of CellTiter-Blue® reagent was added to each well and then incubated at 37°C with 5% CO2 for 3 h before fluorescence reading using a Victor 1420 Multilabel Counter (Wallac). Data were presented as percentage compared with control. For colony-formation assay, treated cells were plated into 6-well plates (BD Biosciences). After 12–14 d, colonies were stained with methylene-blue solution for 30 min and counted. All experiments were performed in triplicate and at least three independent experiments were performed.
Acknowledgments
The work at our laboratory is supported by Deutsche Krebshilfe (#108553 and #109672) and Deutsche Forschungsgemeinschaft (#Yu 156/2–1). We are grateful to Drs KW Kinzler and B Vogelstein, Ludwig Center at Johns Hopkins, Howard Hughes Medical Institute, Baltimore, for the cell lines HCT116 p53+/+ and HCT116 p53−/−. We thank heartedly Dr. T Berg, university of Leipzig, for providing Poloxin and for critical reading of this manuscript.
Glossary
Abbreviations:
- Plk1
Polo-like kinase 1
- PBD
Polo-box binding domain
- PARP
poly(ADP-ribose) polymerase
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24573
References
- 1.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov. 2010;9:643–60. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 2.Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol. 2009;10:265–75. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 3.Petronczki M, Lénárt P, Peters JM. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev Cell. 2008;14:646–59. doi: 10.1016/j.devcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24:267–76. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 5.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–30. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 6.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825–41. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 8.Taylor S, Peters JM. Polo and Aurora kinases: lessons derived from chemical biology. Curr Opin Cell Biol. 2008;20:77–84. doi: 10.1016/j.ceb.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Keppner S, Proschak E, Kaufmann M, Strebhardt K, Schneider G, Spänkuch B. Biological impact of freezing Plk1 in its inactive conformation in cancer cells. Cell Cycle. 2010;9:761–73. doi: 10.4161/cc.9.4.10644. [DOI] [PubMed] [Google Scholar]
- 10.Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–22. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 11.Lénárt P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–15. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 12.Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, et al. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15:3094–102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- 13.Santamaria A, Neef R, Eberspächer U, Eis K, Husemann M, Mumberg D, et al. Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis. Mol Biol Cell. 2007;18:4024–36. doi: 10.1091/mbc.E07-05-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, et al. In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol Cancer Ther. 2007;6:450–9. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- 15.Sanhaji M, Kreis NN, Zimmer B, Berg T, Louwen F, Yuan J. p53 is not directly relevant to the response of Polo-like kinase 1 inhibitors. Cell Cycle. 2012;11:543–53. doi: 10.4161/cc.11.3.19076. [DOI] [PubMed] [Google Scholar]
- 16.Yuan J, Sanhaji M, Krämer A, Reindl W, Hofmann M, Kreis NN, et al. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol. 2011;179:2091–9. doi: 10.1016/j.ajpath.2011.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reindl W, Yuan J, Krämer A, Strebhardt K, Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol. 2008;15:459–66. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 18.Reindl W, Yuan J, Krämer A, Strebhardt K, Berg T. A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. Chembiochem. 2009;10:1145–8. doi: 10.1002/cbic.200900059. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe N, Sekine T, Takagi M, Iwasaki J, Imamoto N, Kawasaki H, et al. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J Biol Chem. 2009;284:2344–53. doi: 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- 20.Schöffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI) Eur J Cancer. 2010;46:2206–15. doi: 10.1016/j.ejca.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 21.Vose JM, Friedberg JW, Waller EK, Cheson BD, Juvvigunta V, Fritsch H, et al. The Plk1 inhibitor BI 2536 in patients with refractory or relapsed non-Hodgkin's lymphoma: A Phase I, open-label, single dose-escalation study. Leuk Lymphoma. 2013;54:708–13. doi: 10.3109/10428194.2012.729833. [DOI] [PubMed] [Google Scholar]
- 22.Frost A, Mross K, Steinbild S, Hedbom S, Unger C, Kaiser R, et al. Phase i study of the Plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Curr Oncol. 2012;19:e28–35. doi: 10.3747/co.19.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mross K, Dittrich C, Aulitzky WE, Strumberg D, Schutte J, Schmid RM, et al. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naïve patients with unresectable exocrine adenocarcinoma of the pancreas - a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br J Cancer. 2012;107:280–6. doi: 10.1038/bjc.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellis PM, Chu QS, Leighl N, Laurie SA, Fritsch H, Gaschler-Markefski B, et al. A Phase I Open-Label Dose-Escalation Study of Intravenous BI 2536 Together With Pemetrexed in Previously Treated Patients With Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2013;14:19–27. doi: 10.1016/j.cllc.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Schöffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P, et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer. 2012;48:179–86. doi: 10.1016/j.ejca.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Schöffski P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist. 2009;14:559–70. doi: 10.1634/theoncologist.2009-0010. [DOI] [PubMed] [Google Scholar]
- 27.Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, et al. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2008;26:5511–7. doi: 10.1200/JCO.2008.16.1547. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol. 2006;26:2093–108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan R, Tapang P, Leverson JD, Albert D, Giranda VL, Luo Y. Small interfering RNA-mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res. 2005;65:2698–704. doi: 10.1158/0008-5472.CAN-04-2131. [DOI] [PubMed] [Google Scholar]
- 30.Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, et al. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol Cancer Ther. 2010;9:2079–89. doi: 10.1158/1535-7163.MCT-10-0095. [DOI] [PubMed] [Google Scholar]
- 31.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 32.Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–22. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 33.Lénárt P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–15. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 34.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–8. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- 35.McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, et al. p53-dependent repression of polo-like kinase-1 (PLK1) Cell Cycle. 2010;9:4200–12. doi: 10.4161/cc.9.20.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem. 2004;279:25549–61. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- 37.Yang X, Li H, Zhou Z, Wang WH, Deng A, Andrisani O, et al. Plk1-mediated phosphorylation of Topors regulates p53 stability. J Biol Chem. 2009;284:18588–92. doi: 10.1074/jbc.C109.001560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu B, Mahmud H, Maass AH, Yu B, van Gilst WH, de Boer RA, et al. The Plk1 inhibitor BI 2536 temporarily arrests primary cardiac fibroblasts in mitosis and generates aneuploidy in vitro. PLoS One. 2010;5:e12963. doi: 10.1371/journal.pone.0012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayo LD. Directing p53 to induce autophagy. Cell Cycle. 2012;11:3353–4. doi: 10.4161/cc.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qian Y, Jung YS, Chen X. DEC1 and MIC-1: new players of p53-dependent cell fate decision. Cell Cycle. 2012;11:3525–6. doi: 10.4161/cc.21962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiang CM. p53-Aurora A mitotic feedback loop regulates cell cycle progression and genomic stability. Cell Cycle. 2012;11:3719–20. doi: 10.4161/cc.22113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Höpker K, Hagmann H, Khurshid S, Chen S, Schermer B, Benzing T, et al. Putting the brakes on p53-driven apoptosis. Cell Cycle. 2012;11:4122–8. doi: 10.4161/cc.21997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Landsverk KS, Patzke S, Rein ID, Stokke C, Lyng H, De Angelis PM, et al. Three independent mechanisms for arrest in G2 after ionizing radiation. Cell Cycle. 2011;10:819–29. doi: 10.4161/cc.10.5.14968. [DOI] [PubMed] [Google Scholar]
- 44.Levine AJ. The evolution of the p53 family of genes. Cell Cycle. 2012;11:214–5. doi: 10.4161/cc.11.2.18899. [DOI] [PubMed] [Google Scholar]
- 45.Vicente-Dueñas C, González-Herrero I, García Cenador MB, García Criado FJ, Sánchez-García I. Loss of p53 exacerbates multiple myeloma phenotype by facilitating the reprogramming of hematopoietic stem/progenitor cells to malignant plasma cells by MafB. Cell Cycle. 2012;11:3896–900. doi: 10.4161/cc.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–73. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 47.Steelman LS, Martelli AM, Nicoletti F, McCubrey JA. Exploiting p53 status to enhance effectiveness of chemotherapy by lowering associated toxicity. Oncotarget. 2011;2:109–12. doi: 10.18632/oncotarget.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D’Orazi G, Givol D. p53 reactivation: the link to zinc. Cell Cycle. 2012;11:2581–2. doi: 10.4161/cc.21020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andreassen PR, Lacroix FB, Lohez OD, Margolis RL. Neither p21WAF1 nor 14-3-3sigma prevents G2 progression to mitotic catastrophe in human colon carcinoma cells after DNA damage, but p21WAF1 induces stable G1 arrest in resulting tetraploid cells. Cancer Res. 2001;61:7660–8. [PubMed] [Google Scholar]
- 50.Vogel C, Kienitz A, Hofmann I, Müller R, Bastians H. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene. 2004;23:6845–53. doi: 10.1038/sj.onc.1207860. [DOI] [PubMed] [Google Scholar]
- 51.Allison SJ, Milner J. Loss of p53 has site-specific effects on histone H3 modification, including serine 10 phosphorylation important for maintenance of ploidy. Cancer Res. 2003;63:6674–9. [PubMed] [Google Scholar]
- 52.Allison SJ, Milner J. Remodelling chromatin on a global scale: a novel protective function of p53. Carcinogenesis. 2004;25:1551–7. doi: 10.1093/carcin/bgh212. [DOI] [PubMed] [Google Scholar]
- 53.Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7–12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 54.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt M, Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updat. 2007;10:162–81. doi: 10.1016/j.drup.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Liu-Sullivan N, Zhang J, Bakleh A, Marchica J, Li J, Siolas D, et al. Pooled shRNA screen for sensitizers to inhibition of the mitotic regulator polo-like kinase (PLK1) Oncotarget. 2011;2:1254–64. doi: 10.18632/oncotarget.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kreis NN, Sommer K, Sanhaji M, Krämer A, Matthess Y, Kaufmann M, et al. Long-term downregulation of Polo-like kinase 1 increases the cyclin-dependent kinase inhibitor p21(WAF1/CIP1) Cell Cycle. 2009;8:460–72. doi: 10.4161/cc.8.3.7651. [DOI] [PubMed] [Google Scholar]
- 58.Kreis NN, Sanhaji M, Krämer A, Sommer K, Rödel F, Strebhardt K, et al. Restoration of the tumor suppressor p53 by downregulating cyclin B1 in human papillomavirus 16/18-infected cancer cells. Oncogene. 2010;29:5591–603. doi: 10.1038/onc.2010.290. [DOI] [PubMed] [Google Scholar]
- 59.Sanhaji M, Friel CT, Kreis NN, Krämer A, Martin C, Howard J, et al. Functional and spatial regulation of mitotic centromere-associated kinesin by cyclin-dependent kinase 1. Mol Cell Biol. 2010;30:2594–607. doi: 10.1128/MCB.00098-10. [DOI] [PMC free article] [PubMed] [Google Scholar]