

Abstract
The role of PPARγ in cancer therapy is controversial, with studies showing either pro-tumorigenic or antineoplastic effects. This debate is very clinically relevant, because PPARγ agonists are used as antidiabetic drugs. Here, we evaluated if the effects of PPARγ on tumorigenesis are determined by the cell type in which PPARγ is activated. Second, we examined if the metabolic changes induced by PPARγ, such as glycolysis and autophagy, play any role in the tumorigenic process. To this end, PPARγ was overexpressed in breast cancer cells or in stromal cells. PPARγ-overexpressing cells were examined with respect to (1) their tumorigenic potential, using xenograft models, and (2) regarding their metabolic features. In xenograft models, we show that when PPARγ is activated in cancer cells, tumor growth is inhibited by 40%. However, when PPARγ is activated in stromal cells, the growth of co-injected breast cancer cells is enhanced by 60%. Thus, the effect(s) of PPARγ on tumorigenesis are dependent on the cell compartment in which PPARγ is activated. Mechanistically, stromal cells with activated PPARγ display metabolic features of cancer-associated fibroblasts, with increased autophagy, glycolysis and senescence. Indeed, fibroblasts overexpressing PPARγ show increased expression of autophagic markers, increased numbers of acidic autophagic vacuoles, increased production of L-lactate, cell hypertrophy and mitochondrial dysfunction. In addition, PPARγ fibroblasts show increased expression of CDKs (p16/p21) and β-galactosidase, which are markers of cell cycle arrest and senescence. Finally, PPARγ induces the activation of the two major transcription factors that promote autophagy and glycolysis, i.e., HIF-1α and NFκB, in stromal cells. Thus, PPARγ activation in stromal cells results in the formation of a catabolic pro-inflammatory microenvironment that metabolically supports cancer growth. Interestingly, the tumor inhibition observed when PPARγ is expressed in epithelial cancer cells is also associated with increased autophagy, suggesting that activation of an autophagic program has both pro- or antitumorigenic effects depending on the cell compartment in which it occurs. Finally, when PPARγ is expressed in epithelial cancer cells, the suppression of tumor growth is associated with a modest inhibition of angiogenesis. In conclusion, these data support the “two-compartment tumor metabolism” model, which proposes that metabolic coupling exists between catabolic stromal cells and oxidative cancer cells. Cancer cells induce autophagy, glycolysis and senescence in stromal cells. In return, stromal cells generate onco-metabolites and mitochondrial fuels (L-lactate, ketones, glutamine/aminoacids and fatty acids) that are used by cancer cells to enhance their tumorigenic potential. Thus, as researchers design new therapies, they must be conscious that cancer is not a cell-autonomous disease, but rather a tumor is an ecosystem of many different cell types, which engage in metabolic symbiosis.
Keywords: CDK inhibitors, PPARγ agonist therapy, Warburg effect, autophagy, cancer associated fibroblasts, catabolism, glycolysis, inflammatory microenvironment, lactate, mitophagy, oncometabolite, p16(INK4A), p21(WAF1/CIP1), premature aging, senescence
Introduction
Peroxisome proliferator-activated receptor γ (PPARγ) belongs to the nuclear hormone receptor superfamily. It was originally identified as a key regulator of the differentiation of adipocytes and of glucose metabolism. Indeed, synthetic PPARγ ligands (the thiazolidinediones, TZDs) are clinically used for the treatment of type 2 diabetes, to enhance insulin sensitivity. Later, it was found that PPARγ is also expressed in immune cells and in tumor cells.
The role of PPARγ in cancer is controversial, with studies showing either antineoplastic or protumorigenic effects. High expression levels of PPARγ significantly correlate with long-term survival in breast cancers,1,2 in bladder3 and in head and neck squamous cell carcinomas.4 Loss-of-function mutations in PPARγ have been found in approximately 10% of colon cancer patients,5 and in follicular thyroid cancer, a chromosomal translocation between PAX8 and PPARγ leads to a dominant-negative onco-fusion-protein that greatly stimulates cell proliferation.6
In experimental rat models of nitrosomethylurea-induced7 or 7,12-dimethylbenz(a)anthracene (DMBA)-induced8 mammary tumorigenesis, PPARγ ligands significantly reduced tumor burden. PPARγ expression or PPARγ ligands have strong anti-proliferative and anti-growth properties in colon9 and breast cancers10 in vitro and in vivo. Induction of cell cycle arrest, apoptosis and cell differentiation, as well as inhibition of angiogenesis are all possible mechanism(s) to explain the antineoplastic activities of PPARγ.
On the other hand, several studies indicate that activation of PPARγ may have tumor-promoting effects. Treatment with PPARγ agonists increases polyp numbers in the ApcMin mouse model of colon cancer,11,12 and PPARγ increases β-catenin levels in the ApcMin background,11 suggesting that the genetic environment of the host may dictate the outcome of PPARγ activation.
A better understanding of the role of PPARγ in cancer is crucial, especially because PPARγ agonists are extensively used clinically as antidiabetic drugs. Because of their putative anti-proliferative effects, several clinical trials have recently investigated the use of PPARγ agonists for the treatment of solid tumors, including breast, prostate and liposarcomas. Unfortunately, these trials have failed to demonstrate the efficacy of PPARγ agonists as single agents in advanced malignancies. Notably, certain PPARγ ligand derivatives have been withdrawn from the market, due to an increased incidence of hepatitis and bladder cancers. These findings underscore the complexity of action of PPARγ in cancer patients.
In this study, we have attempted to evaluate the mechanism(s) underlying the multifaceted roles of PPARγ in tumorigenesis. Our hypothesis is that the outcome of tumorigenesis depends on the cell type in which PPARγ is activated. Indeed, we present convincing data demonstrating that activation of PPARγ in breast cancer cells inhibits tumor growth, consistent with clinical data in patients, showing that high PPARγ expression predicts favorable prognosis. Conversely, activation of PPARγ in stromal cells generates a permissive microenvironment that favors tumor growth, consistent with the data showing that PPARγ has a pro-tumorigenic role, depending on the host microenvironment.
Mechanistically, we show that PPARγ induces autophagy, glycolysis and senescence in stromal cells, generating a catabolic pro-inflammatory microenvironment that metabolically supports tumor growth. Interestingly, the tumor growth inhibition observed when PPARγ is expressed in epithelial cancer cells is also associated with increased autophagy. Thus, our results suggest that activation of an autophagic program has both pro- or anti-tumorigenic effects, depending on the cellular compartment in which it operates.
Results
Fibroblasts overexpressing PPARγ show increased autophagy, with downregulation of Cav-1 expression
Human immortalized hTERT-fibroblasts were transduced with a lentiviral vector encoding PPARγ or the empty vector (EV) control. After selection, cells were subjected to immunoblot analysis with specific antibodies to validate PPARγ expression (Fig. 1A).

Figure 1. Fibroblasts overexpressing PPARγ show increased autophagy, with downregulation of Cav-1 expression. (A) Human immortalized hTERT-fibroblasts were transduced with a lentiviral vector encoding PPARγ or the empty vector (EV) control. After selection, cells were subjected to immunoblot analysis with specific antibodies to validate PPARγ overexpression. β-actin is shown as an equal loading control. (B and C) EV control or PPARγ expressing fibroblasts were maintained in normoxia (B) or under hypoxia for 6 h (C). Then, immunoblot analysis was performed with antibodies directed against a panel of autophagy markers. (B) PPARγ induces autophagy, as assessed by the upregulation of Beclin-1, BNIP3, cathepsin B and LC3, relative to EV control fibroblasts. (C) Hypoxic stress further promotes LC3 upregulation and activation, as judged by increased cleavage of LC3 in PPARγ-expressing fibroblasts, relative to controls. For all, β-actin was used as an equal loading control. (D) Acridine orange staining was used to detect acidic vesicular organelles (AVOs), which represent autophagic vacuoles. EV or PPARγ-expressing fibroblasts were stained with acridine orange, mounted and immediately analyzed using a confocal microscope. The presence of orange vesicles is highly increased in PPARγ-overexpressing fibroblasts relative to EV controls, demonstrating that PPARγ overexpression promotes the formation of autophagic vacuoles. Original magnification, 40×. (E) EV control and PPARγ-overexpressing fibroblasts were starved for 8 h and analyzed by immunoblotting, using antibodies directed against Cav-1. PPARγ induces the downregulation of Cav-1 expression, as compared with EV control. β-actin was used as an equal loading control. Note that two exposures of the same blot are shown to better illustrate Cav-1 downregulation.
To evaluate if PPARγ promotes autophagy, control and PPARγ fibroblasts were analyzed by immunoblotting with antibodies directed against a panel of autophagy markers. Figure 1B shows that PPARγ expression induces an autophagic program, as assessed by the upregulation of Beclin-1, BNIP3, cathepsin B and LC3, relative to EV control fibroblasts. To evaluate if a hypoxic stress further promotes LC3 activation, control and PPARγ fibroblasts were maintained in hypoxia for 6 h and analyzed by immunoblotting with LC3 antibodies. Figure 1C shows that hypoxia further promotes LC3 activation, as judged by the increased cleavage of LC3 in PPARγ expressing cells relative to controls.
To independently confirm that PPARγ expression induces autophagy, EV control or PPARγ-expressing fibroblasts were stained with acridine orange and analyzed using a confocal microscope. Acridine orange staining is used to detect acidic vesicular organelles (AVOs), which represent autophagic vacuoles. Figure 1D shows that the presence of orange vesicles is highly increased in PPARγ-overexpressing fibroblasts relative to EV controls, demonstrating that PPARγ overexpression promotes the formation of autophagic vacuoles.
Studies have shown that a loss of caveolin-1 (Cav-1) in stromal cells is a biomarker of poor clinical outcome in human breast cancers, and that Cav-1 is degraded by autophagy. Thus, a loss of Cav-1 is a functional reporter of an autophagic and lethal tumor microenvironment. To evaluate if PPARγ overexpression modulates Cav-1 expression, control and PPARγ-overexpressing fibroblasts were starved for 8 h and analyzed by immunoblotting using antibodies directed against Cav-1. Figure 1E shows that PPARγ induces the downregulation of Cav-1 expression, as compared with EV control.
Fibroblasts overexpressing PPARγ show increased activation of NFκB and HIF-1α pathways
To assess the possible mechanism(s) driving increased autophagy in PPARγ-overexpressing fibroblasts, we evaluated the activation of the two main transcription factors that promote autophagy, i.e., NFκB and HIF-1α, using immunoblotting with specific antibodies. Figure 2A shows that PPARγ overexpression promotes the activation of NFκB, as judged by increased levels of phospho-NFκB, relative to control cells. Total NFκB is also increased, in parallel. In addition, Figure 2B shows that PPARγ overexpression stabilizes HIF-1α expression relative to control cells. These results indicate that PPARγ may promote autophagy via the activation of both NFκB and HIF-1α.

Figure 2. Fibroblasts overexpressing PPARγ show increased activation of NFκB and HIF-1α signaling pathways. EV-control and PPARγ-overexpressing fibroblasts were analyzed by immunoblotting with antibodies directed against phospho-NFκB and HIF-1α. (A) PPARγ overexpression promotes the phosphorylation and activation of NFκB. Total NFκB levels were also increased. (B) PPARγ overexpression stabilizes HIF-1α expression. In both panels, β-actin was used as an equal loading control.
Fibroblasts overexpressing PPARγ are more glycolytic and show decreased mitochondrial activity
Activation of the HIF-1α pathway is linked to decreased mitochondrial activity and to a glycolytic phenotype. We have also observed that the expression of BNIP3 is elevated in PPARγ-overexpressing fibroblasts (Fig. 1B). BNIP3 is a marker of mitophagy (mitochondrial autophagy). Thus, we hypothesized that fibroblasts overexpressing PPARγ may display impaired mitochondrial activity. We first evaluated the mitochondrial activity of control and PPARγ-overexpressing fibroblasts during normoxia or under hypoxia by immunoblotting with a panel of antibodies directed against OXPHOS complexes.
Figure 3A shows that PPARγ overexpression decreases the expression of OXPHOS complex I during normoxia and of complexes II and III under hypoxia, relative to control cells. To independently assess mitochondrial activity, control and PPARγ-overexpressing fibroblasts were stained with MitoTracker. MitoTracker accumulates in functional mitochondria with an active membrane potential, thus correlating with mitochondrial activity. Figure 3B shows that fibroblasts expressing PPARγ exhibit decreased MitoTracker staining relative to empty vector controls, consistent with impaired mitochondrial activity. We next asked if the decreased mitochondrial activity is associated with a shift toward a glycolytic metabolism. To this end, L-lactate concentrations were measured in the cell culture media derived from control and PPARγ-overexpressing fibroblasts and maintained under conditions of hypoxia for 12 h. Figure 3B shows that fibroblasts overexpressing PPARγ display a 70% increase in L-lactate accumulation, relative to control EV fibroblasts.

Figure 3. Fibroblasts overexpressing PPARγ show decreased mitochondrial activity, with a shift toward glycolysis. (A) EV-control and PPARγ-overexpressing fibroblasts were maintained in normoxia or under hypoxia for 24 h and analyzed by immunoblotting, with a panel of antibodies directed against OXPHOS markers. Note that PPARγ overexpression decreases the expression of the OXPHOS complex I in normoxia, and of complexes II and III under hypoxia, relative to control cells. β-actin was used as an equal loading control. (B) EV control and PPARγ-overexpressing fibroblasts were stained with MitoTracker to visualize mitochondrial activity. MitoTracker accumulates in functional mitochondria with an active membrane potential, thus correlating with mitochondrial activity. DAPI was used to visualize nuclei (blue). Fibroblasts expressing PPARγ exhibit decreased MitoTracker staining (red), relative to EV-controls, consistent with impaired mitochondrial activity. Original magnification, 60×. (C) Lactate assay. EV-control and PPARγ-overexpressing fibroblasts were maintained under hypoxia for 12 h. Then, L-lactate concentrations were measured in the cell culture media and normalized for total cell number. Note that fibroblasts overexpressing PPARγ display a 70% increase in L-lactate accumulation, relative to control EV fibroblasts. p values are as shown.
Fibroblasts overexpressing PPARγ display a senescent phenotype
We and others have previously shown that autophagy and senescence are a continuum of the same biological process, and that autophagic fibroblasts are more prone toward undergoing senescence. It is also known that increased glycolysis is observed in senescent fibroblasts. As PPARγ-overexpressing fibroblasts display increased autophagy and glycolysis, we evaluated if PPARγ expression promotes a senescent phenotype. To this end, control and PPARγ-overexpressing fibroblasts maintained during normoxia or under hypoxia were analyzed by immunoblotting with a panel of antibodies directed against cell cycle regulators.
Figure 4A shows that upon hypoxic stress, PPARγ overexpression increases the expression of the two well-established CDK inhibitors, namely p21(WAF1/CIP1) and p16(INK4A), relative to control cells. p21(WAF1/CIP1) and p16 are markers of senescence and cell cycle arrest. To directly measure senescence, empty vector (Lv-105) control and PPARγ-overexpressing fibroblasts were maintained under hypoxia and stained with β-gal. Figure 4B shows that the number of β-gal-positive senescent cells is elevated by 5-fold in PPARγ-overexpressing fibroblasts, relative to controls. Representative images of β-gal staining are shown in Figure 4C, to illustrate that β-gal staining is highly elevated in PPARγ fibroblasts, indicative of a senescent phenotype.

Figure 4. Fibroblasts overexpressing PPARγ show a senescent phenotype. (A) EV control and PPARγ-overexpressing fibroblasts were maintained in normoxia or under hypoxia for 24 h and analyzed by immunoblotting with a panel of antibodies directed against cell cycle regulators. Note that upon hypoxic stress, PPARγ overexpression increases the expression of the CDK inhibitors p21(WAF1/CIP1) and p16(INK4A), relative to control cells. p21 and p16 are markers of senescence and cell cycle arrest. β-actin was used as an equal loading control. (B) To measure senescence, EV-control and PPARγ-overexpressing fibroblasts were maintained under hypoxia for 24 h and stained with β-gal. The number of β-gal-positive cells per field was scored and represented graphically. The number of β-gal-positive senescent cells is elevated by 5-fold in PPARγ-overexpressing fibroblasts, relative to empty vector (Lv-105) controls. P values are as shown. (C) Representative images showing β-gal staining in control and PPARγ-overexpressing fibroblasts. β-gal staining is virtually absent in control cells, and strongly present in PPARγ fibroblasts, indicative of a senescent phenotype. (D) To measure cell hypertrophy, EV-control and PPARγ-overexpressing fibroblasts were maintained under hypoxia for 12 h, and the protein was divided by the total cell number. Note that “cell hypertrophy” is increased by 85% in PPARγ-overexpressing fibroblasts, relative to empty vector (Lv-105) controls, consistent with a senescent phenotype.
It is believed that senescent cells are hypertrophic, as they have an increased volume, consistent with their flattened morphology. To independently evaluate senescence, cell hypertrophy was measured by dividing the total protein amount per total cell number of control and PPARγ-overexpressing fibroblasts maintained under hypoxia. Figure 4D shows that cell hypertrophy is increased by 85% in PPARγ-overexpressing fibroblasts, relative to empty vector controls, consistent with a senescent phenotype.
Fibroblasts overexpressing PPARγ promote breast cancer tumor growth, without increased angiogenesis
To determine if PPARγ expression in stromal cells favors breast cancer tumor growth, we employed a xenograft model. To this end, MDA-MB-231 breast cancer cells were co-injected with control or PPARγ fibroblasts into the flanks of nude immunodeficient mice. Tumor growth rates were monitored for 3–4 weeks (Fig. 5A), demonstrating that fibroblasts overexpressing PPARγ significantly promote tumor growth, relative to control cells. Tumor volumes were measured after tumor excision, 24 d post-injection. Figure 5B shows that fibroblasts overexpressing PPARγ induce a 60% increase in tumor volume, relative to Lv-105 control fibroblasts. To evaluate if the stromal expression of PPARγ promotes tumor growth via increased angiogenesis, vascular density (number of CD31-positive vessels per field) was scored in tumor sections derived from control and PPARγ-fibroblast xenografts. Figure 5C shows that the stromal PPARγ-induced tumor growth is not due to increased angiogenesis. Thus, our data indicate that PPARγ expression in stromal cells induces a catabolic microenvironment, with increased autophagy, glycolysis and senescence, which metabolically support tumor growth.

Figure 5. Fibroblasts overexpressing PPARγ promote breast cancer tumor growth, without increased angiogenesis. MDA-MB-231 breast cancer cells were co-injected with EV or PPARγ fibroblasts into the flanks of athymic nude mice. (A) Tumor growth rates are shown. Note that fibroblasts overexpressing PPARγ significantly promote tumor growth, relative to control cells. (B) Tumor volumes were measured after tumor excision, 24 d post-injection. Fibroblasts overexpressing PPARγ induce a 60% increase in tumor volume, relative to Lv-105 control fibroblasts. p values are as shown. n = 10 tumors per experimental group. (C) Tumor angiogenesis. Tumor frozen sections were immunostained with anti-CD31 antibodies. Vascular density quantification (number of CD31-positive vessels per field) indicates that the enhanced tumor growth is not due to increased angiogenesis.
MDA-MB-231 cells overexpressing PPARγ display increased autophagy and decreased mitochondrial activity
To evaluate if the effects of PPARγ on metabolic phenotypes are compartment-specific, PPARγ was also overexpressed in MDA-MB-231 breast cancer cells via a lentiviral approach. Empty vector (EV) control cells were generated in parallel. Immunoblot analysis with specific antibodies validated PPARγ overexpression (Fig. 6A).

Figure 6. MDA-MB-231 cells overexpressing PPARγ display increased autophagy and decreased mitochondrial activity. (A) MDA-MB-231 breast cancer cells were transduced with a lentiviral vector encoding PPARγ or the empty vector (EV) control. After selection, cells were subjected to immunoblot analysis with specific antibodies to validate PPARγ expression. β-actin is shown as an equal loading control. (B) PPARγ-overexpressing MDA-MB-231 cells and EV controls were subjected to starvation for 2–6 h and analyzed by immunoblotting using antibodies directed against a panel of autophagy markers. Note that PPARγ induces autophagy in breast cancer cells by upregulating the expression levels of Beclin-1, BNIP3, cathepsin B, Lamp1 and LC3. β-actin is shown as an equal loading control. (C) EV control and PPARγ-overexpressing MDA-MB-231 cells were maintained under hypoxia for 12 h and analyzed by immunoblotting with a panel of antibodies directed against OXPHOS complex markers. Note that PPARγ decreases the expression of OXPHOS complexes I–V in breast cancer cells, suggesting a severe impairment in mitochondrial respiration. β-actin is shown as an equal loading control. (D) EV control and PPARγ-overexpressing MDA-MB-231 cells were maintained under hypoxia for 12 or 24 h and stained with MitoTracker (red) to visualize mitochondrial activity. Nuclei were stained with DAPI (blue). Note that cancer cells harboring PPARγ show reduced MitoTracker staining, relative to control cells, indicating decreased mitochondrial activity. Original magnification, 60×.
To evaluate if PPARγ induces autophagy in cancer cells, control and PPARγ-overexpressing MDA-MB-231 cells were subjected to starvation and analyzed by immunoblotting, using antibodies directed against a panel of autophagy markers. Figure 6B shows that PPARγ induces autophagy in breast cancer cells by upregulating the expression levels of Beclin-1, BNIP3, cathepsin B, Lamp1, and LC3. We next assessed if the expression of PPARγ in breast cancer cells impairs mitochondrial function. To this end, control and PPARγ-overexpressing MDA-MB-231 cells were maintained under hypoxia and analyzed by immunoblotting with a panel of antibodies directed against OXPHOS markers. Figure 6C shows that PPARγ decreases the expression of OXPHOS complexes I-V in breast cancer cells, indicating a severe impairment of mitochondrial respiration. To independently validate these results, control and PPARγ-overexpressing MDA-MB-231 cells were maintained under hypoxia for 12 or 24 h and stained with MitoTracker. MitoTracker was used to visualize mitochondrial activity, as it accumulates in functional mitochondria with an active membrane potential. Figure 6D shows that cancer cells harboring PPARγ display reduced MitoTracker staining, relative to control cells, suggesting decreased mitochondrial activity.
MDA-MB-231 cells overexpressing PPARγ show decreased tumor growth
To evaluate whether the effects of PPARγ on tumor growth are compartment-specific, we next employed a xenograft model of MDA-MB-231 cells (control or expressing PPARγ), injected into the flanks of athymic nude mice. The tumor volume is shown in Figure 7A, demonstrating that breast cancer cells overexpressing PPARγ show reduced tumor growth relative to EV control cells. To assess the role of tumor angiogenesis, frozen tumor sections were immunostained with anti-CD31 antibodies. Vascular density (number of CD31-positive vessels per field) was scored, and suggests that angiogenesis is decreased by 20% in PPARγ tumors (Fig. 7B).
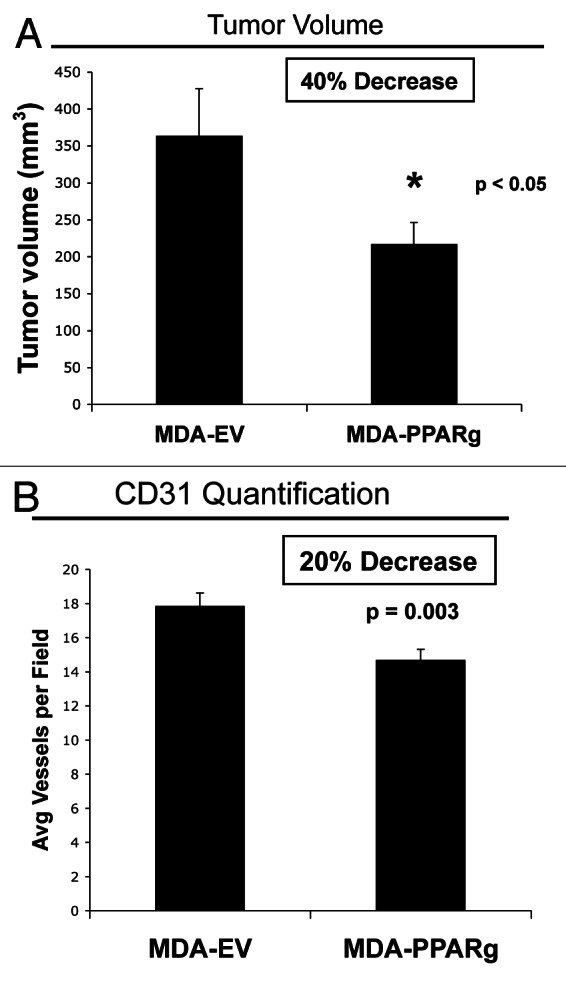
Figure 7. MDA-MB-231 cells overexpressing PPARγ show decreased tumor growth. MDA-MB-231 breast cancer cells harboring EV or PPARγ were injected into the flanks of athymic (immunodeficient) nude mice. (A) Tumor volume is shown after tumor excision 3 weeks post-injection. Note that breast cancer cells overexpressing PPARγ show reduced tumor growth, relative to EV control cells. p values are as shown. n = 10 tumors per experimental group. (B) Tumor angiogenesis. Tumor frozen sections were immunostained with anti-CD31 antibodies. Vascular density quantification (number of CD31-positive vessels per field) indicates that angiogenesis is decreased by 20% in PPARγ tumors.
Discussion
The role of PPARγ in tumorigenesis is controversial, with studies showing either pro-tumorigenic or antineoplastic effects. This debate has very relevant clinical implications, because PPARγ agonists are currently used as antidiabetic drugs. The aim of this study was to test the hypothesis that the effects of PPARγ on tumorigenesis are determined by the cell type in which PPARγ is activated. Here, we show that when PPARγ is activated in epithelial cancer cells, tumor growth is inhibited. However, when PPARγ is activated in stromal cells, tumor growth is enhanced (Fig. 8). Thus, the consequences of PPARγ activation on tumorigenesis are dependent on the cell compartment in which PPARγ is activated.

Figure 8. The role of PPARγ in cancer is compartment-specific. The effects of PPARγ activation on tumorigenesis are dependent on the cell compartment in which PPARγ is activated. When PPARγ is activated in epithelial cancer cells, tumor growth is inhibited. However, when PPARγ is activated in stromal cells, tumor growth is enhanced. (A) Our findings indicate that PPARγ activation in stromal cells plays a role in the formation of a catabolic pro-inflammatory microenvironment that supports cancer growth. Indeed, fibroblasts overexpressing PPARγ share similar metabolic features with cancer-associated fibroblasts, are autophagic, senescent and glycolytic and produce large amounts of L-lactate to feed adjacent cancer cells and actively support their growth. (B) Interestingly, PPARγ activation in epithelial breast cancer cells leads to tumor suppression. Mechanistically, we show that in epithelial breast cancer cells, PPARγ potently induces an autophagic program, that suppresses tumor growth. Thus, PPARγ-mediated activation of an autophagic program has pro- or anti-tumorigenic effects, depending on the cellular compartment in which it occurs.
This cell compartment-specific concept fully integrates a novel framework that we have recently proposed to explain cancer metabolism, termed “two-compartment tumor metabolism.”13-17 In this model, stromal cells and epithelial cancer cells are metabolically coupled in the tumor microenvironment, with stromal cells playing an essential role in supporting and “feeding” aggressive cancer cells.18-21 Cancer cells use oxidative stress to induce catabolic metabolism in cancer-associated fibroblasts (CAFs), driving autophagy, glycolysis, mitochondrial dysfunction and senescence.22-25 We have also termed this process the “reverse Warburg effect” to indicate that aerobic glycolysis is enhanced in stromal cells, and not in epithelial cancer cells, as was previously thought.26-36
We have previously defined the metabolic features of cancer-associated fibroblasts linked with increased tumor growth and metastatic potential, and demonstrated that cancer-associated fibroblasts are glycolytic, autophagic and senescent.37-43 As a consequence, CAFs generate metabolites and mitochondrial fuels (such as L-lactate, ketones, glutamine/other amino acids and fatty acids) that are taken up by epithelial cancer cells to “boost” their mitochondrial capacity and proliferative potential.44-46 Using a genetic approach, we have demonstrated that induction of glycolysis or autophagy in stromal cells, via the overexpression of glycolytic enzymes (such as PKM1 and PKM2) or autophagic proteins (such as ATG16L1, Cathepsin B and BNIP3) promotes the growth and/or metastasis of co-injected breast cancer cells in xenograft models.40-42 Interestingly, autophagic fibroblasts also display senescent features, as assessed by increased β-gal staining and increased expression of the CDK-inhibitor p21(WAF1/CIP1). Conversely, overexpression of CDK-inhibitors p16, p19 and p21 leads to senescent fibroblasts that are also highly catabolic and generate L-lactate and/or ketone bodies, and greatly favors the growth of co-injected breast cancer cells in vivo.40-42 Thus, senescence, glycolysis and autophagy are a continuum of the same biological spectrum, all generating a “fertile” tumor microenvironment that sustains breast cancer tumor growth.47-58
Our findings indicate that PPARγ activation in stromal cells plays a role in the formation of the catabolic microenvironment that supports cancer growth. Indeed, fibroblasts overexpressing PPARγ share similar metabolic features with cancer-associated fibroblasts, as they are autophagic, senescent and glycolytic and produce large amounts of L-lactate that feeds cancer cells and actively supports their growth (Fig. 8A). It is well known that PPARγ activation promotes glycolysis and is associated with increased glucose uptake via the upregulation of the glucose transporter GLUT1 and GLUT4.59,60 In fact, PPARγ ligands are used as insulin sensitizers and antidiabetic drugs. Thus, PPARγ-dependent increases in glycolysis in stromal cells may be one of the mechanism(s) driving increased tumor growth. Consistent with increased glycolysis, we demonstrate that PPARγ activation in fibroblasts leads to impaired mitochondrial function, as assessed by the decreased expression of the OXPHOS complexes and mitochondrial membrane potential.
Our findings are consistent with previous studies showing that PPARγ activation and PPARγ ligands induce autophagy in breast cancer cells.61,62 The exact mechanism(s) whereby PPARγ promotes autophagy and glycolysis will require further elucidation, but we show here that PPARγ induces the activation of NFκB and HIF-1α in stromal cells, two key transcription factors that control autophagy and glycolysis. Consistent with our current findings, PPARγ activation was shown to induce autophagy via HIF-1α and BNIP3 in MDA-MB-231 breast cancer cells.61 Interestingly, in SW13 adrenocortical cancer cells, the PPARγ ligand rosiglitazone was shown to induce autophagy in a PPARγ-independent fashion, via an increase in oxidative stress and disruption of mitochondrial membrane potential.63
We also show that stromal cells activated by PPARγ display the downregulation of Cav-1. Previous studies have shown that a loss of Cav-1 in stromal cells is a biomarker of early tumor recurrence, metastasis, decreased survival, therapy resistance and overall poor clinical outcome in breast cancer patients.49-52,54,64-68 Follow-up studies have demonstrated that Cav-1 is degraded by autophagy, and that a loss of Cav-1 is a functional reporter of an autophagic and lethal tumor microenvironment.30,46 Thus, induction of autophagy by PPARγ may be one the mechanism(s) leading to a loss of Cav-1 in fibroblasts.
The relationship between PPARγ and senescence remains relatively unexplored. Studies have shown that PPARγ inhibits the expression and activity of SIRT1,69 an inhibitor of senescence and aging. These results suggest that PPARγ may promote a senescent phenotype. In support of this idea, our data indicate that fibroblasts with activated PPARγ display an increase in β-gal staining and in the expression levels of p16(INK4A) and p21(WAF1/CIP1), clearly indicating that PPARγ promotes a senescent phenotype.
Interestingly, PPARγ activation in epithelial breast cancer cells leads to tumor suppression (Fig. 8B). Mechanistically, we show that in epithelial breast cancer cells, PPARγ potently induces an autophagic program that suppresses tumor growth. Thus, activation of an autophagic program has pro- or anti-tumorigenic effects depending on the cell compartment in which it occurs. One of the known mechanisms by which PPARγ suppresses tumor growth is the inhibition of angiogenesis. In our model, when PPARγ is activated in epithelial cancer cells, the suppression of tumor growth is associated with an inhibition of angiogenesis. However, when PPARγ is activated in stromal cells, we observed an increased tumorigenic potential, without any significant changes in angiogenesis. Hence, we conclude that PPARγ-dependent inhibition of angiogenesis depends also on which cell type PPARγ is activated.
In conclusion, activation of PPARγ in stromal cells induces a 60% increase in tumor growth, whereas activation of PPARγ in breast cancer cells induces a 40% decrease in tumor growth. Thus, the “net effects” of PPARγ agonists toward tumorigenesis may effectively cancel each other out. Also, our results suggest that extreme caution should be taken in the clinical use of PPARγ agonists, and that the tumor-promoting or tumor-inhibiting effects of PPARγ may depend on the genetic and epigenetic background of the host.
Materials and Methods
Materials
Antibodies used were as follows: anti-PPARγ (H-100) (Santa Cruz, sc-7196); anti-β-Actin (Sigma-Aldrich, A5441); anti-Beclin-1 (Novus Biologicals, NBP1–00085); anti-BNIP3 (Abcam, ab10433); anti-Cathepsin B (FL-339) (Santa Cruz, sc-13985); anti-Lamp-1 (E-5) (Santa Cruz, sc-17768); anti-LC3 (Abcam, ab48395); anti-HIF-1α (BD Transduction Laboratories, 610959); anti-p21(WAF1/CIP1) (H-164) (Santa Cruz, sc-756); anti-p16(INK4A) (H-156) (Santa Cruz, sc-759); anti-Cav-1 (2297) (BD Transduction Laboratories, 610406); anti-OXPHOS (MitoSciences, MS601); anti-p-NFκB (Cell Signaling, 3037); anti-NFκB (Cell Signaling, 3034).
Cell culture
Human skin fibroblasts immortalized with human telomerase reverse transcriptase (hTERT-BJ1, originally purchased from Clontech) and human GFP-positive MDA-MB-231 breast cancer cells (gift of Dr. Fatatis, Drexel University) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin (100 units/mL)/streptomycin (100 µg/mL) (PS), in a 37°C, 5% CO2 incubator. For hypoxia experiments, cells were maintained at 37°C with 0.5% O2. To induce starvation, the media was replaced with Hank’s Balanced salt solution (HBSS), containing 40 mM Hepes and 1% PS.
Lentiviral transduction
Lentiviral plasmids (all from GeneCopoeia) encoding PPARγ (EX-Q0148-Lv105) or the empty vector (EX-NEG-Lv105) were transfected into 293Ta lentiviral packaging cells using Lenti-Pac HIV Expression Packaging Kit (all from GeneCopoeia), following the manufacturer’s protocol. After 48 h, the cells were infected with the resulting virus-containing media, previously collected, centrifuged and filtered. Infected cells were selected with 1.5 µg/mL (hTERT-fibroblasts) or 2 µg/mL (MDA-MB-231) puromycin.
Western blot analysis
Cells were scraped in OG lysis buffer (10mM TRIS-HCl pH7.5, 150mM NaCl, 1% TritonX-100 and 60 mM n-octyl-glucoside), supplemented with protease (Roche Diagnostic) and phosphatase (Sigma) inhibitors. Samples were incubated on a rotating platform for 40 min at 4°C and then centrifuged for 10 min at 13,000 g at 4°C to remove insoluble debris. To detect NFκB and cell cycle-related proteins, cells were harvested in RIPA buffer (50 mM TRIS-HCl pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, with protease and phosphatase inhibitors) and incubated for 10 min on ice. Samples were sonicated for 10 sec and centrifuged at 13,000 g at 4°C. Protein concentration was quantified using the BCA kit (23225, Thermo Scientific). To detect HIF-1α, cells were lysed in UREA buffer (6.7 M urea, 10% glycerol, 1% SDS, 10mM TRIS-HCl pH 6.6, 1% TritonX-100 plus protease and phosphatase inhibitors), homogenized, incubated on ice for 10 min and then centrifuged at 12,000 x g at 4°C. Protein content was determined using the Bradford assay (BioRad). Protein extracts were separated by SDS-PAGE (10 to 15% acrylamide), and transferred to nitrocellulose membranes. Membranes were stained with Ponceau S. All subsequent washing buffers contained 10 mM TRIS-HCl pH 8.0, 150 mM NaCl, and 0.05% Tween 20, which was supplemented with 4% nonfat dry milk for the blocking solution and 1% BSA (Sigma) for antibody dilution. To detect phosphospecific antibodies, membranes were blocked with 5% BSA. Horseradish peroxidase-conjugated secondary antibodies were used to visualize bound primary antibodies with chemiluminescence (ECL) substrate (Thermo Scientific).
Mitochondrial activity
Mitochondrial activity was evaluated with MitoTracker (CMTMRos, M7510, Invitrogen). Cells were seeded on glass coverslips in 12-well plates in complete media. The next day, the media was replaced with DMEM, 10% NuSerum (BD Biosciences) and 1% PS. After 48 h, cells were incubated with pre-warmed MitoTracker solution (diluted in serum free DMEM to a final concentration of 25nM) for 12 min at 37°C. Then, cells were washed three times with PBS with 0.1 mM CaCl2 and 1 mM MgCl2 (PBS/CM), fixed with 2% PFA, rinsed, incubated with DAPI (D3571, Invitrogen) and mounted with Prolong Gold Anti-Fade mounting reagent (P36930, Invitrogen). Images were collected with a Zeiss LSM510 meta-confocal system with a 60× objective.
Detection of acidic vesicular organelles (AVOs) with acridine orange
To detect AVOs, the acridine orange vital staining (A3568, Molecular Probes) was used. Approximately 80,000 cells were plated onto glass coverslips in 12-well plates in complete media. After 48 h, cells were washed with PBS/CM and stained for 15 min at 37°C with 100 ng/mL acridine orange solution. After washing, cells were incubated with Hoechst 33342 nuclear stain (Molecular Probes), rinsed and mounted with Slow-fade Anti-fade Reagent (Molecular Probes). Samples were analyzed immediately using a Zeiss LSM510 meta-confocal microscope, with a 40x objective.
Lactate assay
L-lactate concentrations were quantified using the EnzyChromTM L-Lactate Assay Kit (ECLC-100, BioAssay System), according to the manufacturer’s instructions. Fibroblasts were seeded in 12-well plates in complete media. The next day, the media was changed to DMEM with 2% FBS. After 48 h, the media was collected, and the L-lactate concentration was measured. Values were normalized for the total cell number.
Senescence-associated β-galactosidase staining
Senescent fibroblasts were detected using the senescence β-Galactosidase Staining Kit (9860, Cell Signaling). Briefly, cells were plated into 6-well plates in complete media. The day after, the media was replaced with DMEM with 10% NuSerum. After 48 h, the cells were washed twice with PBS and fixed with fixative solution for 15min. Then, cells were rinsed with PBS and incubated overnight at 37°C in a dry incubator (without CO2), with the β-gal staining solution. After incubation, the cells were observed under a microscope. The number of β-gal-positive cells per field was scored and represented graphically.
Animal studies
MDA-MB-231 cancer cells (106 cells) alone or admixed with fibroblasts (3 x 105 cells) were resuspended in 100 μl of sterile PBS and injected into the flanks of athymic nude mice (NCRNU, Taconic Farms; 6–8 weeks of age). Tumor growth was monitored for 4 weeks post-injection; the mice were sacrificed, and tumors were dissected and measured using calipers. Tumor volume was calculated using the formula (X2Y)/2, where X and Y are the short and long dimensions, respectively, of the tumor. Tumors were either fixed with 10% formalin or flash-frozen in liquid nitrogen-cooled isopentane.
Quantification of tumor angiogenesis
Immunohistochemical staining for CD31 was performed on frozen tumor sections via a three-step biotin-streptavidin-horseradish peroxidase method. Frozen sections were fixed in 4% PFA, washed, blocked with 10% rabbit serum and incubated with a rat anti-mouse CD31 antibody (BD Biosciences) overnight at 4°C. Then, sections were incubated with biotinylated secondary antibodies (Vector Labs) followed by streptavidin-HRP (Dako). Immunoreactivity was revealed with 3,3′diaminobenzidine.
Statistical analysis
Data were analyzed with the Student’s t-test. p values lower than 0.05 were considered statistically significant.
Acknowledgments
F.S. was the recipient of a Young Investigator Award from the Breast Cancer Alliance. U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.).
This work was supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK and an Advanced ERC Grant from the European Research Council.
Also, these developments were made possible through the resources of Thomas Jefferson University.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24289
References
- 1.Jiang Y, Zou L, Zhang C, He S, Cheng C, Xu J, et al. PPARgamma and Wnt/beta-Catenin pathway in human breast cancer: expression pattern, molecular interaction and clinical/prognostic correlations. J Cancer Res Clin Oncol. 2009;135:1551–9. doi: 10.1007/s00432-009-0602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki T, Hayashi S, Miki Y, Nakamura Y, Moriya T, Sugawara A, et al. Peroxisome proliferator-activated receptor gamma in human breast carcinoma: a modulator of estrogenic actions. Endocr Relat Cancer. 2006;13:233–50. doi: 10.1677/erc.1.01075. [DOI] [PubMed] [Google Scholar]
- 3.Mylona E, Giannopoulou I, Diamantopoulou K, Bakarakos P, Nomikos A, Zervas A, et al. Peroxisome proliferator-activated receptor gamma expression in urothelial carcinomas of the bladder: association with differentiation, proliferation and clinical outcome. Eur J Surg Oncol. 2009;35:197–201. doi: 10.1016/j.ejso.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Theocharis S, Klijanienko J, Giaginis C, Rodriguez J, Jouffroy T, Girod A, et al. Peroxisome proliferator-activated receptor-γ in mobile tongue squamous cell carcinoma: associations with clinicopathological parameters and patients survival. J Cancer Res Clin Oncol. 2011;137:251–9. doi: 10.1007/s00432-010-0882-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, et al. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/S1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 6.Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, et al. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected] Science. 2000;289:1357–60. doi: 10.1126/science.289.5483.1357. [corrected] [DOI] [PubMed] [Google Scholar]
- 7.Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, et al. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999;59:5671–3. [PubMed] [Google Scholar]
- 8.Pighetti GM, Novosad W, Nicholson C, Hitt DC, Hansens C, Hollingsworth AB, et al. Therapeutic treatment of DMBA-induced mammary tumors with PPAR ligands. Anticancer Res. 2001;21(2A):825–9. [PubMed] [Google Scholar]
- 9.Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med. 1998;4:1046–52. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- 10.Elstner E, Müller C, Koshizuka K, Williamson EA, Park D, Asou H, et al. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A. 1998;95:8806–11. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, et al. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–7. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 12.Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, et al. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med. 1998;4:1058–61. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 13.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 15.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle. 2010;9:2201–19. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 17.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010;2:185–99. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 19.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Mitochondrial oxidative stress drives tumor progression and metastasis: should we use antioxidants as a key component of cancer treatment and prevention? BMC Med. 2011;9:62. doi: 10.1186/1741-7015-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213. doi: 10.1186/bcr2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, et al. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11:1445–54. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Outschoorn UE, Goldberg A, Lin Z, Ko YH, Flomenberg N, Wang C, et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol Ther. 2011;12:924–38. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle. 2011;10:2521–8. doi: 10.4161/cc.10.15.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10:2504–20. doi: 10.4161/cc.10.15.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle. 2012;11:3956–63. doi: 10.4161/cc.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone body utilization drives tumor growth and metastasis. Cell Cycle. 2012;11:3964–71. doi: 10.4161/cc.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–51. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP. Mitochondrial biogenesis drives tumor cell proliferation. Am J Pathol. 2011;178:1949–52. doi: 10.1016/j.ajpath.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784–93. doi: 10.4161/cc.10.11.15674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297–306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salem AF, Howell A, Sartini M, Sotgia F, Lisanti MP. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1α, autophagy and ketone body production. Cell Cycle. 2012;11:4167–73. doi: 10.4161/cc.22316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salem AF, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle. 2012;11:4174–80. doi: 10.4161/cc.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salem AF, Whitaker-Menezes D, Lin Z, Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, et al. Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle. 2012;11:2545–56. doi: 10.4161/cc.20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599–610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11:2285–302. doi: 10.4161/cc.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11:2272–84. doi: 10.4161/cc.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carito V, Bonuccelli G, Martinez-Outschoorn UE, Whitaker-Menezes D, Caroleo MC, Cione E, et al. Metabolic remodeling of the tumor microenvironment: migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle. 2012;11:3403–14. doi: 10.4161/cc.21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiavarina B, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Tanowitz HB, Pestell RG, et al. Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle. 2012;11:3280–9. doi: 10.4161/cc.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE, Witkiewicz AK, Birbe RC, Howell A, et al. Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther. 2011;12:1101–13. doi: 10.4161/cbt.12.12.18703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther. 2009;8:1071–9. doi: 10.4161/cbt.8.11.8874. [DOI] [PubMed] [Google Scholar]
- 50.Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol Ther. 2010;10:135–43. doi: 10.4161/cbt.10.2.11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Bilal E, et al. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle. 2011;10:1794–809. doi: 10.4161/cc.10.11.15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–17. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu KN, Queenan M, Brody JR, Potoczek M, Sotgia F, Lisanti MP, et al. Loss of stromal caveolin-1 expression in malignant melanoma metastases predicts poor survival. Cell Cycle. 2011;10:4250–5. doi: 10.4161/cc.10.24.18551. [DOI] [PubMed] [Google Scholar]
- 55.Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle. 2009;8:2420–4. doi: 10.4161/cc.8.15.9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salem AF, Al-Zoubi MS, Whitaker-Menezes D, Martinez-Outschoorn UE, Lamb R, Hulit J, et al. Cigarette smoke metabolically promotes cancer, via autophagy and premature aging in the host stromal microenvironment. Cell Cycle. 2013;12:818–25. doi: 10.4161/cc.23722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanchez-Alvarez R, Martinez-Outschoorn UE, Lamb R, Hulit J, Howell A, Gandara R, et al. Mitochondrial dysfunction in breast cancer cells prevents tumor growth: understanding chemoprevention with metformin. Cell Cycle. 2013;12:172–82. doi: 10.4161/cc.23058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanchez-Alvarez R, Martinez-Outschoorn UE, Lin Z, Lamb R, Hulit J, Howell A, et al. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle. 2013;12:289–301. doi: 10.4161/cc.23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao X, Su G, Brown SN, Chen L, Ren J, Zhao P. Peroxisome proliferator-activated receptors gamma and alpha agonists stimulate cardiac glucose uptake via activation of AMP-activated protein kinase. J Nutr Biochem. 2010;21:621–6. doi: 10.1016/j.jnutbio.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 60.Choi SS, Cha BY, Iida K, Lee YS, Yonezawa T, Teruya T, et al. Artepillin C, as a PPARγ ligand, enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Biochem Pharmacol. 2011;81:925–33. doi: 10.1016/j.bcp.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 61.Zhou J, Zhang W, Liang B, Casimiro MC, Whitaker-Menezes D, Wang M, et al. PPARgamma activation induces autophagy in breast cancer cells. Int J Biochem Cell Biol. 2009;41:2334–42. doi: 10.1016/j.biocel.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rovito D, Giordano C, Vizza D, Plastina P, Barone I, Casaburi I, et al. Omega-3 PUFA ethanolamides DHEA and EPEA induce autophagy through PPARγ activation in MCF-7 breast cancer cells. J Cell Physiol. 2013;228:1314–22. doi: 10.1002/jcp.24288. [DOI] [PubMed] [Google Scholar]
- 63.Cerquetti L, Sampaoli C, Amendola D, Bucci B, Masuelli L, Marchese R, et al. Rosiglitazone induces autophagy in H295R and cell cycle deregulation in SW13 adrenocortical cancer cells. Exp Cell Res. 2011;317:1397–410. doi: 10.1016/j.yexcr.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 64.El-Gendi SM, Mostafa MF, El-Gendi AM. Stromal caveolin-1 expression in breast carcinoma. Correlation with early tumor recurrence and clinical outcome. Pathol Oncol Res. 2012;18:459–69. doi: 10.1007/s12253-011-9469-5. [DOI] [PubMed] [Google Scholar]
- 65.Koo JS, Park S, Kim SI, Lee S, Park BW. The impact of caveolin protein expression in tumor stroma on prognosis of breast cancer. Tumour Biol. 2011;32:787–99. doi: 10.1007/s13277-011-0181-6. [DOI] [PubMed] [Google Scholar]
- 66.Qian N, Ueno T, Kawaguchi-Sakita N, Kawashima M, Yoshida N, Mikami Y, et al. Prognostic significance of tumor/stromal caveolin-1 expression in breast cancer patients. Cancer Sci. 2011;102:1590–6. doi: 10.1111/j.1349-7006.2011.01985.x. [DOI] [PubMed] [Google Scholar]
- 67.Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174:2035–43. doi: 10.2353/ajpath.2009.080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simpkins SA, Hanby AM, Holliday DL, Speirs V. Clinical and functional significance of loss of caveolin-1 expression in breast cancer-associated fibroblasts. J Pathol. 2012;227:490–8. doi: 10.1002/path.4034. [DOI] [PubMed] [Google Scholar]
- 69.Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T. SIRT1 is regulated by a PPARγ-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010;38:7458–71. doi: 10.1093/nar/gkq609. [DOI] [PMC free article] [PubMed] [Google Scholar]