

Abstract
Neuronal survival is dependent upon the retinoblastoma family members, Rb1 (Rb) and Rb2 (p130). Rb is thought to regulate gene repression, in part, through direct recruitment of chromatin modifying enzymes to its conserved LXCXE binding domain. We sought to examine the mechanisms that Rb employs to mediate cell cycle gene repression in terminally differentiated cortical neurons. Here, we report that Rb loss converts chromatin at the promoters of E2f-target genes to an activated state. We established a mouse model system in which Rb-LXCXE interactions could be induciblely disabled. Surprisingly, this had no effect on survival or gene silencing in neuronal quiescence. Absence of the Rb LXCXE-binding domain in neurons is compatible with gene repression and long-term survival, unlike Rb deficiency. Finally, we are able to show that chromatin activation following Rb deletion occurs at the level of E2fs. Blocking E2f-mediated transcription downstream of Rb loss is sufficient to maintain chromatin in an inactive state. Taken together our results suggest a model whereby Rb-E2f interactions are sufficient to maintain gene repression irrespective of LXCXE-dependent chromatin remodeling.
Keywords: Rb, E2f, neuronal quiescence, cell cycle, transcription
Introduction
The retinoblastoma protein (Rb) plays an essential role in regulating cell proliferation by the repression of E2f transcription factors.1 Rb/E2f interactions are best characterized for their role in regulating the G1/S transition; however, growing evidence is mounting to suggest a more diverse role for the pathway in both cell cycle and non-cell cycle gene regulation.1 In addition to directly inhibiting transcription through interaction with E2fs, Rb is able to actively repress gene expression through the recruitment of co-factors.2 Rb is thought to mediate many of these repressive effects through its conserved LXCXE binding domain.2
The study of viral oncoproteins, such as E1A and SV40 Large T-antigen, led to the identification of the conserved LXCXE motif that mediates interactions with Rb.3,4 The LXCXE domain was shown to be essential for the inactivation of Rb by direct interaction with oncoproteins.5,6 Further studies identified similar LXCXE-interaction domains found in these viral proteins in other cellular constituents.2 Seminal studies showed that factors such as HDAC1, which contain an LXCXE-interacting domain, interact with Rb and are essential for its repressive function under certain contexts.7 Subsequent studies identified LXCXE interacting factors that modulate transcriptional activation by histone methylation and nucleosome remodeling.8,9 Other non-transcriptional LXCXE interactions have also been revealed, such as cyclin D and BRCA1, which are important for Rb function and tumor suppression.10-12 Understanding how these LXCXE interactions shape Rb/E2f activity physiologically is imperative in our knowledge of how Rb loss contributes to pathological events such as tumorigenesis and neurodegeneration.1,13
The development of a murine transgenic knock-in mutant in which Rb is defective for LXCXE interactions has challenged initial studies detailing its importance in Rb-mediated gene repression. The in vivo observation that LXCXE interactions are important under conditions of stress, such as senescence and following DNA damage, suggest unique contexts where this domain is required.14-16 Examination of fully differentiated retinal neurons deficient for the LXCXE-binding domain of Rb did not reveal any proliferative or differentiation defects;14 however, the use of germline homozygosity may have allowed for compensation by other Rb family members. Furthermore, retinal neurons display a proliferative competence suggesting potential differences in gene-repression when compared with a proliferation-incompetent quiescent cell type such as cortical neurons.17-19 It is still unknown how Rb mediates long-term gene repression in a population that is incapable of proliferation.
Given our previous observation that Rb is essential for mediating cell cycle gene repression and survival in post-mitotic neurons, we sought to examine the mechanism utilized by Rb to regulate this repression.19 Cortical neurons present an excellent system to study quiescence, as they are terminally differentiated cells and should therefore employ a consistent non-transient mechanism to repress cell cycle-related transcription. Using our previously established acute model for Rb deletion in vitro, we observe an activation of chromatin at E2f-regulated gene promoters following Rb loss. We next examined the dependence on LXCXE-dependent interactions for gene repression and cell cycle regulation, as this domain has been broadly implicated in Rb-mediated chromatin remodeling. We surprisingly report that Rb-LXCXE interactions are dispensable for long-term regulation of cell cycle gene repression and neuronal survival both in vitro and in vivo. To address the dependence of chromatin activation on Rb and E2f, we broadly inhibited E2f activation in the absence of Rb. The inhibition of E2f activation was sufficient to prevent chromatin activation, suggesting that this process is dependent on E2f-activation and not Rb-mediated repression in post-mitotic cortical neurons. In this study, we highlight the sufficiency of Rb-E2f interactions in maintaining neuronal quiescence and survival.
Results
Rb loss induces an active chromatin state at E2f target genes
Our previous study had identified an essential role for Rb in the survival of post-mitotic cortical neurons.19 We observed specific induction of cell cycle-related gene targets and disruption of neuronal quiescence upon acute removal of Rb.19 Given the previously described role for Rb in the regulation of chromatin dynamics, we were interested to see the impact of Rb deletion on the chromatin landscape in quiescent cortical neurons.
To ask whether Rb plays a role in regulating chromatin dynamics, RbFlox/Flox neurons were infected with either GFP- or Cre-expressing lentiviruses, as previously described,19 then fixed and harvested at 6 d in vitro (DIV). We performed chromatin immunoprecipitation with antibodies directed toward acetylated histone 3 (Acetyl-H3) and tri-methylated lysine 9 on histone 3 (H3K9me3), as these are two well-characterized Rb-associated chromatin marks.2 Using the PCNA and CyclinA2 promoters as established E2f-regulated gene products,1 we performed quantitative real-time PCR to assess the enrichment of Acetyl-H3 and H3K9me3 at these promoters in the presence or absence of Rb (Fig. 1B and C). In accordance with an increase at the protein level by western blot (Fig. 1A), Rb deletion induces the chromatin-activation mark Acetyl-H3 at both the PCNA and CycA2 promoters, and a significant decrease in the repressive H3K9me3 mark (Fig. 1B–C). This evidence supports the notion that Rb plays a role in the regulation of gene repression at the level of chromatin in quiescent cortical neurons.
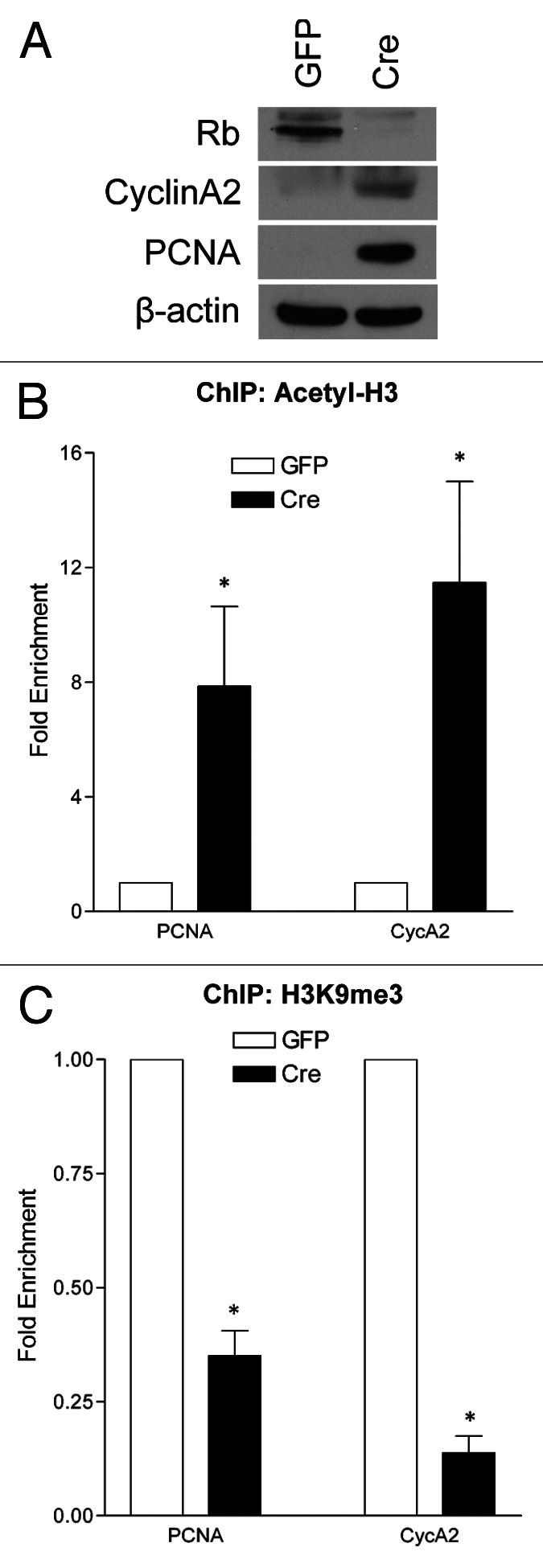
Figure 1. Acute Rb removal results chromatin activation at E2f-regulated promoters. qRT-PCR analysis of the PCNA and CycA2 promoters in GFP- or Cre-infected cortical neurons (RbFlox/Flox) at 6 DIV from chromatin immunoprecipitated DNA. (A) Western blot analysis of total protein extracted from control (GFP) and Rb-deficient (Cre) cortical neurons at 6DIV. (B) Chromatin was immunoprecipitated with an antibody directed toward acetylated histone 3 (Acetyl-H3). (C) Chromatin was immunoprecipitated using an antibody that recognizes trimethylated lysine 9 on histone 3 (H3K9me3). Error bars represent SEM (n = 3, where n represents a ChIP performed on neurons isolated from a unique embryo), significance was determined using a two-tailed Student’s t-test where *p < 0.05.
The Rb LXCXE-binding domain is dispensable for neuronal quiescence
We next sought to determine a more specific role for Rb in the regulation of chromatin dynamics in cortical neurons. We focused on the Rb LXCXE binding motif for its previously established role in chromatin regulation by interaction with specific factors involved in histone acetylation and histone methylation.2 Cortical neurons present an excellent model to study the role of Rb in gene repression, as they are (1) a proliferation incompetent terminally quiescent cell type and (2) have limited secondary consequences due to their inability to divide. In order to address this question, we established an acute Rb LXCXE-deficiency model (RbΔL) (Fig. S1A). This specific RbΔL mutant has been extensively characterized to be deficient in interacting with known LXCXE-dependent factors.14,15 Our model employs RbFlox/ΔL with the addition of Cre, by viral or transgenic methods, inducing acute removal of the floxed allele, leaving the mutant RbΔL protein to function. This model prevents any compensation due to the long-term absence of Rb or the LXCXE binding domain. Functionality of this model was confirmed by PCR analysis for excision of the Rb floxed allele and presence of the LXCXE binding domain mutation (Fig. S1B).
Upon acute deletion of Rb in cortical neurons, we previously reported a disruption of quiescence as evidenced by the ectopic induction of cell cycle-related genes.19 We hypothesized that if Rb maintains the long-term repression of these factors through interaction with LXCXE-dependent chromatin-modifying enzymes, that a form of Rb defective in these interactions would be insufficient to maintain neuronal quiescence. We examined expression of the cell cycle-related factor CycA2 by western blot at 10DIV (Fig. 2A). There was a significant induction following complete Rb deletion (RbFlox/Flox); however, one RbΔL allele (RbFlox/ΔL) was sufficient to maintain CycA2 protein levels at control (RbFlox/+) levels (Fig. 2A). Similarly, when we examined cortical neurons at 10DIV by immunofluorescence, we did not see any significant increase in Ki67+ cells in RbΔL cells compared with control (Fig. 2B).

Figure 2. Rb regulates neuronal quiescence in an LXCXE-independent manner. (A) Western blot analysis of total protein extracted from control, Rb-deficient and RbΔL cortical neurons at 10 DIV (B) Cortical neurons were fixed at 10DIV and stained for Ki67. Percentage of Ki67+ was quantified using DAPI to represent total cell number. Error bars represent SEM (n = 3, where n represent an independent culture with multiple technical replicates); statistical significance was determined using a one-way ANOVA, where *p < 0.05 was considered statistically significant. (C) Animals of indicated genotypes were injected with tamoxifen (180 mg/kg/day i.p., 3 d) and euthanized 4 wk following the final injection. Representative pictures of ectopic expression of the cell cycle-related factors Ki67 (arrows) and Cyclin E (arrows) in neurons in the cortex of CamKCreERT2; RbFlox/Floxmice compared with CamKCreERT2; RbFlox/+ and CamKCreERT2; RbFlox/ΔLmice. Error bars represent SEM (n = 3, where n represent the cortex from a unique animal with multiple technical replicates); statistical significance was determined using a one-way ANOVA, where *p < 0.05 was considered statistically significant. Scale bar: 50 µM.
We next examined the expression of cell cycle-related genes in vivo. Due to the technical time limitations on maintaining cultured cortical neurons, we hypothesized that lengthening our analysis may reveal gene expression defects in RbΔL neurons. An inducible CamkCreERT2 model was used to delete the floxed Rb allele upon tamoxifen administration in adult post-mitotic cortical neurons (Fig. 2C).19,20 Animals were injected with tamoxifen and examined 4 wk later, a time at which we previously showed massive neurodegeneration upon Rb deletion.19 Animals deficient in Rb expression displayed ectopic expression of the cell cycle-related factors Ki67 and CycE, whereas RbΔL and control neurons did not show any significant changes (Fig. 2C). This suggests that the Rb LXCXE-binding domain is dispensable for the long-term repression of cell cycle-related factors. In addition, we employed a conditional model to address the ability of RbΔL to establish neuronal quiescence by removing Rb expression in embryonic neural precursors (Fig. S2). Consistent with previous results, the complete loss of Rb leads to ectopic proliferation in the developing dorsal cortex at E15.5,21 as evidenced by Ki67 expression, whereas RbΔL and controls did not display ectopic Ki67 (Fig. S2).
The Rb LXCXE-binding domain is dispensable for neuronal survival
The most profound phenotype observed upon acute Rb deletion in cortical neurons was neurodegeneration. Cortical neurons present a unique contrast to other neuronal systems, as they are unable to tolerate loss of Rb.17 To examine whether survival may be impacted independently of quiescence in our LXCXE mutation model; we performed studies to assess neuronal survival. We examined neurons at 10DIV for induction of apoptosis by both microscopic assessment of condensed nuclei and western blot analysis for activated caspase-3 (AC-3) (Fig. 3A and B). We did not observe any significant changes in survival by either of these two techniques in RbΔL cells when compared with control (Fig. 3A and B). To assess the potential for a more long-term contribution of RbΔL on survival, we utilized our CamKCreERT2 model. Mice were examined for neuronal survival by quantifying the percentage of neurons (NeuN+) (Fig. 3C). We observed massive neurodegeneration following Rb deletion, whereas no reduction in the number of neurons was observed in RbΔL brains when compared with control (Fig. 3C). Overall, these results suggest that Rb interactions with the LXCXE-binding motif are dispensable for neuronal survival.

Figure 3. Rb regulates neuronal survival in an LXCXE independent manner. (A) Western blot analysis of total protein extracted from control, Rb-deficient and RbΔL cortical neurons at 10 DIV (B) Cortical neurons of the indicated genotypes were infected with lentiviral Cre and fixed at 10 DIV, and condensed nuclei were examined by DAPI staining. Error bars represent SEM (n = 3, where n represent an independent culture with multiple technical replicates); statistical significance was determined using a one-way ANOVA, where *p < 0.05 was considered statistically significant. (C) Animals of indicated genotypes were injected with tamoxifen (180 mg/kg/day i.p., 3 d) and euthanized 4 wk following the final injection. Representative pictures of NeuN immunofluorescence in the cortex of CamKCreERT2; RbFlox/Floxmice compared with CamKCreERT2; RbFlox/+ and CamKCreERT2; RbFlox/ΔLmice. Scale bar: 100 µm. (D) NeuN+ cells were quantified in the cortex, and percentages were obtained by normalizing to total DAPI cells for indicated genotypes. Error bars represent SEM (n = 3, where n represents the cortex from a unique animal with multiple technical replicates); statistical significance was determined using a one-way ANOVA, where *p < 0.05 was considered statistically significant.
Induction of an active chromatin state occurs downstream of E2f-activation
No defects in the maintenance of quiescence or survival were found in cortical neurons in our acute RbΔL mutant model. In relation to our initial observation, that in the absence of Rb chromatin is remodeled to an activate state, we asked whether this process was solely dependent on Rb or relied on E2f activation downstream of Rb loss. The role of the Rb/E2f pathway in chromatin remodeling is not limited to pocket protein-mediated repression but has also been shown to include E2f-induced chromatin activation.22-24 To test this, we utilized a dominant-negative DP1 construct (DP1Δ103–126).19,25,26 As E2f inhibiton was sufficient to rescue neuronal survival following Rb loss,19 we now asked whether DP1Δ103–126 was also sufficient to prevent chromatin remodeling. We hypothesized that if Rb/E2f-mediated gene repression relies solely on an interaction between Rb and E2f, that this construct would prevent any remodeling. If chromatin remodeling was dependent on Rb itself, the absence of these factors would result in changes to the chromatin landscape in the presence of DP1Δ103–126. Western blot analysis revealed a restoration of PCNA and CycA2 protein to control levels (GFP) in DP1Δ103–126 infected RbFlox/Flox cortical neurons (Cre and DP1Δ103–126)(Fig. 4A). Chromatin immunoprecipitation was performed for Acetyl-H3 and H3K9me3 on RbFlox/Flox cortical neurons infected with LV-GFP, LV-Cre or LV-Cre and DP1Δ103–126. We examined the enrichment for these chromatin marks at the promoters of PCNA and CycA2 (Fig. 4B and C). We did not observe any significant differences in either mark at the PCNA or CycA2 promoters in LV-GFP or LV-Cre/LV-DP1Δ103–126 (Fig. 4B and C). These results indicate that the change in chromatin activation state that is observed upon Rb removal is dependent on E2f-activation and not purely the removal of Rb. Our data therefore highlights the sufficiency of an interaction between Rb and E2f as a driving force in the maintenance of neuronal quiescence and survival.
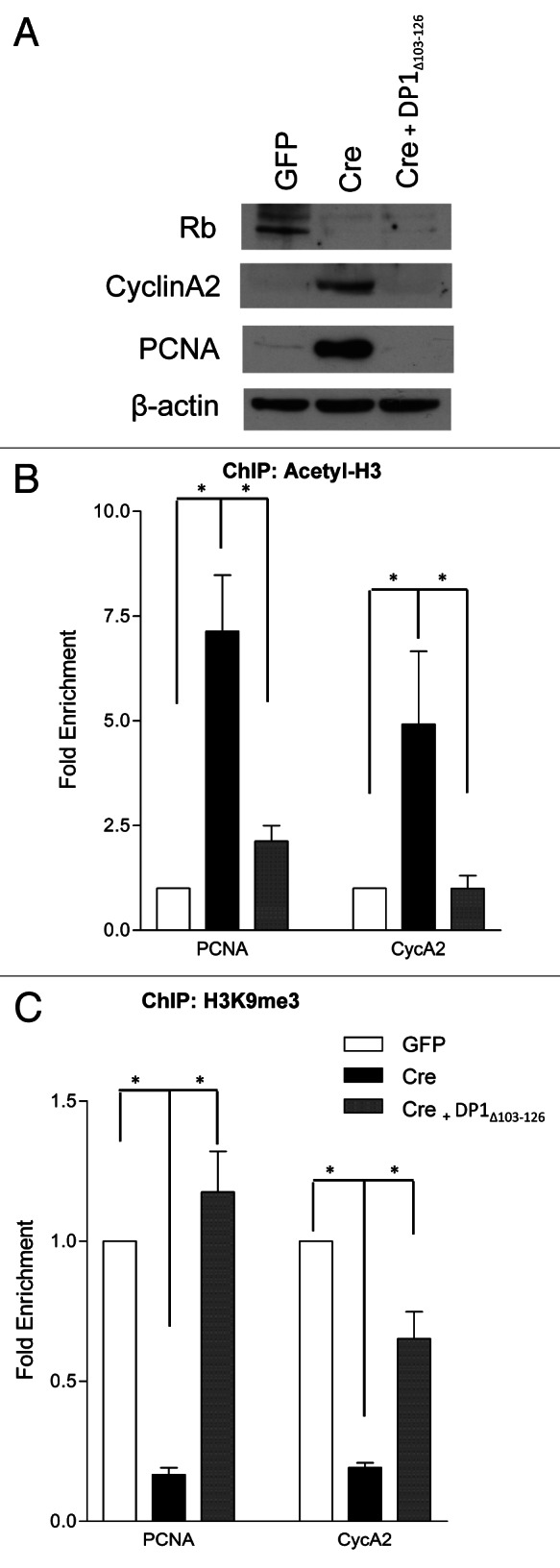
Figure 4. Chromatin activation in the absence of Rb occurs at the level of E2f. qRT-PCR analysis of the PCNA and CycA2 promoters in GFP, Cre or Cre/ DP1Δ103–126 infected cortical neurons (RbFlox/Flox) at 6DIV from chromatin immunoprecipitated DNA. (A) Western blot analysis of total protein extracted from control (GFP), Rb-deficient (Cre) and Rb-deficient/dominant negative DP1 (Cre and DP1Δ103–126) cortical neurons at 6 DIV. (B) Chromatin was immunoprecipitated with an antibody directed toward acetylated histone 3 (Acetyl-H3). (C) Chromatin was immunoprecipitated using an antibody that recognizes tri-methylated lysine 9 on histone 3 (H3K9me3). Error bars represent SEM (n = 3, where n represents a ChIP performed on neurons isolated from a unique embryo); statistical significance was determined using a one-way ANOVA, where *p < 0.05 was considered statistically significant.
Discussion
Our studies demonstrate a number of important conclusions regarding the role of Rb/E2f in the regulation of gene expression in post-mitotic cortical neurons. We have shown that acute loss of Rb results in remodeling of chromatin at E2f-responsive promoters to an active state. Surprisingly, we observe that long-term gene repression by Rb occurs independently of the ability to interact with LXCXE motifs that are found in many chromatin-regulating enzymes. Using an acute model, we did not observe any changes, in vitro or in vivo, in the capacity of RbΔL to mediate physiological changes or gene repression. Finally, we provide evidence that E2f-activation downstream of Rb deletion is essential in chromatin remodeling. In the absence of Rb, a dominant-negative DP1Δ103–126 mutant is able to maintain chromatin in an inactive state. These findings suggest that cell cycle gene repression by Rb/E2f in cortical neurons occurs at the level of E2f.
The initial finding that viral oncoproteins required the LXCXE motif in order to inactivate Rb suggested a critical role for this domain in Rb function. Accumulating evidence is now emerging that it is largely dispensable and only plays context-specific roles in Rb function.14,16,27 An elegant study in the liver, using acute transgenic and viral models, showed that the Rb LXCXE-binding domain is not required for basal transcription.16 In the liver, Rb-LXCXE interactions were only required after treatment with a genotoxic agent, where it was shown that these animals upregulate cell cycle-related targets and, as a result, initiate hepatocellular carcinomas.16 This study complemented initial work revealing that RbΔL mutation, in a p53-null background, resulted in more aggressive tumor formation due to genomic instability created by defects in chromosome condensation.28 Rb-LXCXE interactions have also been shown to be essential for stress-responsive G1 arrest or senescence.14,29 RbΔL mutants showed specific defects in H3K9me3 recruitment at heterochromatin, a hallmark of senescence.14,15,29 Furthermore, studies have implicated Rb in the recruitment of H3K4 demethylases, Jarid1a and Jarid1b, specifically during senescence.30 These studies suggest a unique mechanistic role for Rb in stress-associated cell cycle arrest compared with cell cycle exit during terminal differentiation.14,30 Both processes are Rb-dependent; however, senescence uniquely requires LXCXE-dependent factors.14,30 Though senescence and cortical neuronal differentiation are thought to be permanent cell cycle withdrawal events, our data addresses the differential mechanism employed by Rb in the latter. Studies in retinal neurons may not represent an ideal neuronal population to study terminal quiescence, as these neurons have been shown to de-differentiate and proliferate.14,17 Furthermore, our acute model adds a level of specificity to help prevent compensation from other pocket protein family members.
Our data supports the notion that Rb-mediated gene repression occurs independently of LXCXE interactions, though it does not exclude the possibility that factors interacting through other Rb-domains influence gene repression. Our dominant-negative DP1Δ103–126 model strengthens the conclusion that chromatin remodeling does occur downstream of Rb regardless of LXCXE or non-LXCXE factors. In this experiment, we completely removed the Rb protein, thus all other accessory factors. The addition of DP1Δ103–126 was sufficient to block chromatin activation, suggesting that Rb-interacting factors are either (1) not present in cortical neurons or (2) present but dispensable in this context. Studies have implicated the switch between repressive (histone deacetylase containing) and activator (histone acetyltransferase containing) complexes during the switch to E2f-activation.31 Little is known about the specific role for activator E2fs (E2f1–3) in recruitment of these complexes. Studies in Drosophila have shown that E2f activation is a two-step process, with the initial disruption of Rb-dependent repressor complexes and then recruitment of E2f-activation complexes.32 Evidence has been presented to suggest the relationship between E2f-activation and histone acetyltransferase recruitment;22-24,33 however, further studies probing these interactions in an in vivo physiological context would be extremely informative.
In conclusion, our results reveal a specific role for Rb-E2f interactions in the maintenance of gene repression in cortical neurons. We found that Rb-mediated gene repression occurs independently of its LXCXE-binding domain. We did not observe any physiological or gene expression changes when neurons had to acutely rely on RbΔL. Our use of a dominant-negative DP1Δ103–126 construct revealed that chromatin remodeling after Rb deletion occurs at the level of E2f. These findings reveal the need for constitutive inhibition of E2f in order to maintain gene repression and neuronal survival. It also highlights the differential role Rb plays in terminally differentiated quiescent neurons when compared with previous studies investigating stress-induced cell cycle arrest.14-16,19 Most importantly, our results provide evidence that in terminally quiescent neurons, Rb/E2f function is more dependent on E2f activity than previously thought.
Materials and Methods
Animals
All experiments were approved by the University of Ottawa’s Animal Care Ethics Committee adhering to the Guidelines of the Canadian Council on Animal Care. RbFlox 34 and Foxg1-Cre35 were maintained on an FVBN background, while Rb ∆LXCXE 15 and CamKCreERT2 (EMMA ID: 02125)20 mice were maintained on a C57Bl/6 background. Animals were genotyped according to standard protocols with previously published primers. For in vitro experiments RbFlox/Flox animals were crossed, or RbFlox/∆LXCXE to RbFlox/+. All Cre-expressing animals used were heterozygotes for Cre expression. CamKCreERT2;RbFlox/+ mice were crossed with RbFlox/∆LXCXE mice to generate experimental animals, which at 5–6 wk of age, were given tamoxifen (Sigma) (180 mg/kg/day i.p., 3 d) and euthanized 4 wk after the final injection.
Primary cortical neurons
Embryonic cortical neurons were isolated by standard procedures.36 Neurons were infected at the time of plating with a pWPXLD lentiviral vector expressing control GFP, GFP-tagged Cre recombinase or dominant-negative DP1Δ103–126 at a multiplicity of infection (MOI) of 2. For immunofluorescence, cells were grown on coverslips for 10DIV then fixed with 4% paraformaldehyde (PFA) and stained for Ki67 (Cell Marque, Cat No. 275-16), DAPI and visualized with AlexaFluor 488 secondary antibody (Invitrogen, Cat No. A11055). Statistical differences were determined using a one-way ANOVA, where p < 0.05 was considered statistically significant.
Chromatin immunoprecipitation
Cortical neurons were cross-linked with 1% formaldehyde for 10 min at room temperature. Neurons were lysed [50 mM TRIS-HCl, 1% SDS, 10mM EDTA, Protease Inhibitor Cocktail (Sigma)], sonicated for 30 min [30 sec (on), 45 sec (off)] using a BioRuptor® (Diagenode) and centrifuged at 14,000 × g to remove cellular debris. Each immunoprecipitation was performed using 2 µg of antibody directed to acetylated histone 3 (Millipore, Cat No. 06-599), tri-methylated lysine 9 on histone 3 (Abcam, Cat No. ab8898) or normal rabbit immunoglobulin G (Millipore, Cat No. 12-370). Immunocomplexes were captured using Dynabeads® Protein A (Invitrogen) and washed extensively. Cross-links were reversed overnight, followed by treatment with RNase A at 37°C for 1 h and proteinase K at 65°C for 30 min. Purified DNA was analyzed by real-time PCR using PerfeCTa SYBR Green FastMix Reaction Mix (Quanta Biosciences) and a Rotor-Gene RG-3000 (Corbett Research). Primer sequences are available upon request and were designed to encompass E2f binding regions in the PCNA and CycA2 promoters. Expression values were obtained from three immunoprecipitations from three independent cultures, and significance was determined using a one-way ANOVA, where p < 0.05 was considered statistically significant.
Western blots
Protein was isolated from cultured cortical neurons and western blot analyses performed as previously described,21 with antibodies directed toward cleaved caspase-3 (Cell Signaling, Cat No. 9664S), Rb (PharMingen, Cat No. 554136), Cyclin A2 (Abcam, Cat No. ab7965), PCNA (Millpore, Cat No. MAB424) and β-actin (Sigma, Cat No. A-5316).
Tissue processing, immunohistochemistry and cell quantification
Brains were perfused and fixed as previously described.21,36 Sections were collected as 14 μm coronal cryosections on slides. For immunohistochemistry, sections underwent antigen retrieval in Target Retrieval Solution (Dako) and incubated overnight at 4°C with the following primary antibodies: NeuN (EMD Millipore Corporation, Cat. No. MAB377), Tuj1 (Covance, Cat No. MMS-435P), Ki67 (Cell Marque, Cat No. 275-16) and Cyclin E (Santa Cruz, Cat No. sc-481). Sections were incubated in blocking solution containing donkey anti-rabbit AlexaFluor488 (Jackson Immunoresearch, Cat. No. 711-545-152), donkey anti-mouse Cy3 (Jackson Immunoresearch, Cat No. 715-165-150) and DAPI. All images were acquired using a Zeiss Axioobserver D1. For cell quantification, a minimum of three sections containing the frontal cortex were analyzed per brain and the percentage of positive (NeuN, Ki67 or Cyclin E) cells among the total DAPI+ cells were quantified. Statistical differences were determined using a one-way ANOVA, where p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Jason G. MacLaurin and Linda Jui for excellent technical assistance. This work was supported by CIHR grants to R.S.S. M.G.A. was supported by awards from Ontario Graduate Scholarship and Heart and Stroke Foundation of Ontario. R.V. was supported by postdoctoral fellowships from the Vision 2010 strategic plan of the University of Ottawa, the Alzheimer Society of Canada, the Heart and Stroke Foundation of Canada and by travel awards from the Léon Fredericq Funds (University of Liège, Belgium) and Wallonie–Bruxelles International . Imaging equipment was supported by the Centre for Stroke Recovery.
Glossary
Abbreviations:
- AC-3
activated caspase-3
- Acetyl-H3
acetylated histone 3
- CycA2
Cyclin A2
- DAPI
4',6-diamidino-2-phenylindole
- DIV
days in vitro
- DNA
deoxyribonucleic acid
- DP1
dimerization partner 1
- E1A
early region 1A
- GFP
green fluorescent protein
- H3K9me3
trimethylated histone 3 on lysine 9
- H3K4
histone 3 lysine 4
- HDAC
histone deacetylase
- LV
lentivirus
- LXCXE
leucine- any amino acid- cysteine- any amino acid- glutamate
- ΔL
LXCXE-binding disruption mutant
- NeuN
neuronal nuclei
- PCNA
proliferating cell nuclear antigen
- PCR
polymerase chain reaction
- Rb
retinoblastoma
- SV40
simian virus 40
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24527
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24527
References
- 1.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dick FA. Structure-function analysis of the retinoblastoma tumor suppressor protein - is the whole a sum of its parts? Cell Div. 2007;2:26. doi: 10.1186/1747-1028-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang HG, Rikitake Y, Carter MC, Yaciuk P, Abraham SE, Zerler B, et al. Identification of specific adenovirus E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J Virol. 1993;67:476–88. doi: 10.1128/jvi.67.1.476-488.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–83. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 5.Dyson N, Guida P, McCall C, Harlow E. Adenovirus E1A makes two distinct contacts with the retinoblastoma protein. J Virol. 1992;66:4606–11. doi: 10.1128/jvi.66.7.4606-4611.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HY, Ahn BY, Cho Y. Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. EMBO J. 2001;20:295–304. doi: 10.1093/emboj/20.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–5. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–5. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 9.Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, et al. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79:119–30. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- 10.Dowdy SF, Hinds PW, Louie K, Reed SI, Arnold A, Weinberg RA. Physical interaction of the retinoblastoma protein with human D cyclins. Cell. 1993;73:499–511. doi: 10.1016/0092-8674(93)90137-F. [DOI] [PubMed] [Google Scholar]
- 11.Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993;73:487–97. doi: 10.1016/0092-8674(93)90136-E. [DOI] [PubMed] [Google Scholar]
- 12.Fan S, Yuan R, Ma YX, Xiong J, Meng Q, Erdos M, et al. Disruption of BRCA1 LXCXE motif alters BRCA1 functional activity and regulation of RB family but not RB protein binding. Oncogene. 2001;20:4827–41. doi: 10.1038/sj.onc.1204666. [DOI] [PubMed] [Google Scholar]
- 13.Rashidian J, Iyirhiaro G, Aleyasin H, Rios M, Vincent I, Callaghan S, et al. Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc Natl Acad Sci USA. 2005;102:14080–5. doi: 10.1073/pnas.0500099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talluri S, Isaac CE, Ahmad M, Henley SA, Francis SM, Martens AL, et al. A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol Cell Biol. 2010;30:948–60. doi: 10.1128/MCB.01168-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isaac CE, Francis SM, Martens AL, Julian LM, Seifried LA, Erdmann N, et al. The retinoblastoma protein regulates pericentric heterochromatin. Mol Cell Biol. 2006;26:3659–71. doi: 10.1128/MCB.26.9.3659-3671.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bourgo RJ, Thangavel C, Ertel A, Bergseid J, McClendon AK, Wilkens L, et al. RB restricts DNA damage-initiated tumorigenesis through an LXCXE-dependent mechanism of transcriptional control. Mol Cell. 2011;43:663–72. doi: 10.1016/j.molcel.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ajioka I, Martins RA, Bayazitov IT, Donovan S, Johnson DA, Frase S, et al. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell. 2007;131:378–90. doi: 10.1016/j.cell.2007.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci. 1998;18:2801–7. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrusiak MG, Vandenbosch R, Park DS, Slack RS. The retinoblastoma protein is essential for survival of postmitotic neurons. J Neurosci. 2012;32:14809–14. doi: 10.1523/JNEUROSCI.1912-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erdmann G, Schütz G, Berger S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 2007;8:63. doi: 10.1186/1471-2202-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferguson KL, Vanderluit JL, Hébert JM, McIntosh WC, Tibbo E, MacLaurin JG, et al. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J. 2002;21:3337–46. doi: 10.1093/emboj/cdf338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ait-Si-Ali S, Polesskaya A, Filleur S, Ferreira R, Duquet A, Robin P, et al. CBP/p300 histone acetyl-transferase activity is important for the G1/S transition. Oncogene. 2000;19:2430–7. doi: 10.1038/sj.onc.1203562. [DOI] [PubMed] [Google Scholar]
- 23.Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, Chan HM, et al. E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol Cell Biol. 2004;24:4546–56. doi: 10.1128/MCB.24.10.4546-4556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trouche D, Cook A, Kouzarides T. The CBP co-activator stimulates E2F1/DP1 activity. Nucleic Acids Res. 1996;24:4139–45. doi: 10.1093/nar/24.21.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, et al. Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci. 2000;20:3104–14. doi: 10.1523/JNEUROSCI.20-09-03104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu CL, Classon M, Dyson N, Harlow E. Expression of dominant-negative mutant DP-1 blocks cell cycle progression in G1. Mol Cell Biol. 1996;16:3698–706. doi: 10.1128/mcb.16.7.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan HM, Smith L, La Thangue NB. Role of LXCXE motif-dependent interactions in the activity of the retinoblastoma protein. Oncogene. 2001;20:6152–63. doi: 10.1038/sj.onc.1204793. [DOI] [PubMed] [Google Scholar]
- 28.Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, et al. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010;24:1351–63. doi: 10.1101/gad.1917610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- 30.Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci USA. 2012;109:8971–6. doi: 10.1073/pnas.1119836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004;117:2173–81. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- 32.Frolov MV, Stevaux O, Moon NS, Dimova D, Kwon EJ, Morris EJ, et al. G1 cyclin-dependent kinases are insufficient to reverse dE2F2-mediated repression. Genes Dev. 2003;17:723–8. doi: 10.1101/gad.1031803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trouche D, Kouzarides T. E2F1 and E1A(12S) have a homologous activation domain regulated by RB and CBP. Proc Natl Acad Sci USA. 1996;93:1439–42. doi: 10.1073/pnas.93.4.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 35.Hébert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- 36.Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, et al. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol. 2001;155:207–16. doi: 10.1083/jcb.200105137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.