

Abstract
Estrogen receptors (ERs) and p53 can interact via cis-elements to regulate the angiogenesis-related VEGFR-1 (FLT1) gene, as we reported previously. Here, we address cooperation between these transcription factors on a global scale. Human breast adenocarcinoma MCF7 cells were exposed to single or combinatorial treatments with the chemotherapeutic agent doxorubicin and the ER ligand 17β-estradiol (E2). Whole-genome transcriptome changes were measured by expression microarrays. Nearly 200 differentially expressed genes were identified that showed limited responsiveness to either doxorubicin treatment or ER ligand alone but were upregulated in a greater than additive manner following combined treatment. Based on exposure to 5-fuorouracil and nutlin-3a, the combined responses were treatment-specific. Among 16 genes chosen for validation using quantitative real-time PCR, seven (INPP5D, TLR5, KRT15, EPHA2, GDNF, NOTCH1, SOX9) were confirmed to be novel direct targets of p53, based on responses in MCF7 cells silenced for p53 or cooperative targets of p53 and ER. Promoter pattern searches and chromatin IP experiments for the INPP5D, TLR5, KRT15 genes supported direct, cis-mediated p53 and/or ER regulation through canonical and noncanonical p53 and ER response elements. Collectively, we establish that combinatorial activation of p53 and ER can induce novel gene expression programs that have implications for cell-cell communications, adhesion, cell differentiation, development and inflammatory responses as well as cancer treatments.
Keywords: 17-beta estradiol, MCF7 cells, cis-element, doxorubicin, estrogen receptor, non-canonical response elements, nutlin, p53, synergistic cooperation, synergy
Introduction
The transcriptional activity of a sequence-specific transcription factor (TF) can be modulated in many ways including post-transcriptional and post-translational modifications, interactions with components of the basal transcription machinery or specific cofactors as well as the chromatin state.1,2 Equally important is the “quality” of the response element sequences and the cooperation/interaction with other transcription factors.1,2
The tumor suppressor p53, which has been described as the “guardian of the genome,” controls several biological outcomes that include cell cycle, growth, apoptosis, senescence, angiogenesis and genome stability.3,4 Also, it can regulate many other cellular processes such as autophagy, energy metabolism, mTOR signaling, immune responses, cell motility/migration and cell-cell communication, in part through modulation of several microRNA genes.5-7
The estrogen receptors (ERs) are nuclear receptor transcription factors that exert hormonal responses through the activation of proliferation pathways. While ERs are master regulators essential for development and maintenance of normal sexual and reproductive functions, they can also play a role in the cardiovascular, musculoskeletal, immune and central nervous systems.8-10
These two diverse networks exhibit crosstalk that can be due to direct interaction between p53 and the ERs, with the more frequently described outcome being repression of p53 activity,11-14 although p53 can also inhibit ERα.15,16 The inhibitory crosstalk, which can be mediated by physical interactions between the two proteins, can be relieved by stress-dependent post-translational modifications of p53.12,14 The p53/ER interactions can also result in mutual positive regulation at the level of target gene expression level.17,18 Most of the studies addressing p53/ER interaction were performed in breast cancer cell lines, implicating regulation of the activity and expression of p53 and ERs in tumorigenesis. This was supported by findings of a correlation between the presence of wild type p53 and ER-positive breast cancer and a correlation between mutant p53 and ER-negative breast cancer.19,20 The two transcription factors can also share co-regulators, such as p300 and MDM2. Both inhibition21 and positive regulation22 of ERα can result from the p53 negative regulator MDM2.
We recently identified transcriptional cooperation between activated p53 and ligand-bound ERs at the promoter of the VEGFR-1/FLT1 gene.23,24 The functional interaction appeared to occur through noncanonical cis-promoter REs for both transcription factors located in close proximity within the target promoter, where the p53 was a half-site created by an infrequent single nucleotide polymorphism.25-27 Neither p53 nor ER alone could significantly upregulate FLT1, but the combination resulted in synergistic activation.24 We proposed that noncanonical p53 REs consisting of ½ or ¾ sites can expand the p53 target network providing for moderate or weak p53 responsiveness, but at the same time providing the opportunity of conditional, context-dependent transactivation.5,25,27 Also, in the case of ERs the structural organization of the response element (ERE) has been shown to influence the binding affinity as well as the modulation of the expression of target genes. The consensus half-site ERE is considered the minimal target site for ERs, and other transcription factors as well as cofactors can promote binding and transcriptional modulation.28-30
Based on our finding at the FLT1 locus, we have taken a global approach to address whether similar scenarios might exist elsewhere in the genome using breast adenocarcinoma-derived MCF7 cells. Whole-genome expression changes were determined following combinations of exposures to doxorubicin (DOX), a genotoxic chemotherapeutic drug commonly used in cancer therapy that induces p53, and the ER ligand 17β-estradiol (E2). We identified 201 genes for which combined DOX/E2 treatment led to greater than additive upregulation. The genes were involved in cellular differentiation/development, extracellular matrix, cell adhesion and inflammation responses. For 10 out of 16 genes examined further, the synergistic transactivation was validated using quantitative real-time PCR. Using MCF7 cells with reduced p53 expression, we demonstrated that p53 participates directly in the modulation of their expression and in the cooperation with ER, and we discovered three new p53 target genes (GDNF, KRT15, SOX9). The cis-mediated cooperation at the level of the promoter of three of the 16 genes was interrogated by chromatin immunoprecipitation. KRT15 expression appeared to be regulated in cis through p53 and ERα response elements.
Results
Genome-wide transcriptome analyses identify a combinatorial effect of p53 and ERs activation in response to DOX and E2
We established the utility of our MCF7 cell system for detecting p53 and/or ER responses following treatment with DOX and/or the ER ligand E2. The chemotherapeutic agent 5-fluorouracil (5FU) and the non-genotoxic MDM2 inhibitor nutlin-3a31 were included to further support p53-specific effects on gene expression. The ERα protein levels in total extracts did not change after any of the 10-h stimuli used, while p53 protein was stabilized by DOX, 5FU and nutlin-3a but not by E2 (Fig. 1A). Both pathways were activated based on qPCR analysis of expression of the standard p53 target p21/CDKN1A and the ERα target pS2/TFF1 genes (Fig. 1B and C). p21 was induced to similar levels by DOX and 5FU, while E2 had no effect on expression. Nutlin-3a treatment resulted in higher relative p21 expression that was increased 1.5-fold with the addition of E2 (Fig. 1C). pS2/TFF1 was upregulated only in the presence of E2 and as a function of its concentration (10−7 or 10−9 M) with no further increase with DOX, 5FU or nutlin-3a (Fig. 1B). Under these conditions, there was no apparent toxicity for the p53 activator drugs or E2 alone while the combination of a p53 activator with E2 increased the overall cell index value, consistent with a role for estradiol in promoting proliferation (Fig. S1)
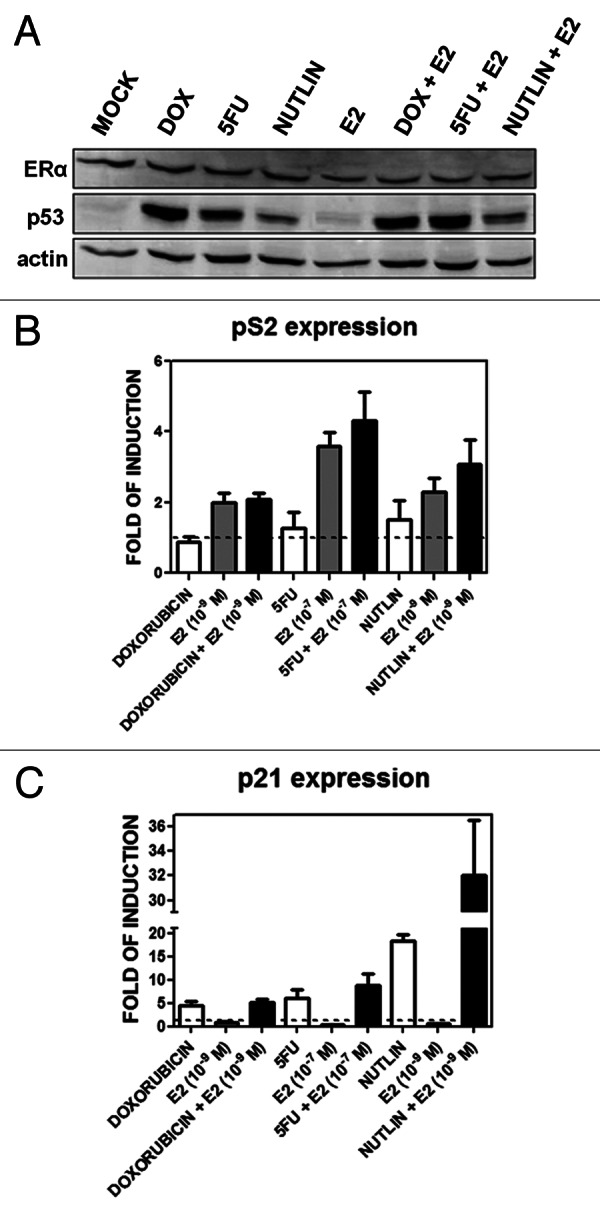
Figure 1. p53 and ERα protein levels and transactivation activities upon DOX, 5FU, nutlin-3a, E2 single or combined treatments. (A) Western blot analysis showing p53 and ERα protein levels 10 h after the indicated treatments at the following doses: DOX, 1.5 µM; 5FU, 375 µM, nutlin-3a, 10 µM; E2, 10−9 M. (B and C) qPCR results for the p53 target gene p21 (B) and the ERα target gene pS2/TFF1 (C). Presented in the bar graphs are fold-induction relative to the mock condition and the standard errors of three biological and two technical replicates for each condition. GAPDH, B2M and β-actin housekeeping genes served as internal controls.
Global gene expression profiling and statistical analysis of the microarray were performed as described in “Materials and Methods.” MCF7 cells cultured in estrogen-depleted media were subjected to single or combined treatments with DOX (1.5 µM) and E2 (at a pharmacological concentration 10−7 M, or a more physiological concentration 10−9 M). Gene ontology (GO), pathway enrichment and network analyses were conducted using DAVID (http://david.abcc.ncifcrf.gov/)32 as well as the Ingenuity Pathway Analysis (IPA, www.ingenuity.com).
Differences in transcriptome responses were identified in relation to the E2 doses (Fig. 2A and B). The lower E2 concentration (10−9 M) resulted in the same number of up- and downregulated DEGs, whereas the pharmacological concentration (10−7 M) was generally more repressive. Both concentrations of E2 resulted in differentially expressed genes (DEGs) exhibiting functional clusters enrichment that reflect expected estrogen-induced differentiation, proliferation, survival, hormonal responses and inhibition of p53 and SMARCB1 (Table S1A and B). Unexpected functional clusters were observed after 10−7 M E2, including positive regulation of apoptosis and negative regulation of cell growth as well as inhibition of SP1 (Table S1B). Therefore, we decided to focus our analysis on 10−9 M E2, since it resulted in a signature much closer to that of typical estrogen responses (Table S1A).
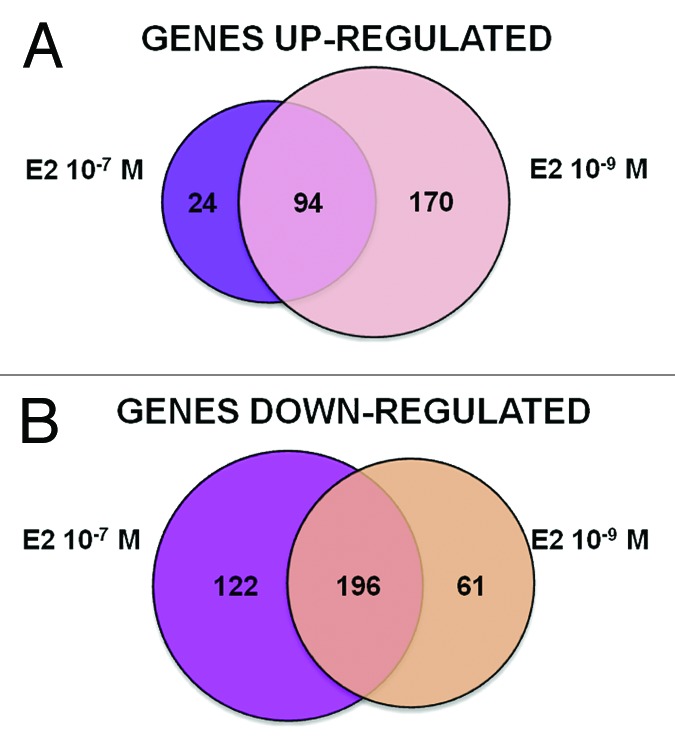
Figure 2. Graphical overview of E2 treatment-specific transcriptome changes. Differentially expressed genes (DEGs) were identified by Agilent microarray feature extraction, bioinformatics and statistical analyses, as described in the “Materials and Methods” section. Presented are Venn diagrams showing the number of upregulated (A) or downregulated (B) DEGs specific or in common between the different treatments with E2, 10−9 M and 10−7 M.
The clusters identified with DOX DEGs were consistent with genotoxic stress and p53 pathway activation, including cell cycle and apoptosis regulation, modulation of transcription, regulation of DNA damage checkpoints, BRCA1 functions and ATM signaling (Table S1C).
Next, we focused on the DOX + E2 (10−9 M) treatments to examine crosstalk between p53 and ERs. The overall transcriptome changes were heavily influenced by both treatments (Table S1D), although a greater overlap was observed between DEGs for DOX and DOX + E2 for both upregulated (66%) and downregulated (75%) genes (Fig. 3A and B). There was much less overlap between E2 and DOX + E2 DEGs (24% and 13% for the upregulated and downregulated groups, respectively). Stem cell pluripotency appeared as a distinctive IPA pathway (Table S1D). Interestingly, 66 upregulated and 167 downregulated DEGs were uniquely identified following DOX + E2 treatment. Conversely, for 380 upregulated and 369 repressed DOX DEGs the differential expression was not observed in the double treatment. Only 29 upregulated and 57 repressed DEGs were in common for the DOX and E2 single treatments, of which 27 up- and 54 downregulated genes were also DEGs with the double treatment (Fig. 3A and B). The functional annotation clusters obtained with these gene groups are summarized in Table S1E–H, although the small numbers limited the statistical power.
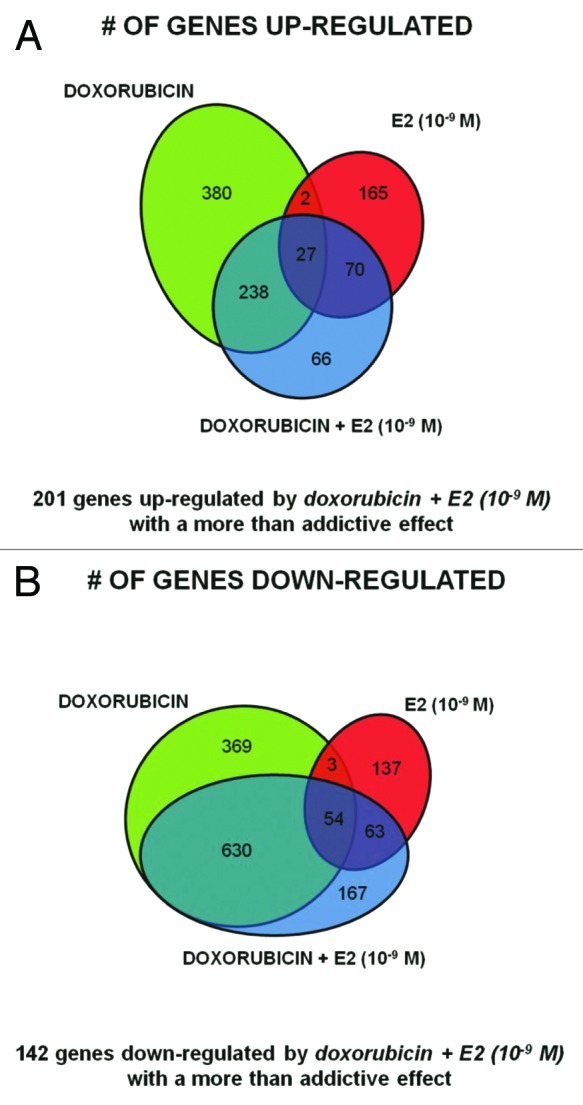
Figure 3. Specific gene signatures of the DOX+E2 combination treatment. Venn diagrams showing upregulated genes (A) or downregulated genes (B) comparing DOX, E2 and DOX + E2 DEGs. The number of genes differentially expressed in common or unique after doxorubicin or E2 (10−9 M) treatment or after their combination is indicated.
Cooperative p53, ER-mediated upregulation of genes involved in differentiation, cell-cell communication, adhesion and inflammatory response
As described in the “Materials and Methods,” we adopted a conservative approach based on the algebraic sum of logarithmic (log2) fold-change in expression. Statistical analysis for synergistic impact of combined treatments is presented in Table S3.
Notably, 201 upregulated and 142 downregulated genes met these criteria and exhibited a greater than additive response following the combined p53/ER-inducing treatments (Fig. 3). Analysis revealed enrichment for cell-cell communication, cell adhesion, development/differentiation and inflammatory response pathways (Table S1I) for the upregulated genes, while cell cycle and mitosis functions were enriched in the repressed group (Table S1J). We chose to pursue further the genes from the upregulated group, especially since repression via cis elements has yet to be established for p53 and ERα interactions (Table S3).
From the group of 201 genes exhibiting more than additive upregulation after combined DOX+E2 treatment (bold, Table S3), 16 that represented the main biological functions were selected for further analysis (Table S1I). Some are usually expressed in a different biological environment than breast cells (TEX14, SOX9, INPP5D), and others belong to biological pathways that can expand the p53/ER transcriptional master network (TFF3, CA5A, CDH26, NOTCH1, GDNF, INPP5D) (see Table S2A for references). For some, a direct or indirect functional interaction with p53 (NOTCH1, IGF2, TLR5 PML, INPP5D, EPHA2), with ER (NOTCH1, CDH26), or with other selected genes (IGF2 and H19, NOTCH1 and SOX9) has already been proposed (Table S2A). A summary of functional interactions predicted by text mining of the literature is shown in Figure S2 (http://stitch.embl.de/).33
Quantitative real-time PCR (qPCR) was performed to confirm the microarray results after DOX treatment with or without the addition of E2 (Fig. 4A). The trend of the microarray results was confirmed for 14/16 genes upon DOX and/or E2 treatment. T-test analysis on the log2 of the values obtained for relative expression confirmed for 10/16 genes the synergistic effect (p < 0.05) of DOX + E2 combination (Fig. 4A; Table S2A).

Figure 4. Treatment-selective transcriptional cooperation between p53-inducing stimuli and estradiol. qPCR reactions for the 16 chosen genes were performed using 384-well plates in a final volume of 10 µl using TaqMan® Gene Expression Assays with 3 biological and 2 technical replicates for each condition. GAPDH, B2M and β-actin housekeeping genes served as internal controls. Asterisks indicate statistically significant, more than additive effects in the combined treatment as described in the “Materials and Methods.” The same RNAs used in the microarray experiments were tested in (A and B), where the experiment served also as a validation of the array results, while all results in (C) were obtained from independent treatment and RNA extractions.
Expression of the 16 genes was also investigated following treatment with 5FU, another commonly used genotoxic agent that results in p53 activation. The responses clearly differed between DOX and 5FU (Fig. 4A and B). Only CDH26, INPP5D, NOTCH1 were responsive to 5FU (Fig. 4B); of these INPP5D and NOTCH1 were also DOX-responsive. The synergistic effects observed after DOX + E2 administration were also observed for H19, INPP5D and, in part, also for GDNF after 5FU + E2 (10−7 M) (Fig. 4B; Table S2B). Unlike for DOX, the combined treatment did not affect TLR5 or EPHA2, which are p53 target genes.34,35 Thus, the E2 enhancing effects on expression differ between two different inducers of p53.
Nutlin-3a treatment can synergistically cooperate with E2, but only on a subset of genes
Unlike genotoxic stress, nutlin-3a can directly activate p53. It targets the complex p53-MDM2, which results in p53 stabilization and activation without apparently inducing any kind of genotoxic stress.31 Given the difference in mechanism of p53 activation, we investigated possible interactions between E2 (10−9 M) and p53 following nutlin-3a treatment.
Among the 16 genes described above, the following six were upregulated by nutlin-3a treatment alone (fold-induction > 1.5; Fig. 4C) based on qPCR: EPHA2, INPP5D, KRT15, NOTCH1, SOX9, TEX14. The KRT15 gene was not responsive to DOX or 5FU (Fig. 4A and B), possibly indicating a differential effect of genotoxic post-translational modifications on p53-targeted expression. Only EPHA2, H19 and INPP5D showed a greater than additive effect for nutlin-3a + E2 (Fig. 4C; Table S2C). The synergy was also found for the H19 and INPP5D genes with E2 + DOX or 5FU and for EPHA2 with DOX + E2 (Figs. 4A and B).
Silencing of p53 in MCF7 cells establishes a direct role of p53 in doxorubicin responsiveness of the target genes
We validated direct p53 inducible expression of the novel genes using a stable MCF7 cell line expressing shRNA to p53.36 As shown in Figure 5A, the p53 protein level in MCF7-p53i is greatly reduced based on western blot analysis and gene expression of the p53 target gene p21, as compared with the control cells (“MCF7 vector”) after DOX treatment. Neither the p53 nor the ERα mRNA levels are changed after 10-h treatment with DOX or nutlin-3a (Fig. 5A). Expression of 8 of the 16 genes was determined at 10 h after DOX or nutlin-3a treatment of MCF7-p53i and -vector cells cultured in normal medium (Fig. 5B). EPHA2, GDNF, NOTCH1 and INPP5D were induced after either treatment of the MCF7 vector cells but were non-responsive or only slightly responsive in MCF7-p53i cells. The other five genes did not show any p53-specific responsiveness, although TLR5 is a p53 target.35

Figure 5. Changes in p53 and ERα protein levels and relative expression. Presented are results for p21, p53 and ER genes and of eight selected genes after 10 or 24 h DOX (1.5 µM) or nutlin-3a (10 µM) treatment in MCF7 vector and p53i. (A and C), left panel: western blot analysis showing p53 and ERα protein levels after 10 (A) and 24 h (C) of treatment. (A and C) right panel: qPCR results for the p53 target gene p21, the p53 and ERα (ESR1) genes after 10 (A) and 24 (C) hours of treatment. (B and D) qPCR results for the indicated eight genes after 10 or 24 h of treatment (left panels, DOX; right panels, nutlin). The fold-induction relative to the mock condition for MCF7-vector or MCF7-p53i is presented (H2O for DOX treatment or DMSO for nutlin-3a treatment).
We also examined DOX and nutlin-3a responses after 24 h. Both treatments enhanced expression of p53. However, DOX repressed ERα levels both at the protein and mRNA level, which would affect estradiol responses including the transcriptional cooperation with p53 at that time point (Fig. 5C). There was p53-dependent induction for seven of the eight genes following either treatment (Fig. 5D). DOX treatment led to residual induction of several of the genes in the MCF7-p53i cells, while only INPP5D was slightly responsive upon nutlin-3a treatment (Fig. 5D). This was presumably due to the low amount of p53 expression. CDH26 gene expression offers another example of treatment dependencies, as the gene was not regulated by p53 at either time point with either DOX or nutlin-3a, but was inducible by 5FU treatment alone (Fig. 4B and Fig. 5B and D).
The transcriptional responsiveness of INPP5D, TLR5 and KRT15 is associated with p53 and ER response elements
The biological impact and expression responses due to p53 plus estradiol led us to investigate in depth the promoter regions of the INPP5D, TLR5 and KRT15 genes for the presence of canonical and noncanonical p53 and ER response elements. An in silico search identified two distinct regions within the promoter of each of these genes (called A and B in Fig. 6) containing at least one putative ½-site p53 RE and one putative ½-site ERE (Fig. 6A).

Figure 6. Predicted p53 REs and EREs and relative occupancy of p53 and ER at TLR5A, INPP5D and KRT15 promoter regions. (A) Sequence, organization and position of mapped p53 and ER target sites. Promoters of selected genes were evaluated combining three approaches (see “Materials and Methods” for details). Dashed arrows mark ERE half sites, while tail-to-tail solid arrows denote the p53 RE half site. The chromosomal position, strand and the distance from the transcriptional start sites are also indicated. Two promoter fragments (denoted as #A and #B) were examined separately for each gene. (B–E) Chromatin immunoprecipitation and quantitative real-time PCR analyses. ChIP assays were performed using either an antibody against p53 (DO-1, Santa Cruz) or ERα (H-184) or control IgG (sc-2025). PCR was performed in 384-well plates in a final volume of 10 µl using primers designed to amplify regions containing validated REs and ERE for established p53 and ERα target genes (B), or to generate amplicons centered around the identified p53 REs and EREs in TLR5 (C), INPP5D (D) or KRT15 (E).
The promoters were also examined by ChIP qPCR for p53 and ER occupancy. As expected, there was p53 occupancy at the canonical p53 target REs of the p21, PUMA and BAX genes (Fig. 6B). Interestingly, E2 led to p53 recruitment at these promoters. p53 occupancy at the promoter regions was also found for the INPP5D, TLR5 (fragment A) and KRT15 genes (Fig. 6C–E) following DOX treatment. However, we were only able to detect ERα occupancy at the KRT15 promoter for fragment B (Fig. 6E) as well as the canonical ERα target pS2 (Fig. 6A). It appears that there is independent occupancy by the two transcription factors, in that the binding of one is not required for the recruitment of the other.
Histone marks associated with DOX and/or E2 treatment
While transcriptional synergy was established, it could not be ascribed to levels of p53 or ER binding, at least for the sites examined. Since changes in chromatin around regulatory regions of transcribed genes can modulate the activity and cooperativity between transcription factors, we analyzed chromatin status at the TLR5, INPP5D, KRT15 genes as well as at the control genes CDKN1A and TFF1. Promoter regions containing putative or known p53 REs and EREs along with regions encompassing the transcription start site (TSS) were examined for changes in histone tail post-translational modifications as well as total histones employing ChIP approaches and the same experimental conditions used to address p53 and ER occupancy.
Treatment with DOX resulted in a significant increase of the dimethylation H3K9me2 mark, which is associated with repression, for all tested genes. The increases were generally restricted to regions upstream of the TSS, but in the case of INPP5D and KRT15 were visible also at TSS. However, E2 treatment alone led to only a small increase in H3K9me2 at some sites and E2 was capable of reducing the DOX effect (Fig. 7A). No major changes were observed for the H3K4me2 mark, which is associated with active transcription. However, DOX treatment resulted in a slight increase at the TSS for TFF1 and INPP5D. E2 treatment was associated with an increase at TFF1 and CDKN1A TSS (Fig. 7B).

Figure 7. Treatment-induced histone methylation changes at CDKN1A, TFF1, TLR5, INPP5D and KRT15 promoter regions. Chromatin immunoprecipitation assays were performed using antibodies against H3K9me2 (07–441, Millipore) (A) or H3K4me2 (07–030) (B). IgG was used as control (sc-2027, Santa Cruz). Two or three regions of the promoter containing established or predicted p53 REs and EREs and the TSS were examined by quantitative PCR analysis. The distance from TSS of the promoter portions is indicated (see also Fig. 6A). Presented for each amplicon are average and standard deviation of changes relative to the mock condition. The colors of the bars indicate the promoter regions that were amplified and match the boxes that are placed in the schematic drawing of the genes on the top of each figure. The distance from TSS of the promoter regions that were examined is indicated.
There were increases associated with DOX and DOX + E2 treatments in H3 and H4 acetylation marks, corresponding mainly to open chromatin, in the region surrounding the p53 RE present −2.2 Kb from CDKN1A TSS (Fig. 8A and B). The E2 treatment led to an increase in H3 acetylation at TFF1 TSS and H4 acetylation both at the TSS and in the ERE-containing sequence located ~250 bp upstream from TSS. In both genes, these modifications are consistent with the enhanced transcription observed after DOX or E2 treatments. DOX counteracted the effect of E2 on these marks in TFF1. No significant changes were observed for the TLR5 and INPP5D genes, except for a consistent decrease in acetylation for INPP5D after combined treatment (Fig. 8A and B). For KRT15, the E2 and DOX + E2 treatments led to an increase in acetyl marks, especially near the TSS.

Figure 8. Treatment-induced histone acetylation changes at CDKN1A, TFF1, TLR5, INPP5D and KRT15 promoter regions. Chromatin immunoprecipitation assays were performed using antibodies against pan-H3Ac (06–599, Millipore) (A) or pan-H4Ac (06–866) (B), as described for Figures 6 and 7. The colors of the bars indicate the promoter regions that were amplified and match the boxes that are placed in the schematic drawing of the genes on the top of each figure. The distance from TSS of the promoter regions that were examined is indicated.
The total levels of H3 were also examined (Fig. S3). They appeared to be reduced near the TSS of the CDKN1A and TFF1 genes with all treatments For KRT15, the same trend was observed for all three regions analyzed. However, no changes were observed for the promoters of TLR5 and INPP5D, and an apparent increase was detected at TSS, particularly after DOX treatment.
Overall, our results indicate that all the genes analyzed were in an active chromatin state even in the mock condition, which is consistent with their basal expression levels. The treatments had an impact on several histone marks, although there was not a specific signature apparent for the double treatment.
Discussion
We have addressed the consequences of DOX and E2 on whole genome transcriptomes using p53 wild type and ERα-positive MCF7 cells as an experimental model of luminal-A subtype breast adenocarcinoma.37 Based on our previous work, we anticipated genes for which the inducible transcription factors p53 and ERs could act collaboratively in cis at their respective REs. Regardless of the mode of interaction, identifying genes for which there is a synergistic p53/ER response is expected to inform treatments of breast or other cancers. Therapeutic protocols often include modulation of either or both transcription factors, using p53-inducing drugs, such as DOX or 5FU, as well as ER antagonists or inhibitors of estrogen synthesis (www.chemocare.com).9,38 Other examples of crosstalk between different drugs in breast and other cancer types have been recently reported.39-41 Those findings exemplify the relevance of examining the impact of combinatorial treatments at the genome level.
DOX treatment resulted in dramatic changes in gene expression with 647 upregulated and 1056 downregulated genes and enrichment for the p53-pathway activation. While, strongly influenced by DOX, combined treatment with E2 resulted in 66 genes uniquely responsive and a total of 201 upregulated genes with greater than additive changes. Based on ontology and pathway analysis, the upregulated genes with greater than additive responses were enriched for cell-cell communication, epithelial cell differentiation and inflammatory response. Greater than additive downregulation was observed for 142 genes with enrichment for cell cycle, mitosis and metabolic functions (Table S1). Thus, we have identified interesting candidates for increased responses to genotoxic plus estrogen treatments.
We chose to focus on 16 upregulated genes in order to better understand the greater than additive responses toward p53 and estrogen inducing agents. While DOX or 5FU treatment resulted in similar p53 levels and p21 induction, there were marked differences in expression after single treatments, as well as when combined with estradiol (summarized in Table S4). Previous studies have established cell type-specific responses to DOX and 5FU as well as other drugs.42 Notably, only H19 and INPP5D consistently exhibited transcriptional cooperation between E2 and DOX, 5FU and nutlin treatments. Using p53-deficient MCF7 cells, the dependency on p53 was examined for eight genes and confirmed for the newly identified p53 target genes GDNF, KRT15, and SOX9 as well as the previously reported TLR5,35 INPP5D,43 NOTCH161 and EPHA2.34 CDH26 was 5FU-responsive (Fig. 4B), but a requirement for p53-dependent induction was not confirmed in our cell system, highlighting once again the specificity of drug response.
Given our earlier results with the FLT1 gene,23,24 we examined three of the 16 genes for the possibility of cis interactions by assessing p53 and ER occupancy. p53 bound directly to p53-related target sequences in the promoters of the TLR5, INPP5D and KRT15 genes. We further confirmed their p53-dependent induction after DOX and nutlin-3a treatment using MCF7 cells silenced for p53. TLR5 gene is involved in innate immunity.35 INPP5D affects regulation of inositol signaling43,44 and showed a more than additive transactivation in all three combined treatments. KRT15 is an intermediate filament type I protein responsible for the mechanical integrity of epithelial cells,45 and its expression is regulated by E2. Direct evidence for possible functional interaction between p53 and ER via cis-elements was only established for the KRT15 gene, which also showed ERα occupancy upon E2 single treatment. There are several reasons that might explain a lack of detectable ERα occupancy upon combined treatments, if it truly contributes to the greater than additive gene responses. Included is the possibility of a role for ERβ, which was not examined. Also, the type of interaction that can occur between p53 and ERα might differ between genes. ERα can in fact bind other transcription factors in an ERE-independent manner.9,10 Furthermore, non-genomic estrogen signaling pathways9,10 must also be considered for their contribution to the observed transcriptional cooperation. This might be particularly relevant in the early phase of E2 responses. The sources of interaction would be interesting to pursue in future structure-function analyses. Importantly we establish that the in-cis p53/ER cooperation involves a p53 half-site and half-site EREs, extending our previous findings beyond the FLT1 gene and model plasmid-based systems.26
p53 and ER occupancy levels were examined and did not correlate directly with the observed cooperation in expression. In our experiments the same time point (10 h) was chosen both for transcriptome and ChIP assays. Possibly, chromatin changes had occurred earlier that would influence the subsequent expression. However, in a comparison of the impact of DOX and DOX + E2 treatments in MCF7 cells, there was also a lack of correlation between p53 occupancy and transactivation levels46 for the case of ChIP analysis at 4 h and qPCR 12 h.
We also investigated changes in chromatin, since drug treatments could elicit epigenetic changes related to transcriptional reprogramming and DNA damage responses. Chromatin could change in a gene-specific manner without a direct correlation to TF occupancy levels of expression. The H3K4me2 mark is usually associated with actively transcribed genes and positioned around the TSS and the promoter area,47 and H3K9me2 is associated with gene silencing, especially when the mark is widespread along the gene. H3K9me2 can also be associated with openness/gene activity when present at the 5′ region of a gene47 and can reflect changes elicited by DNA damage responses.48,49
p53 and ER have been functionally and physically related to proteins involved in chromatin methyl mark changes, such as G9a and LSD1.50-55 However, the outcome of the induced epigenetic changes is variable. For example, G9a, considered the major euchromatin H3K9 methyltransferase, can act both as corepressor and as a coactivator for nuclear receptor functions, in cooperation with CARM1 and p300.50 Notably, both CARM1 and p300 can be recruited by p53 contributing to transcriptional activation.51 Acetylation marks at H3 and H4 histone tails are considered chromatin activation markers. Both p53 and ER can recruit histone acetyltransferases contributing to gene activation.51,56,57
Thus, the complexity of histone tail epigenetic changes cannot be easily related to alterations of transcription. However, the results obtained allowed us to propose that all genes analyzed are in an active chromatin state already in the mock condition. While treatments had an impact on histone marks, a specific signature of increased promoter openness after double treatment was not evident.
There are other mechanisms that can account for transcriptional cooperation that would be interesting to pursue. Functional interactions with p53 could involve other members of the large superfamily of nuclear receptors, including, for example, the glucocorticoid or androgen receptors, connected through a multi-protein mediator complex. Furthermore, our initial studies suggest that for a subset of promoters, crosstalk with ER could be affected by p63 and p73 members of the p53 family.26 p53 splice variants and various kinds of p53 stress or ER activators might be expected to affect the ER/p53 synergistic responses. p53 activators can vary in their impact on p53 post-translational modifications and alter transcriptome responses.58,59 It is important to note that, while p53 has been implicated, there may be other reasons for the genotoxic stress/ER synergistic responses.
Overall, we have found extensive transcriptional cooperation between ERs and p53 across the genome. Given the importance of activators of these two genes in cancer treatments, these findings provide opportunities for investigations of treatments involving many newly identified targets of synergy, although the mechanisms of synergy remain to be established. The findings are also relevant to understanding combined ER hormonal responses and any of the many4,6 stresses that can induce p53 as well as general biological and cancer implications. Although it is difficult to predict phenotypic outcome, the relevance of potential p53/ER biological outcomes is apparent. For example, among the 16 genes examined in depth, H19,60 NOTCH1,61 SYNM,62 TLR563 and cadherins64 are found either overexpressed or downregulated in breast cancer. The synergy might lead to increased aggressiveness or tumor metastasis (such as EMT) or, alternatively, influence inhibition of classical tumor hallmarks such as proliferation. EPHA2 has been reported to play a role in angiogenesis and tumor neovascularization as well as being a positive mediator of UV-induced and largely p53-independent apoptosis, but it can also affect oncogenesis in melanocytes.65,66 Other genes, such as PML,67 INPP5D68 and APC269 are thought to be tumor suppressor genes. Cadherins are usually downregulated in tumors,64 whereas IGF2 is often overexpressed in many types of cancers and thought to be an oncogene.70
Finally, our findings suggest the opportunity to identify additional luminal breast tumor markers. Expression of some of the 16 selected genes is usually weak or moderate in breast tissues (Human Protein Atlas).71 Understanding the functional roles that altered expression of those genes can play in different tissues could also aid in understanding the role that they may have in tumorigenesis.
Materials and Methods
Cell lines and treatments
The human breast adenocarcinoma-derived MCF7 cell line (p53 wild type; ERα, ERβ-weakly positive) was obtained from ICLC and maintained in Dulbecco’s modified Eagle’s (DMEM), 10% FBS, 2 mM glutamine, 100 units/ml penicillin and 100 µg/ml streptomycin. Estrogen-depleted medium consisted of DMEM without phenol red supplemented with 10% charcoal filtered FBS. MCF7 cells stably expressing an shRNA targeting p53 (MCF7-p53i), or control cells (MCF7-vector), were kindly provided by Dr Agami.36 Media and reagents were supplied by BioWhittaker® or Invitrogen. MCF7 cells were treated with 1.5 µM doxorubicin (DOX) or 375 µM 5-fluorouracil (5FU) or 10 µM nutlin-3a for p53 stabilization, +/−10−9/10−7 M 17β-estradiol. Stock solutions were dissolved in 100% DMSO for 5FU (0.5 M) and nutlin-3a (10 mM), H2O for DOX (10 mM) and 100% EtOH for E2 (10−3 M). DOX, 5FU and E2 were purchased from Sigma-Aldrich®; Nutlin-3a was obtained from Alexis® Biochemicals (Enzo Life Sciences). All treatments were done with cells at 70–80% confluence.
Antibodies and western blot analysis
Antibodies used for ChIP assays and western blotting analysis were: p53 (DO-1), ERα (H-184), Actin (I-19 or C-11) and IgG (sc-2025 or sc-2027) (Santa Cruz Biotechnology®) Anti-dimethyl-Histone H3 (Lys9) (07–441), anti-dimethyl-Histone H3 (Lys4) (07–030), anti-acetyl-Histone H3 (06–599), anti-acetyl-Histone H4 (06–866), anti-Histone H3 (06–755) antibodies (Millipore). Proteins were extracted using RIPA buffer supplemented with protease inhibitors and quantified using the BCA assay (Thermo Scientific, Pierce Protein Research Products). Proteins separated on 12% SDS-PAGE gels were transferred to a nitrocellulose membrane (GE Healthcare) using an iBlot® Dry Blotting System (Invitrogen™, Life Technology) and checked by Ponceau S staining. Membranes were blocked using 5% skim milk + PBS-Tween20 (0.1%) for 1 h at RT and probed with primary antibodies in 1% skim milk + PBS-Tween20. Immune complexes were visualized using Amersham ECL™ Advance WB Detection Kit (GE Healthcare) or SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). The relative molecular mass of the immunoreactive bands was determined using PageRuler™ Plus Prestained Protein Ladder (Fermentas).
Microarray hybridization and scanning, data acquisition and analysis
Cells were seeded and treated on 10 cm Petri dishes. Total RNA was extracted from 3–7 biological replicates using the Agilent Total RNA Isolation Mini Kit (Agilent Technologies) according to the manufacturer’s protocol. RNA was quantified using the NanoDrop spectrophotometer (NanoDrop Technologies), and quality was checked by gel electrophoresis as well as Agilent 2100 Bioanalyzer. Details on labeling, hybridization, analysis of TIFF images by Agilent Feature Extraction and the R software environment for statistical computing and the Bioconductor library of biostatistical packages are provided with the Gene Expression Omnibus (GEO) (www.ncbi.nlm.nih.gov/geo/) submission (GSE24065). Briefly, hybridization, blocking and washing were performed according to Agilent protocol “One-Color Microarray-Based Gene Expression Analysis (Quick Amp Labeling).” Hybridized microarray slides (Agilent-014850 Whole Human Genome Microarray 4 × 44 K G4112F-Probe Name version) were then scanned with an Agilent DNA Microarray Scanner (G2505C) at 5-micron resolution with the manufacturer’s software (Agilent ScanControl 8.1.3).
The scanned TIFF images were analyzed numerically for data extraction, background correction and flagging of non-uniform features using the Agilent Feature Extraction Software version 10.7.7.1 according to the Agilent standard protocol GE1_107_Sep09. The output of Feature Extraction was analyzed with the R software environment for statistical computing and the Bioconductor library of biostatistical packages. Probes with low signals were removed in order to filter out the constantly unexpressed genes and keep only probes flagged as present in the majority of replicates in at least one condition. Signal intensities across arrays were normalized with the quantile normalization algorithm. In order to select differentially expressed genes, every condition corresponding to a treatment was first compared with the mock treatment. Three thresholds were set in order to select differentially expressed genes for each comparison: (1) t-test unpaired unequal variance p value < 0.01; (2) rank product percentage of false positive (pfp) < 0.0572; (3) absolute log2 (fold change) > log2(2).
Using the DAVID resource,32 a functional annotation clustering analysis (enrichment score ≥ 1.5, medium classification stringency) was performed on the lists of differentially expressed genes corresponding to each treatment.
Genes upregulated by the concomitant treatment of doxorubicin and E2 (10−9 M) with more than an additive effect were identified among those satisfying the condition log2[FCdouble treatment] > 2 (a parameter allowing us for a more reasonable validation) subtracting the 2-fold changes corresponding to the single treatments to the fold change corresponding to the double treatment and selecting those with a positive result: (log2[FCdouble treatment] – log2[FCDOX] – log2[FCE2]) > 0.1 (Table S2).
Quantitative real-time PCR (qPCR)
One μg of total RNA was reverse transcribed in 20 µl of reaction using the “RevertAidTM First Strand cDNA Synthesis Kit” (Fermentas) or TaqMan reverse transcription reagents from Applied Biosystems. qPCR was performed using 384-well plates in a final volume of 10 μl either on a CFX384 Touch™ Real-Time PCR Detection Systems (Bio-Rad) or on the ABI prism HT7900 system (Applied Biosystems). KAPA Probe FAST qPCR Kit/TaqMan Universal PCR Master Mix (Applied Biosystems) or KAPA SYBR® FAST qPCR Kit (Kapa Biosystems, Resnova) was used to perform the reaction together with TaqMan® Gene Expression Assays (Applied BiosystemTM, Life Technology) or primers purchased from Eurofins (MWG, Operon). Relative mRNA quantification was obtained using the comparative Ct method (ΔΔCt), where glyceraldehyde 3-phosphate dehydrogenase (GAPDH), β-2microglobulin (B2M) or β-actin genes served as internal controls. Calculations were performed using QbasePLUS software (Biogazelle) that uses the geNorm method73 to evaluate the expression stability of candidate reference genes.
A statistical analysis considering the log2 of the fold-induction was used to confirm the synergistic effect. The means of two normally distributed populations composed of log2[FCdouble treatment] and log2[FCDOX] + log2[FCE2] were analyzed using a t-test approach (p < 0.05). The logarithmic values can flatten the differences between the fold change values on one hand but, on the other hand, can make the results of our analysis more robust. The sum of logarithms is comparable to the multiplication of the fold changes and the subtraction of logarithms to the ratio of the fold-changes.
Promoter pattern searches
An in silico analysis was performed in order to identify putative canonical or non-canonical p53 and ERα response elements (REs) couples with a maximum distance of around 500 bp within the promoters of the selected genes. Three different approaches were used and combined: (1) pattern matching analysis (½ p53 RE: RRRCWWGYYY; ½ ERα RE: (A)GGTCA, TGACC(T) or GGCTA), (2) RSAT analysis74 and (3) R tool analysis using TransFac matrixes.
Chromatin immunoprecipitation (ChIP) assay
MCF7 cells were cultured in estrogen-depleted conditions in a 150-mm Petri dish and treated for 10 h with DOX and/or the physiological concentration of E2 (10−9 M). The procedure for crosslinking, sonication, IP and analysis followed a previously described protocol.23,24,35
Supplementary Material
Acknowledgments
We thank Dr Valentina Adami for technical assistance with the microarray experiments. This work was partially supported by the Italian Association for Cancer Research, AIRC (#IG9086 to AI), by CIBIO start-up funds and by the Intramural Research Program of the NIEHS (to D.M. and M.A.R.: Z01 ES065079). M.L. is a PhD Fellow of the International Doctorate in Biomolecular Sciences, University of Trento. Y.C. is supported by a Marie-Curie/Autonomous-Province-of-Trento (PAT) co-fund grant (#40101712). V.dS. was supported by a Fellowship from Pezcoller Foundation, Trento.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were diclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24309
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24309
References
- 1.Espinosa JM. Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene. 2008;27:4013–23. doi: 10.1038/onc.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Y, Tsai C-J, Ma B, Nussinov R. Mechanisms of transcription factor selectivity. Trends Genet. 2010;26:75–83. doi: 10.1016/j.tig.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 4.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menendez D, Inga A, Resnick MA. Potentiating the p53 network. Discov Med. 2010;10:94–100. [PubMed] [Google Scholar]
- 6.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 7.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 8.Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, et al. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol Rev. 2006;58:773–81. doi: 10.1124/pr.58.4.8. [DOI] [PubMed] [Google Scholar]
- 9.Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–70. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–31. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 11.Konduri SD, Medisetty R, Liu W, Kaipparettu BA, Srivastava P, Brauch H, et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci USA. 2010;107:15081–6. doi: 10.1073/pnas.1009575107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu W, Ip MM, Podgorsak MB, Das GM. Disruption of estrogen receptor α-p53 interaction in breast tumors: a novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res Treat. 2009;115:43–50. doi: 10.1007/s10549-008-0044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu W, Konduri SD, Bansal S, Nayak BK, Rajasekaran SA, Karuppayil SM, et al. Estrogen receptor-α binds p53 tumor suppressor protein directly and represses its function. J Biol Chem. 2006;281:9837–40. doi: 10.1074/jbc.C600001200. [DOI] [PubMed] [Google Scholar]
- 14.Sayeed A, Konduri SD, Liu W, Bansal S, Li F, Das GM. Estrogen receptor α inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer Res. 2007;67:7746–55. doi: 10.1158/0008-5472.CAN-06-3724. [DOI] [PubMed] [Google Scholar]
- 15.Liu G, Schwartz JA, Brooks SC. p53 down-regulates ER-responsive genes by interfering with the binding of ER to ERE. Biochem Biophys Res Commun. 1999;264:359–64. doi: 10.1006/bbrc.1999.1525. [DOI] [PubMed] [Google Scholar]
- 16.Yu C-L, Driggers P, Barrera-Hernandez G, Nunez SB, Segars JH, Cheng S-y. The tumor suppressor p53 is a negative regulator of estrogen receptor signaling pathways. Biochem Biophys Res Commun. 1997;239:617–20. doi: 10.1006/bbrc.1997.7522. [DOI] [PubMed] [Google Scholar]
- 17.Fernández-Cuesta L, Anaganti S, Hainaut P, Olivier M. Estrogen levels act as a rheostat on p53 levels and modulate p53-dependent responses in breast cancer cell lines. Breast Cancer Res Treat. 2011;125:35–42. doi: 10.1007/s10549-010-0819-x. [DOI] [PubMed] [Google Scholar]
- 18.Angeloni SV, Martin MB, Garcia-Morales P, Castro-Galache MD, Ferragut JA, Saceda M. Regulation of estrogen receptor-alpha expression by the tumor suppressor gene p53 in MCF-7 cells. J Endocrinol. 2004;180:497–504. doi: 10.1677/joe.0.1800497. [DOI] [PubMed] [Google Scholar]
- 19.Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005;102:13550–5. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olivier M, Langerød A, Carrieri P, Bergh J, Klaar S, Eyfjord J, et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res. 2006;12:1157–67. doi: 10.1158/1078-0432.CCR-05-1029. [DOI] [PubMed] [Google Scholar]
- 21.Duong V, Boulle N, Daujat S, Chauvet J, Bonnet S, Neel H, et al. Differential regulation of estrogen receptor α turnover and transactivation by Mdm2 and stress-inducing agents. Cancer Res. 2007;67:5513–21. doi: 10.1158/0008-5472.CAN-07-0967. [DOI] [PubMed] [Google Scholar]
- 22.Kim K, Burghardt R, Barhoumi R, Lee SO, Liu X, Safe S. MDM2 regulates estrogen receptor α and estrogen responsiveness in breast cancer cells. J Mol Endocrinol. 2011;46:67–79. doi: 10.1677/JME-10-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciribilli Y, Andreotti V, Menendez D, Langen J-S, Schoenfelder G, Resnick MA, et al. The coordinated p53 and estrogen receptor cis-regulation at an FLT1 promoter SNP is specific to genotoxic stress and estrogenic compound. PLoS ONE. 2010;5:e10236. doi: 10.1371/journal.pone.0010236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menendez D, Inga A, Snipe J, Krysiak O, Schönfelder G, Resnick MA. A single-nucleotide polymorphism in a half-binding site creates p53 and estrogen receptor control of vascular endothelial growth factor receptor 1. Mol Cell Biol. 2007;27:2590–600. doi: 10.1128/MCB.01742-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–37. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 26.Menendez D, Inga A, Resnick MA. Estrogen receptor acting in cis enhances WT and mutant p53 transactivation at canonical and noncanonical p53 target sequences. Proc Natl Acad Sci USA. 2010;107:1500–5. doi: 10.1073/pnas.0909129107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menendez D, Krysiak O, Inga A, Krysiak B, Resnick MA, Schönfelder G. A SNP in the flt-1 promoter integrates the VEGF system into the p53 transcriptional network. Proc Natl Acad Sci USA. 2006;103:1406–11. doi: 10.1073/pnas.0508103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carroll JS, Brown M. Estrogen receptor target gene: an evolving concept. Mol Endocrinol. 2006;20:1707–14. doi: 10.1210/me.2005-0334. [DOI] [PubMed] [Google Scholar]
- 29.Gruber CJ, Gruber DM, Gruber IML, Wieser F, Huber JC. Anatomy of the estrogen response element. Trends in Endocrinology &. Metabolism. 2004;15:73–8. doi: 10.1016/j.tem.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 30.Joshi SR, Ghattamaneni RB, Scovell WM. Expanding the paradigm for estrogen receptor binding and transcriptional activation. Mol Endocrinol. 2011;25:980–94. doi: 10.1210/me.2010-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat Rev Clin Oncol. 2011;8:25–37. doi: 10.1038/nrclinonc.2010.174. [DOI] [PubMed] [Google Scholar]
- 32.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn M, Szklarczyk D, Franceschini A, Campillos M, von Mering C, Jensen LJ, et al. STITCH 2: an interaction network database for small molecules and proteins. Nucleic Acids Res. 2010;38(Database issue):D552–6. doi: 10.1093/nar/gkp937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin YJ, Wang J, Qiao C, Hei TK, Brandt-Rauf PW, Yin Y. A novel mechanism for p53 to regulate its target gene ECK in signaling apoptosis. Mol Cancer Res. 2006;4:769–78. doi: 10.1158/1541-7786.MCR-06-0178. [DOI] [PubMed] [Google Scholar]
- 35.Menendez D, Shatz M, Azzam K, Garantziotis S, Fessler MB, Resnick MA. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011;7:e1001360. doi: 10.1371/journal.pgen.1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 37.Subik K, Lee J-F, Baxter L, Strzepek T, Costello D, Crowley P, et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer (Auckl) 2010;4:35–41. [PMC free article] [PubMed] [Google Scholar]
- 38.Munster PN, Carpenter JT. Estradiol in breast cancer treatment: reviving the past. JAMA. 2009;302:797–8. doi: 10.1001/jama.2009.1223. [DOI] [PubMed] [Google Scholar]
- 39.Azmi AS, Banerjee S, Ali S, Wang Z, Bao B, Beck FW, et al. Network modeling of MDM2 inhibitor-oxaliplatin combination reveals biological synergy in wt-p53 solid tumors. Oncotarget. 2011;2:378–92. doi: 10.18632/oncotarget.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng XS, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11:367–76. doi: 10.4161/cc.11.2.18813. [DOI] [PubMed] [Google Scholar]
- 41.Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle. 2011;10:1563–70. doi: 10.4161/cc.10.10.15703. [DOI] [PubMed] [Google Scholar]
- 42.Troester MA, Hoadley KA, Parker JS, Perou CM. Prediction of toxicant-specific gene expression signatures after chemotherapeutic treatment of breast cell lines. Environ Health Perspect. 2004;112:1607–13. doi: 10.1289/ehp.7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Q, Dumont DJ. Molecular cloning and chromosomal localization in human and mouse of the SH2-containing inositol phosphatase, INPP5D (SHIP). Amgen EST Program. Genomics. 1997;39:109–12. doi: 10.1006/geno.1996.4374. [DOI] [PubMed] [Google Scholar]
- 44.Kerley-Hamilton JS, Pike AM, Li N, DiRenzo J, Spinella MJ. A p53-dominant transcriptional response to cisplatin in testicular germ cell tumor-derived human embryonal carcinoma. Oncogene. 2005;24:6090–100. doi: 10.1038/sj.onc.1208755. [DOI] [PubMed] [Google Scholar]
- 45.Badock V, Steinhusen U, Bommert K, Wittmann-Liebold B, Otto A. Apoptosis-induced cleavage of keratin 15 and keratin 17 in a human breast epithelial cell line. Cell Death Differ. 2001;8:308–15. doi: 10.1038/sj.cdd.4400812. [DOI] [PubMed] [Google Scholar]
- 46.Bailey ST, Shin H, Westerling T, Liu XS, Brown M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc Natl Acad Sci USA. 2012;109:18060–5. doi: 10.1073/pnas.1018858109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 48.Mund A, Schubert T, Staege H, Kinkley S, Reumann K, Kriegs M, et al. SPOC1 modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic Acids Res. 2012;40:11363–79. doi: 10.1093/nar/gks868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sulli G, Di Micco R, d’Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012;12:709–20. doi: 10.1038/nrc3344. [DOI] [PubMed] [Google Scholar]
- 50.Lee DY, Northrop JP, Kuo MH, Stallcup MR. Histone H3 lysine 9 methyltransferase G9a is a transcriptional coactivator for nuclear receptors. J Biol Chem. 2006;281:8476–85. doi: 10.1074/jbc.M511093200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–48. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 52.Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–40. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128:505–18. doi: 10.1016/j.cell.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, et al. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–8. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 55.Tsai WW, Nguyen TT, Shi Y, Barton MC. p53-targeted LSD1 functions in repression of chromatin structure and transcription in vivo. Mol Cell Biol. 2008;28:5139–46. doi: 10.1128/MCB.00287-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell. 2001;8:57–69. doi: 10.1016/S1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- 57.Hanstein B, Eckner R, DiRenzo J, Halachmi S, Liu H, Searcy B, et al. p300 is a component of an estrogen receptor coactivator complex. Proc Natl Acad Sci USA. 1996;93:11540–5. doi: 10.1073/pnas.93.21.11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010;17:901–11. doi: 10.1038/cdd.2010.35. [DOI] [PubMed] [Google Scholar]
- 59.Poyurovsky MV, Katz C, Laptenko O, Beckerman R, Lokshin M, Ahn J, et al. The C terminus of p53 binds the N-terminal domain of MDM2. Nat Struct Mol Biol. 2010;17:982–9. doi: 10.1038/nsmb.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berteaux N, Lottin S, Monté D, Pinte S, Quatannens B, Coll J, et al. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J Biol Chem. 2005;280:29625–36. doi: 10.1074/jbc.M504033200. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Fu L, Gu F, Ma Y. Notch1 is involved in migration and invasion of human breast cancer cells. Oncol Rep. 2011;26:1295–303. doi: 10.3892/or.2011.1399. [DOI] [PubMed] [Google Scholar]
- 62.Noetzel E, Rose M, Sevinc E, Hilgers RD, Hartmann A, Naami A, et al. Intermediate filament dynamics and breast cancer: aberrant promoter methylation of the Synemin gene is associated with early tumor relapse. Oncogene. 2010;29:4814–25. doi: 10.1038/onc.2010.229. [DOI] [PubMed] [Google Scholar]
- 63.Cai Z, Sanchez A, Shi Z, Zhang T, Liu M, Zhang D. Activation of Toll-like receptor 5 on breast cancer cells by flagellin suppresses cell proliferation and tumor growth. Cancer Res. 2011;71:2466–75. doi: 10.1158/0008-5472.CAN-10-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cowin P, Rowlands TM, Hatsell SJ. Cadherins and catenins in breast cancer. Curr Opin Cell Biol. 2005;17:499–508. doi: 10.1016/j.ceb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 65.Udayakumar D, Zhang G, Ji Z, Njauw CN, Mroz P, Tsao H. EphA2 is a critical oncogene in melanoma. Oncogene. 2011;30:4921–9. doi: 10.1038/onc.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang G, Njauw C-N, Park JM, Naruse C, Asano M, Tsao H. EphA2 is an essential mediator of UV radiation-induced apoptosis. Cancer Res. 2008;68:1691–6. doi: 10.1158/0008-5472.CAN-07-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–70. doi: 10.1016/S0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 68.Luo JM, Liu ZL, Hao HL, Wang FX, Dong ZR, Ohno R. Mutation analysis of SHIP gene in acute leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2004;12:420–6. [PubMed] [Google Scholar]
- 69.Zhou M-N, Kunttas-Tatli E, Zimmerman S, Zhouzheng F, McCartney BM. Cortical localization of APC2 plays a role in actin organization but not in Wnt signaling in Drosophila. J Cell Sci. 2011;124:1589–600. doi: 10.1242/jcs.073916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shetty PJ, Movva S, Pasupuleti N, Vedicherlla B, Vattam KK, Venkatasubramanian S, et al. Regulation of IGF2 transcript and protein expression by altered methylation in breast cancer. J Cancer Res Clin Oncol. 2011;137:339–45. doi: 10.1007/s00432-010-0890-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248–50. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 72.Breitling R, Armengaud P, Amtmann A, Herzyk P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004;573:83–92. doi: 10.1016/j.febslet.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 73.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(H0034):research0034.1–research.11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thomas-Chollier M, Defrance M, Medina-Rivera A, Sand O, Herrmann C, Thieffry D, et al. RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res. 2011;39(suppl):W86–91. doi: 10.1093/nar/gkr377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.