

Abstract
The tumor suppressor p53 is a critical regulator of apoptosis and cell cycle arrest/pro-survival. Upon DNA damage, p53 evokes both cell cycle arrest/pro-survival and apoptosis transcriptional programs. The ultimate cellular outcome depends on the balance of these two programs. However, the p53 downstream targets that mediate this cell fate decision remain to be identified. Using an integrative genomic approach, we identify Rap2b as a conserved p53-activated gene that counters p53-mediated apoptosis after DNA damage. Upon DNA damage, p53 directly binds to the promoter of Rap2b and activates its transcription. The reduction of Rap2b levels by small interference RNA sensitizes cells to DNA damage-induced apoptosis in a p53-dependent manner. Consistent with its pro-survival function, analysis of cancer genomic data reveals that Rap2b is overexpressed in many types of tumors. Anchorage-independent growth assays show that Rap2b has only weak transformation activity, suggesting that it is not an oncogene by itself. Together, our results identify Rap2b as a new player in the pro-survival program conducted by p53 and raise the possibility that targeting Rap2b could sensitize tumor cells to apoptosis in response to DNA damage.
Keywords: gene regulation, chromatin, p53, epigenetics
Introduction
The tumor suppressor p53 is a DNA sequence-specific transcription factor and a stress sensor.1 Upon various stresses, such as DNA damage, oxidative stress and oncogene overexpression, p53 is stabilized, binds to chromatin and activates or represses numerous downstream genes that elicit many cellular outcomes, such as cell cycle arrest, apoptosis, DNA repair, senescence and stem cell differentiation.1-4 Cell cycle arrest and apoptosis are two of the major p53-regulated processes in response to DNA damage stress. The current view is that DNA damage initially activates a p53-mediated cell cycle arrest program, which allows cells more time to repair their damaged DNA and avoid mitotic catastrophe. Once repair is finished, the cells re-enter the cell cycle.1,3,5 Therefore, reversible cell cycle arrest is considered to be pro-survival.5-7 p53 targets, Cdkn1a and 14-3-3 sigma, are involved in cell cycle arrest and potent anti-apoptosis genes.8 Persistent or irreparable DNA damage, however, eventually invokes a p53-mediated apoptotic program that removes the cells from the population.6 Understanding the balance between cell cycle arrest/survival and apoptosis mediated by p53 is of great interest for developing therapeutic strategies to increase the effectiveness of cancer cell killing.
There are several existing models of how the balance between survival and death in p53 signaling is regulated. First, post-translational modifications, such as lysine 120 acetylation (K120ac), can skew the transcriptional output of p53 toward either cell cycle arrest or apoptosis and therefore affect the cell fate decision process of p53.9,10 Second, p53 binding partners, such as Hzf, Slug and hCAS, selectively regulate the expression of cell cycle arrest genes or apoptotic genes.11-14 Third, some p53 targets, for example Glx2 (also called glyoxalase II) and Cdkn1a, regulate the survival of cells after DNA damage but do not affect the expression of other p53 targets.15 Last, the levels of p53 in response to stresses also affect the outcome of various stresses.16 These individual events could also cooperate to regulate the life and death decisions controlled by p53.
Although p53 is a strict tumor suppressor, some of its downstream targets do not have a strict tumor-suppressive function. For example, Mdm2, the negative regulator of p53, is overexpressed in many types of tumors.17-19 Cdkn1a also has been implicated in maintaining the self-renewal of leukemia stem cells through its transient cell cycle arrest and pro-survival function.20 Another pro-survival target of p53, Cox-2, is associated with tumorigenesis, and its inhibition may have potential tumor-inhibitory effects.21 Therefore, tumor cells could “hijack” the pro-survival part in the p53 signaling. The investigation of the pro-survival function of p53 is of great significance in terms of cancer therapy.7,22 By targeting the pro-survival function of p53, the balance between life and death of cancer cells may be shifted toward apoptosis, and the optimal efficacy of chemo- or radiotherapy could be achieved.
Despite the fact that many cell cycle arrest/survival genes and apoptotic genes have been found, the comprehensive gene network of p53 remains to be determined.6,7,23,24 Previous genome-wide studies by us as well as others have identified a large number of p53 downstream targets.25-28 Most of these studies have used a single cell type, and the comparisons between cell types have not been fully performed. The identified genes contain both cell type-specific p53 targets and cell type-nonspecific p53 targets. Cell type-specific p53 targets may mediate cell type-specific functions of p53. For example, the Wnt genes are preferentially induced by p53 in mouse embryonic stem (mES) cells, and they may represent a cell non-autonomous anti-differentiation role of p53 in mES cells.28 In this study, we performed integrated genome-wide studies of p53 signaling in mES and mouse embryonic fibroblast (MEF) cells, and we also employed comparable analyses using genome-wide data sets of p53 in human cells to identify conserved p53 targets between mouse and human. Our goal is to identify new p53 target(s) that is (are) involved in regulating survival and/or apoptosis during DNA damage response. Through these analyses, we identify Rap2b as a new player in the p53 gene network that affects the balance between cell survival and apoptosis after DNA damage. Our result also suggests that targeting Rap2b might sensitize tumor cells to apoptosis induced by DNA damage.
Results
Identification of Rap2b as a novel p53 target using an integrated genome-wide approach
To identify p53 targets in an unbiased manner, we employed an integrated genome-wide approach by combining gene expression microarray and ChIP-chip assays in both mouse embryonic fibroblasts (MEFs) and mouse embryonic stem (mES) cells. This approach has been previously used to identify p53 direct targets in mES cells.4,28 A chemotherapeutic drug, adriamycin (also known as doxorubicin), was used to activate p53, because it induces DNA damage. Untreated cells were controls.
Using gene expression microarray, we detected 2,350 genes (1,260 activated and 1090 repressed) that were significantly (p < 0.001) changed in a p53-dependent manner after adriamycin treatment in MEF cells. In mES cells, 4,067 genes were significantly (p < 0.001) changed after the treatment (Fig. 1A). Among them, 2115 genes were activated, while 1,952 were repressed. Comparison of p53-dependent genes in MEF with those in mES revealed that 1,097 genes were common in both cell types (Fig. 1A; Table S1); 1,253 genes were only expressed in MEF; and 2,970 genes were only expressed in mES cells (Fig. 1A; Tables S2 and S3). This result suggests that a large portion of p53-dependent genes are cell type-specific or respond differently to a single dosage of adriamycin. Consistent with the previous study,28 several Wnt genes, including Wnt8b, Wnt3, Wnt3a and Wnt4, were only detected in mES cells (Table S3), indicating that our comparative analysis was able to distinguish cell type-specific genes from -nonspecific genes.
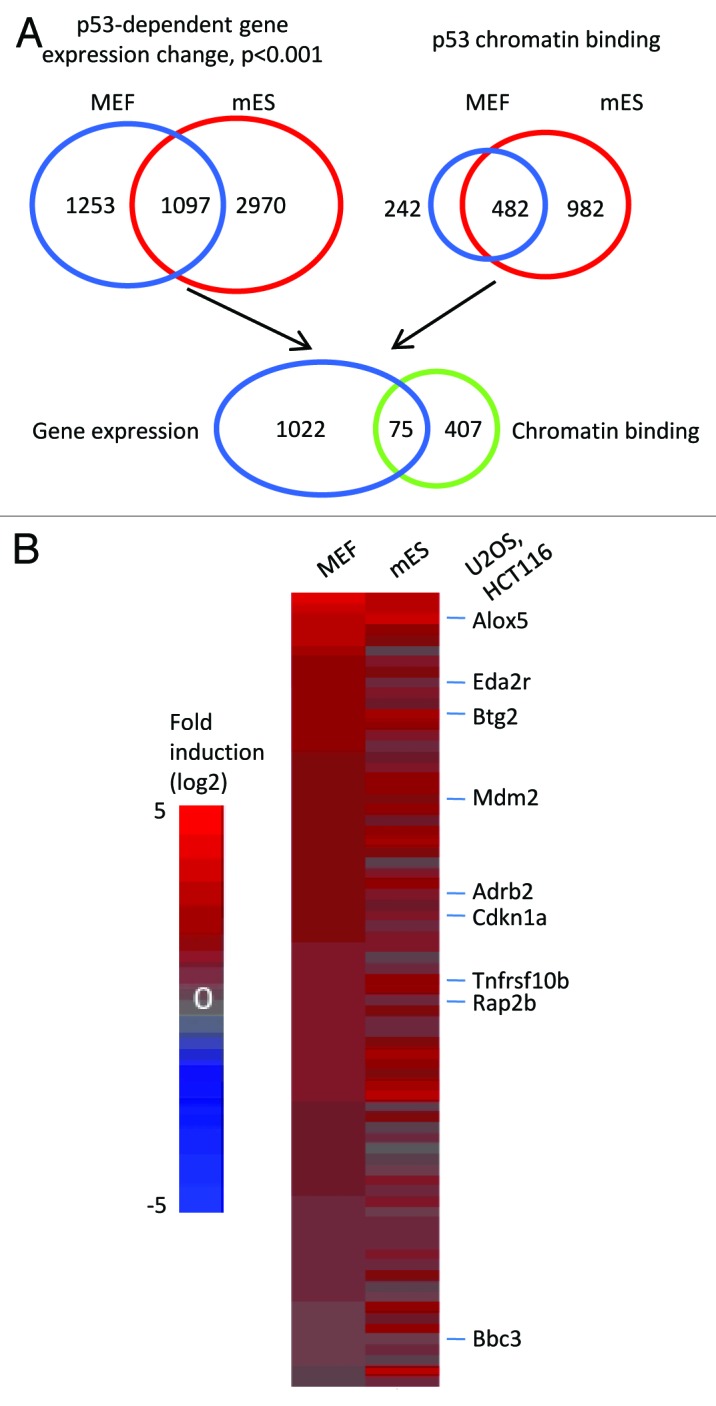
Figure 1. An integrated genome-wide approach to identify p53 direct targets. (A) Schematics of the strategy to identify p53 direct targets in both MEF and mES cells. (B) Heatmap showing the common direct targets of p53 in MEF and mES cells. Genes are rank-ordered according to the fold induction (adriamycin vs. untreated) in MEF cells. Genes shown on the right of the heatmap are common targets between mouse and human cell lines (U2OS and HCT116).26,27
p53 directly and indirectly regulates gene expression. In an indirect manner, p53 regulates the expression of other factors, e.g., noncoding RNAs or other transcription factors, which, in turn, alter the transcription of secondary genes.29,30 Genes regulated by p53 though these types of mechanisms are p53 indirect or secondary targets. We define p53 direct or primary targets as those whose expression is altered in a p53-dependent manner and that are bound by p53. To determine genes bound by p53, ChIP-chip assay was performed using a pan-p53 antibody in adriamycin-treated MEF and mES cells. Using the ChIP-chip assay, we detected 724 genes bound by p53 in MEFs and 1464 genes in mES cells (Table S4). The higher number of genes bound by p53 may reflect the fact that the chromatin of mES cells is more accessible to p53 than is that of MEF cells.31 When comparing these two sets of genes bound by p53, 482 genes were common to both MEFs and mES cells (Fig. 1A; Table S4). We then integrated the gene expression microarray data sets with the ChIP-chip data sets to obtain the common p53 direct targets in both MEF and mES cells. This analysis derived 75 common p53 direct targets in the two cell types (Fig. 1A). Strikingly, all these 75 genes were p53-activated genes (Fig. 1B), suggesting that p53-activated genes, as opposed to p53-repressed genes, are more likely to be cell type-nonspecific. We also separately analyzed p53 direct targets in MEF and mES cells (Fig. S1). A prominent finding was that the portion (17.7%, 72 out of 407) of p53-repressed genes in mES cells was significantly (p = 8.15e-5, Fisher’s exact test) higher than that in MEF cells (4.9%, 7 out of 144) (Fig. S1).
To identify p53 targets conserved between mouse and human, we compared our mouse p53 targets with genome-wide p53 targets identified in two human cell lines U2OS and HCT116 cells.26,27 The comparison detected nine genes, Alox5, Eda2r, Btg2, Mdm2, Adrb2, Cdkn1a (also called p21 or Waf1), Tnfrsf10b (also called DR5), Rap2b and Bbc3 (also called Puma), all of which were common p53 targets in the investigated mouse and human cell types. All these genes except Rap2b have been shown to be p53 downstream targets.32-38 Btg2, Mdm2 and Cdkn1a are known cell cycle arrest genes, while Eda2r, Bbc3 and Tnfrsf10b are apoptotic genes.32-38 Alox5 is involved in p53-mediated senescence through regulating the production of reactive oxygen species.39 Adrb2 is a p53 target in response to ultraviolet (UV) irradiation in melanocytes, but its function is unknown.35 Because Rap2b is the only gene that has not been reported to link to p53 signaling, we decided to further study the role of Rap2b during p53-mediated stress response. It should be noted that our integrated genome-wide approach could have missed some p53 targets due to the sensitivity of the ChIP-chip and gene expression microarray. Nonetheless, this approach is powerful to identify novel p53 targets that may play critical roles in p53 stress responses.
Rap2b is a conserved p53 target in response to DNA damage
Rap2b is one of the members of the Ras super-family. Phylogenetic analysis demonstrated that mouse and human Rap2b have identical amino acid sequences (Fig. 2A). Multi-alignment analysis with Rap2b amino acid sequences from eight species showed that Rap2b is a highly conserved protein (Fig. S2). In some species, including Homo sapiens, Macaca mulatta, Mus musculus, Pan troglodytes and Rattus norvegicus, the amino acid sequences of Rap2b are identical. The high conservation of the Rap2b protein sequence suggests that it may play important role(s) during evolution.

Figure 2. The induction of Rap2b mRNA by DNA damage is p53-dependent. (A) Phylogenetic analysis of mouse and human Ras super-family members. Numbers in the brackets were the amino acid difference. (B–E) Primary MEF and HCT116 cells were treated with adriamycin for various time as indicated. Real-time PCR analyses were performed to measure the induction of Rap2b (B and C) and Cdkn1a (D and E). 28S RNA was used as internal control to normalize the values. To facilitate comparison, normalized values of untreated (0 h) p53+/+ cells were set to 1. Results are displayed as mean ± SEM, for MEF, n = 4; for HCT116, n = 3.
To confirm the result from the integrated genome-wide approach, we used real-time PCR to monitor the induction of Rap2b mRNA in p53 wild type (p53+/+) and p53 knockout (p53−/−) MEF cells treated with adriamycin for various times (Fig. 2B). We also performed similar experiments in p53+/+ and p53−/− HCT116 cells (Fig. 2C). As a comparison, the kinetics of Cdkn1a mRNA induction was measured (Fig. 2D and E). Our results showed that the induction of Rap2b mRNA was p53-dependent. Interestingly, in the absence of adriamycin (0 h point), the mRNA levels of Rap2b were similar in p53+/+ and p53−/− cells, while the levels of Cdkn1a were much higher in p53+/+ cells than in p53−/− cells. Thus, p53 regulates the expression of Rap2b only in the presence of extrinsic DNA damage stress, while it regulates the expression of Cdkn1a both in the absence and presence of extrinsic DNA damage stress. This result suggests that Rap2b may play an important role in p53 signaling only during DNA damage stress.
To test whether the protein levels of Rap2b was also induced by p53 in response to DNA damage, we performed immunoblotting analysis in p53+/+ and p53−/− MEF cells. The result demonstrated that the protein levels of Rap2b also increased in p53+/+ MEFs treated with adriamycin, whereas it did not change in p53−/− MEFs (Fig. 3A). Consistent with mRNA induction analysis (Fig. 2B and C), there was no detectable difference of Rap2b protein levels between p53+/+ and p53−/− MEF cells without adriamycin treatment (0 h point). However, the basal level of Cdkn1a in p53−/− cells was much lower than that in p53+/+ cells. We also tested the induction of Rap2b protein in mES cells. mES cells were first transduced with lentivirus-expressing luciferase short hairpin RNA (shRNA) against luciferase (control) or p53 and then treated with adriamycin (Fig. 3B). Rap2b was induced in mES cells with luciferase shRNA but not in cells with p53 shRNA, demonstrating that Rap2b was induced in a p53-dependent manner in mES cells. As previously shown, the Cdkn1a protein cannot be detected in mES cells, although Cdkn1a mRNA is readily detectable.40 In addition, we observed similar induction of Rap2b in human HCT116 cells (Fig. 3C). Thus, Rap2b protein was induced by adriamycin in a p53-dependent manner in both mouse and human cells.
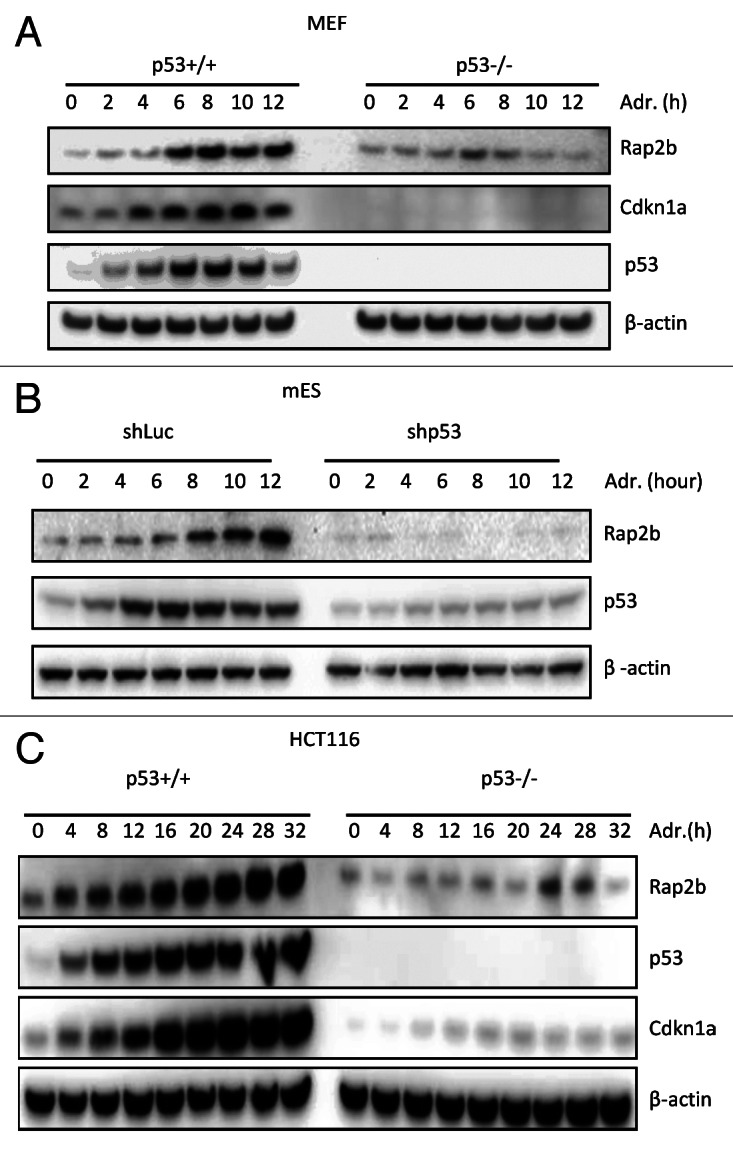
Figure 3. Rap2b protein is induced by p53 in mouse and human cells upon DNA damage. (A) Western blot analyses to measure the protein levels of Rap2b, Cdkn1a, p53 and β-actin in p53+/+ and p53−/− MEFs treated with adriamycin (Adr) for various times. (B) Western blot analyses of Rap2b, p53 and β-actin in mES cells transduced with lentiviruses expressing luciferase shRNA or p53 shRNA. (C) Western blot analyses of Rap2b, Cdkn1a, p53 and β-actin in p53+/+ and p53−/− HCT116 cells treated with Adr.
The induction of Rap2b by p53 was also examined in NIH3T3, MCF7 and IMR90 cells (Fig. S3). In addition, we tested the induction of Rap2b in H1299 cells that express p53 in a tetracycline-inducible manner. We observed that Rap2b was also activated by p53 in this cell type (Fig. S3). Together, the Rap2b gene is induced by p53 in a wide range of mouse and human cells.
Rap2b is induced by other stresses in a p53-dependent manner
Because p53 is regarded as a general stress sensor and it is activated by various stresses, we investigated whether other types of stresses also induce Rap2b through p53.41 To address this question, p53+/+ and p53−/− MEF cells were either untreated or treated with Nutlin or UV followed by western blot analysis with Rap2b and p53 antibodies (Fig. 4A). Of note, Nutlin is a non-genotoxic p53 activator and stabilizes p53 through disrupting the interaction of p53 and Mdm2.42 Nutlin and UV both activated and stabilized p53 in p53+/+ MEF cells, as judged by the increase of p53 protein levels (Fig. 4A). The protein levels of Rap2b increased significantly in p53+/+ cells treated with Nutlin or UV, but not in p53−/− cells. We also observed similar results in p53+/+ and p53−/− HCT116 cells (Fig. 4B). These results indicate that the induction of Rap2b by p53 is not limited to adriamycin.
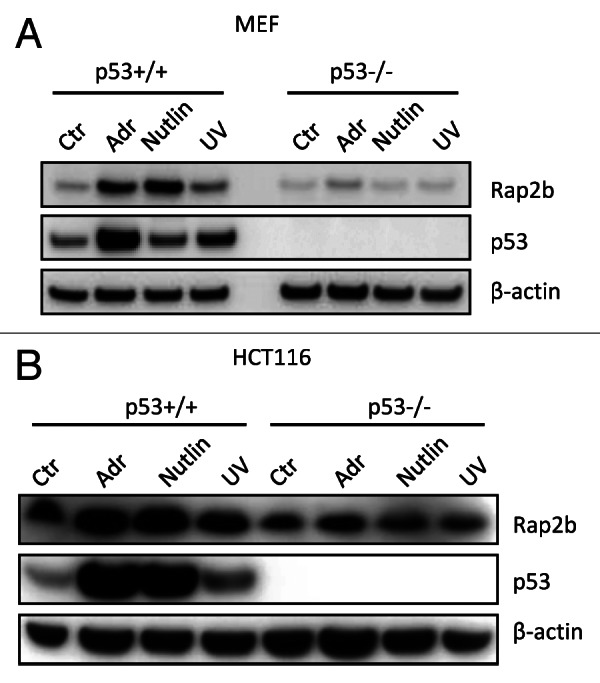
Figure 4. Rap2b was induced by other stresses in a p53-dependent manner. (A) Western blot analyses to evaluate the protein levels of Rap2b, p53, and β-actin in p53+/+ and p53−/− MEF cells untreated or treated with Adr, Nutlin and UV. (B) Western blot analyses to evaluate the protein levels of Rap2b, p53 and β-actin in p53+/+ and p53−/− HCT116 cells untreated or treated with Adr, Nutlin and UV.
p53 binds to the promoter of the Rap2b gene
To study the molecular mechanism of how p53 regulates the expression of Rap2b, we set out to investigate the binding of p53 on the Rap2b gene. Result from ChIP-chip assays in MEF cells showed that there was a binding locus of p53 in the promoter region of the Rap2b gene (Fig. 5A). To confirm this result, we performed conventional chromatin immunoprecipitation assay (ChIP). Guided by the chromosomal coordinates of the p53 peak detected by the ChIP-chip assay, we designed six pairs of real-time PCR primers that spread around the p53 peak (Fig. 5B). ChIP result showed that the recruitment of p53 to the promoter of the Rap2b gene was comparable to that on the Mdm2 gene, a well-known p53 target.34 Before adriamycin treatment, p53 already bound to the promoters of both the Mdm2 and Rap2b genes. Adriamycin treatment enhanced the recruitment of p53 to the promoters (Fig. 5B). Because the mRNA levels of Rap2b in untreated p53+/+ and p53−/− cells were similar (Fig. 2B), it suggests that the binding of p53 in the untreated cells has no transcriptional output. DNA damage not only increases the binding of p53 to chromatin but also switches on p53 to induce Rap2b. The chromatin recruitment of p53 to the promoter of the Rap2b gene was high in both region 4 (R4) and 5 (R5). The length of DNA sequence between R4 and R5 is 116 base pair (bp), a length that is suitable to perform downstream sequence analysis. Importantly, we did not detect the recruitment of p53 to the promoters of Rap2b and Mdm2 in p53−/− MEF cells, demonstrating that the ChIP signal is specific to p53.
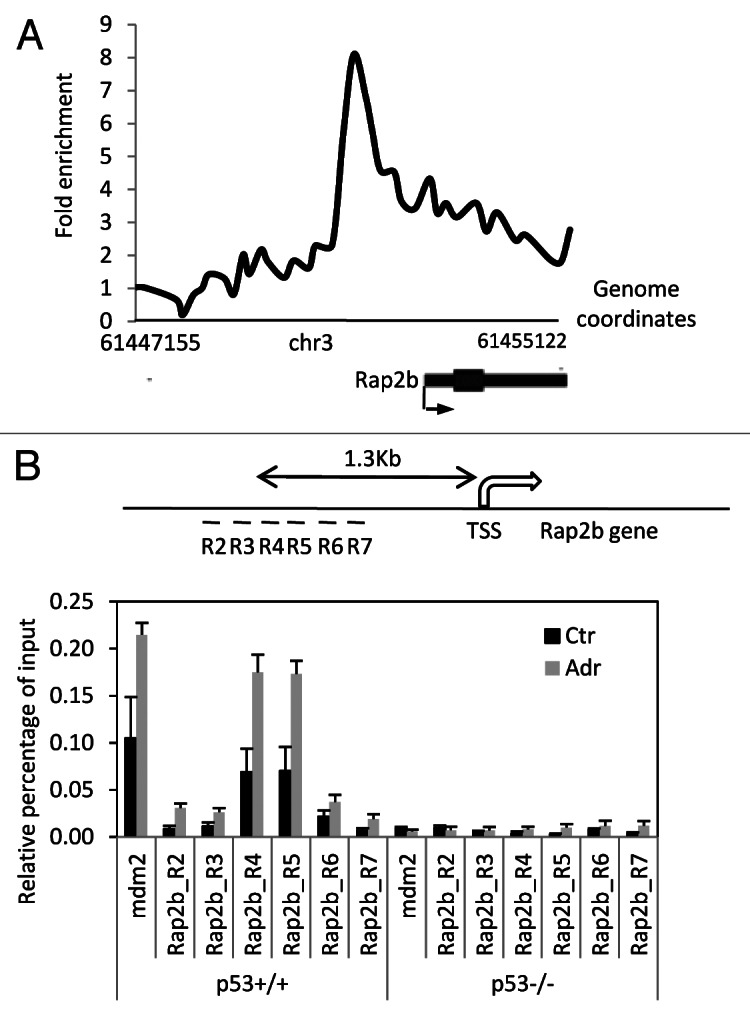
Figure 5. p53 binds to the promoter of the Rap2b gene. (A) A genomic view of p53 chromatin recruitment to the Rap2b gene in MEF cells treated with adriamycin for 8 h. Shown is genomic coordinates of build mm8. (B) Conventional ChIP assay to measure the binding of p53 in the promoter region of the Rap2b gene in MEF cells untreated or treated with adriamycin for 8 h. Results are means ± SEM, n = 3. Regions (R2–R7) were surveyed by real-time PCR.
To further characterize the R4–5 region containing the binding site of p53, we cloned this region into a reporter and performed the luciferase reporter assay (Fig. 6A). Co-transfection of the reporter with an expression vector encoding p53 resulted in about 85-fold induction compared with the reporter alone. The fold induction was even higher than that of a reporter containing the promoter of the Cdkn1a gene, another well-characterized p53-regulated gene.36 Inspection of the cloned sequence (R4–5) revealed that this sequence contained three detectable p53 half sites.43 Half sites 1 and 2 or 2 and 3 form a canonical p53 response element.43 To determine whether these three half sites are critical for the induction of Rap2b, site-directed mutagenesis was performed to disrupt each or the combination of these half sites (Fig. 6A). We observed that the third half site appeared to play a dominant role in the induction of Rap2b, because the disruption of this half site (mut5) decreased the fold induction by 85 percent (p < 0.01). Mutation of either half site 1 or 2 had a much milder effect on the fold induction. The combination of mutation of either half site 1 or 2 with mutation of half site 3 did not result in a further decrease of the induction (p > 0.1). Taken together, the cloned DNA sequence is involved in the Rap2b induction by p53. To test whether the DNA binding domain of p53 was required for the induction of Rap2b, we co-transfected the reporter with an empty vector or a vector expressing wild type p53 or the p53-R175H or p53-R273H mutants (Fig. 6B). Both of these mutants are p53 “hot-spot” mutants and disrupt the DNA binding of p53.44 In our assay, neither of the mutants induced Rap2b, suggesting that the DNA binding domain of p53 is required for the Rap2b induction (Fig. 6B). In addition, we also cloned the promoter region of the human Rap2b gene into a reporter. Our result showed that p53 also activates the human promoter of Rap2b (Fig. 6C). Therefore, these results demonstrate that p53 directly binds to the Rap2b promoter and regulates its expression.

Figure 6. Characterization of the promoter of the Rap2b gene. (A) The promoter sequence containing the p53 binding site in mouse Rap2b gene. The half site of p53 response element was shown below the sequence. TSS, transcription start site; potential half sites were underlined. (B) Luciferase assay to measure the induction of Rap2b by p53. Left: putative p53 response elements in the region 4 to 5 in the promoter of Rap2b. p53 half sites were underlined and shown in bold. Mutated half sites were un-bolded. Right: luciferase assay with a reporter containing either wild type (wt) or mutated promoters of Rap2b. Fold induction (with p53 vs. without p53) were measured. Reporter containing the promoter of Cdkn1a was used as a comparison. (C) A reporter vector containing the mouse Rap2b promoter was co-transfected with an empty vector or a vector expressing wild type p53 (p53WT), p53R175H or p53R273H mutant. Fold induction was measured. (D) Reporter containing wild type human Rap2b promoter was co-transfected with an empty vector or a vector expressing wild type p53. Fold induction was measured. Shown are mean ± SEMs; n > or = 3; **p-value < 0.01.
Knockdown of Rap2b sensitizes cells to DNA damage-induced apoptosis
To explore the role of Rap2b in p53-mediated DNA damage response, we designed two shRNAs that effectively reduced the levels of Rap2b in both p53+/+ and p53−/− HCT116 cells (Fig. 7A). Cells were either untreated or treated with adriamycin followed by flow cytometry analysis with propidium iodide (PI) staining (Fig. 7B; Fig. S4). In the absence of adriamycin treatment (untreated), Rap2b knockdown did not affect the cell cycle distribution of either p53+/+ or p53−/− HCT116 cells (Fig. 7B, upper panels). However, adriamycin led to a higher percentage of the apoptotic (sub-G1) population in Rap2b knockdown cells than in control cells after 48-h adriamycin treatment (Fig. 7B, lower panels). The gain of apoptotic population in Rap2b knockdown cells resulted from the loss of the G2 population, suggesting that Rap2b knockdown sensitizes G2-arrested cells to adriamycin-induced apoptosis. The pro-survival effect of Rap2b was p53-dependent, as the knockdown of Rap2b did not change the cell cycle distribution and apoptotic population in p53−/− HCT116 cells, either in the absence or presence of adriamycin (Fig. 7B; Fig. S4). These data suggest that Rap2b knockdown sensitizes cells to DNA damage-induced apoptosis only when p53 is wild type.

Figure 7. Knockdown of Rap2b sensitize cells to DNA damage-induced apoptosis. (A) Lentivirus-based shRNA to knockdown Rap2b. Relative mRNA levels were measured by real-time PCR. (B) Flow cytometry analysis of the effect of Rap2b knockdown in HCT116, p53+/+ and p53−/− cells in response to adriamycin (Adr) treatment. PI staining was performed and sub-G1 indicated the apoptotic cell population. Results shown are mean ± SEM; n > or = 3; **p < 0.01. (C) Western blot analysis of the protein levels of Rap2b, Cdkn1a, p53, PARP p85 fragment (cleaved PARP), Bax, Puma and β-actin in Rap2b knockdown HCT116, p53+/+ and p53−/− cells with Adr treatment.
Based on the result from RNA interference, we hypothesized that overexpression of Rap2b in cells would protect cells from DNA damage-induced apoptosis. To test this hypothesis, we used a lentivirus-based system to exogenously express Rap2b in both p53+/+ and p53−/− HCT116 cells. Surprisingly, exogenous expression of Rap2b affected neither the cell cycle distribution nor apoptosis in either the p53+/+ or the p53−/− HCT116 cells, indicating that Rap2b by itself is required but not sufficient to protect cells from DNA damage-induced apoptosis (Fig. S5; also see “Discussion”).
Several p53 targets, such as Hzf and Slug, affect the expression of other p53 targets to tip the balance between cell cycle arrest and apoptosis.11,13 To determine whether Rap2b utilizes a similar mechanism, we interrogated the induction of several canonical p53 downstream targets in control cells and Rap2b knockdown cells, either untreated or treated with adriamycin (Fig. 7C). We did not observe an obvious effect of Rap2b knockdown on the induction of these p53 targets, although the levels of an apoptosis marker PARP-p85, the cleaved PARP,45 increased in the knockdown cells. In addition, Rap2b knockdown did not alter the protein levels of p53. Therefore, the effect of Rap2b knockdown on apoptosis either channels through other p53 downstream targets or is through an unknown mechanism.
Rap2b is upregulated in many human tumors and has weak transformation activity
To probe the function of Rap2b in tumors, we interrogated the expression of Rap2b in the Oncomine database. Rap2b was predominantly upregulated in many types of tumors (Fig. S6). Among the 50 studies comparing cancer tissues with normal tissues, 40 studies showed that Rap2b was upregulated in cancer vs. normal tissues (p < 0.001).
Because Rap2b is a member of the Ras super-family and preferentially upregulated in tumors, we tested whether Rap2b has transformation activity. To address this, anchorage-independent growth assays were used. MCF10A cells, a non-tumorigenic breast epithelial cell line, were transduced with lentivirus expressing luciferase (a negative control), H-Ras (V12) (a positive control) and Rap2b (Fig. 8A). After transduction, cells were grown on agarose plates, and the number of colonies was counted at various time points (Fig. 8A). H-Ras (V12), as expected, transformed about 80% of the transduced MCF10A cells. However, Rap2b only increased the number of transformed MCF10A colonies by 2-fold (from about 5% to 10%) compared with the luciferase control, although the increase was statistically significant (Fig. 8B). We also tested the transformation activity of Rap2b in a mouse cell line, NIH3T3 cells and observed similar result (Fig. S7). Together, our results indicate that Rap2b has weak transformation activity, suggesting that Rap2b may not be an oncogene by itself. It probably needs to cooperate with other factors to give tumors sufficient growth or survival advantage (see “Discussion”). To assess whether Rap2b is involved in the proliferation of cancer cells, we knocked down the levels of Rap2b in HCT116 cells and measured the growth of these cells. The reduction of Rap2b levels in HCT116 cells decreases the cell number of HCT116 cells (Fig. 8C). Because we did not observe evident apoptosis (Fig. 7B and C) under unstressed conditions, this result suggests that Rap2b also regulates cell proliferation without exogenous DNA damage stress.

Figure 8. Rap2b has weak a transformation activity and is upregulated in many types of tumors. (A) Western blot analysis of the ectopic expression of Rap2b and H-Ras(V12) in MCF10A cells. (B) Anchorage-independent growth assay to evaluate the transformation activity of Rap2b in MCF10A cells. Numbers of colonies were calculated. Results are displayed as mean ± SEM; n = 6; *p < 0.05; **p < 0.01. (C) Growth curve of HCT116 cells without or without Rap2b knockdown; n = 3; *p < 0.05. (D) A model describing the role of Rap2b in the pro-survival regulation during p53 signaling.
Discussion
Pro-survival targets of p53
In this study, we identify Rap2b as a novel p53 target that mediates survival after DNA damage (Fig. 8D). Previous studies by others have identified several p53 target genes, such as Glx2, Hzf, Cox-2 and IRF2BP2, that counter the apoptosis induced by p53.11,15,46,47 The cellular consequences of the reduction of Rap2b and these other genes are similar in terms of the increase of apoptotic population (Fig. 7). Compared with these previously identified pro-survival genes, Rap2b does not affect the transcriptional output of p53, as judged by several p53 targets (Fig. 7C). In the context of pro-survival function, Rap2b is very similar to Cdkn1a. Reduced levels of Cdkn1a also sensitize cancer cells to DNA damage-induced apoptosis.48 Different from Cdkn1a, Rap2b does not have a role in cell cycle arrest in the absence from DNA damage stress, and it only affects the survival of cells during DNA damage (Fig. S5). This DNA damage specificity may help to keep the pro-survival function of Rap2b in check during normal physiological conditions. In many types of tumors, Rap2b is upregulated, suggesting that other transcription factors could also regulate the expression of Rap2b (Fig. 8A). Due to these special features, Rap2b may represent an excellent target to maximize the pro-apoptotic effect of DNA damage agent, such as adriamycin, in cancer treatment. The exact mechanism underlying the pro-survival function of Rap2b is currently unknown, which merits further study in the future.
The weak transformation activity of Rap2b
Our anchorage-independent growth assays showed that Rap2b had very weak transformation activity. Given this observation and the upregulation of Rap2b in many tumors (Fig. 8C), Rap2b may not be a bona fide oncogene. Recent genome-wide RNA interference screening revealed that many cancer-associated genes are not oncogenes themselves.49 Instead, they help to maintain an oncogenic state, dubbed as nononcogenic addiction.49 Therefore, it is possible that Rap2b is one of these nononcogenes that support the oncogenic status. During tumorigenesis, tumor cells are constantly exposed to intrinsic DNA damage stress, which may be caused by replication stress. The nononcogenic addiction could also potentially explain why Rap2b does not have a full transformation activity but is upregulated in many types of cancer. Therefore, targeting these nononcogenic genes, such as Rap2b, may also achieve therapeutic benefit. Because exogenously expressed Rap2b alone did not protect cells from apoptosis (Fig. S5), high levels of Rap2b in tumors need other factors to protect tumor cells from the apoptosis induced by the intrinsic DNA damage stress. In the future, it will be interesting to study which factor(s) cooperate with Rap2b to protect tumor cells from DNA damage-induced apoptosis. This can be achieved by using an un-biased library screening to identify genes that cooperate with Rap2b to fully transform cells in the future.
The transcriptional gene network of p53
A full picture of the transcriptional gene network of p53 is critical for understanding the molecular mechanisms of the tumor-suppressive function of p53 and the design of an effective therapeutic strategy based on p53 signaling. The list of p53 target genes is still expanding.23,24 Thus far, there is no single p53 target that fully phenocopies p53, suggesting that p53 targets either cooperate to prevent tumors, or that we have not found the right downstream target. Therefore, identification of new p53 targets and investigation of their functions during p53 signaling is of paramount importance. In the current study, we set out to use integrated genome-wide approaches to find highly conserved p53 direct targets among the four cell types. It is intriguing that all the identified common p53 targets in both MEF and mES cells are p53-activated genes (Fig. 1). This result supports a model that p53-repressed genes are more likely to mediate the cell type-specific function of p53 than do the activated genes.23,25 The mechanisms underlying the transcriptional repression by p53 are more complex than those for activation.24,50 p53 can directly repress gene transcription by recruiting co-repressor, such as Sin3a and LSD1, or interfere with the enhancer activity.25,51-53 Or it can indirectly repress its targets by inducing other transcriptional factors, for example, E2F7 or noncoding RNAs, such as lincRNAs or miRNAs, which, in turn, repress its targets.29,30,54 Therefore, p53 may use many different mechanisms to conduct transcriptional repression in different cell types to fulfill its cell type-specific functions. Compared with p53-repressed genes, the regulatory mechanisms of p53-activated genes are relatively homogenous, i.e., they generally involve the promoter. By comparing p53 targets in different cell types, p53-activated genes are more likely to emerge as the common targets, such as Rap2b.
It should be noted that our analyses likely have missed some important p53 targets because of the sensitivity of our genome-wide tools or the cell type-specific nature of these targets.55 For example, we did not detect the induction of Bax, a well-established p53 target in other cell types, by p53 in mES cells, because the mRNA of Bax did not respond to adriamycin treatment in mES cells. Nonetheless, the identification of Rap2b as an important player in p53 signaling demonstrates that these genome-wide approaches are extremely helpful to dissect the complexity of p53 signaling.
Materials and Methods
Cell lines and growth condition
mES (R1E) cells (ATCC) were maintained in 0.1% gelatin-coated plates with mESC growth medium containing knockout DMEM (KO-DMEM), 15% fetal bovine serum (FBS), 100 mM MEM nonessential amino acids, 0.1 mM 2-mercaptoethanol, 2 mM L-glutamine and 1× penicillin-streptomycin (Invitrogen) plus 1,000 units/mL of LIF (Millipore). Primary mouse embryonic fibroblasts (MEFs) were isolated from 13.5-d embryos and cultured in DMEM containing 15% FBS. Only cells with passage number less than three were used. Mice are maintained under the strict guidelines of the Institutional Animal Care and Use Committee (IACUC)-approved protocols of the National Cancer Institute. HCT116 cells were maintained McCoy’s 5A medium with 10% FBS (Cellgro, 10-050-CV). MCF10A cells (ATCC, CRL-10317) were maintained in DMEM/F-12 (Sigma, D8900-10X1L) containing 5% horse serum (Invitrogen, 16050-114), 20 ng/ml EGF (PeproTech, AF-100-15), 0.5 mg/ml hydrocortisone (Sigma, H0888–1G), 100 ng/ml cholera toxin (Sigma, C8052-.5MG) and 10 μg/ml insulin (Sigma, I4011-250MG). Low-passage IMR90 and H1299 cells were grown in DMEM with 10% FBS.
Gene expression microarray and ChIP-chip
Gene expression microarray assays were performed as previously described at the microarray facility of the National Cancer Institute.28 Briefly, 100 ng total RNA were processed according to the protocol from Affymetrix. Mouse Gene ST 1.0 arrays were used. For each condition, triplicate analyses were performed. Data analysis was performed using the Partek Genomics Suite. Probes with intensity less than three were removed from the analysis because of potential probe failures. To identify genes whose induction was p53-dependent after DNA damage, analysis of variance (ANOVA) was used. Genes with a p-value less than 0.001 (adriamycin vs. untreated in p53+/+ cells) were kept for further analysis. We then compared p53+/+ with p53−/− cells in the absence or presence of adriamycin treatment. Genes with p values larger than 0.001 under both conditions were determined as p53-independent genes.
ChIP-chip followed the same procedure as previously described.28 All the procedures were described in Agilent’s mammalian ChIP-on-chip manual (Version 10.0). G4490 (1 × 244 k) arrays were used. Gene expression microarray and ChIP-chip data sets in mES cells were re-analyzed from the previous data sets in Gene Expression Omnibus (GSE16427 and GSE16428). Ten μg of anti-mouse p53 antibody (Santa Cruz, M19) was used for immunoprecipitating mouse p53.
Induction of Rap2b in cells
MEF, mES, HCT116 and IMR90 cells were treated with 0.5 (mES and MEF) or 1 uM (HCT116) adriamycin (Sigma), 25 J/m2 UV or 20 uM Nutlin (Cayman Chemical) for various times as indicated. H1299 cells that inducibly express p53 were treated with 1 μg/ml doxcycline for various times as indicated. Cells were then harvested for protein and mRNA analyses using immunoblotting and reverse transcription and real-time PCR (qRT-PCR), respectively.
Immunoblotting
The cells were lysed in RIPA buffer (50 mM TRIS-HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mM EDTA) plus protease inhibitors, and then sonicated with a Bioruptor (Diagenode). The lysate concentration was determined by the Bradford assay (Bio-Rad). Eighty micrograms of total protein was loaded onto a NuPAGE 4–12% Bis-Tris gel (Invitrogen) and transferred onto a PVDF membrane (Bio-Rad). The membrane was blocked with PBST (1 × PBS with 0.05% Tween 20) plus 5% non-fat milk at room temperature for 1 h, incubated with primary at 4° overnight and secondary antibodies at room temperature for 1 h and then developed using the SuperSignal West Dura Luminol/Enhancer solution (ThermoFisher Scientific). Images were obtained from a G:BOX (Syngene) imaging system. For multiple detections, the membrane was stripped with Restore Plus Western Blot Stripping Buffer (ThermoFisher Scientific). The primary antibodies used in this study were against β-actin (Sigma), mouse p53 (Santa Cruz), human p53 (EMD4Biosciences), Cdkn1a (Santa Cruz), Puma (Calbiochem) and Bax (EMD4Biosciences). The secondary antibodies included anti-rabbit IgG (Jackson ImmunoResearch), anti-mouse IgG (Jackson ImmunoResearch) and anti-goat IgG (Santa Cruz).
Reverse transcription and real-time PCR
Total mRNA was isolated using TRIzol® Reagent (Invitrogen). Reverse transcription was performed with a Multiscribe Reverse Transcriptase kit (Applied Biosystems). Real-time PCR was performed in duplicate with a total volume of 10 μL using a SYBR Green JumpStart Taq ReadyMix (Sigma). Reactions were run in a PCR System 7500HT or 7900HT (Applied Biosystems). Data were analyzed using SDS 1.4 or SDS 2.3 software, respectively (Applied Biosystems). Data normalization was done as described previously.25,28 Real-time PCR primers used in this study are listed in Table S5.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed as described previously.25,28 Anti-mouse p53 antibody (Santa Cruz, M19) was used for ChIPing mouse p53. Real-time PCR was performed using SybrGreen (Sigma). The relative percentage of input was calculated as the IP signal over the input signal.
Luciferase reporter assay
A DNA fragment attR1-CmR-ccdB-attR2 was cloned from pLenti6/V5-DEST (Invitrogen) into pGL4.23[luc2/minP] (Promega) to construct a gateway luciferase vector pGL4.23[luc2/minP]-DEST. A DNA fragment containing the bla promoter and Amp(R) gene was cloned from pLenti6/V5-DEST into the pENTR/D-TOPO vector to construct a pENTR/D-TOPO-bla-Amp vector and XcmI was used to generate a T vector. The Rap2b promoter was cloned into the T vector using forward primer 5′-CCGCTGTGACTTGGAACCA-3′ and reverse primer 5′-GCTTGCCTCTTCCCAAAGCT-3′. A LR recombination reaction was performed to generate a reporter vector pGL4.23-Rap2b(pr) containing the Rap2b promoter. The half-site mutants of p53 binding sites were constructed using a QuikChange® Site-Directed Mutagenesis Kit (Stratagene, #200523). Luciferase reporter assay was performed using Dual-Luciferase® Reporter Assay System (Promega, E1910). In brief, 100 ng pGL4.23-Rap2b(pr) was co-transfected into H1299 cells with or without 10 ng pcDNA3.1-p53 using Lipofectamine™ 2000 (Invitrogen, 11668-019), and the Cdkn1a promoter was used as a positive control. Ten ng pRL-SV40 (Promega, E2231), as an internal control, was co-transfected. Fold induction was calculated to evaluate the relative activities of the Rap2b promoter mutants and expressed as the signal with p53 over the signal without p53. The experiments were repeated at least three times to calculate the average of fold induction. All the primers used in this study are listed in Table S1.
Knockdown of Rap2b
Short hairpin RNA (shRNA) plasmids were constructed by cloning shRNA oligos (Table S1) into the pLKO.1 vector. Lentiviral particles were produced by cotransfecting Lenti-X™ 293T cells (Clontech) with shRNA plasmid, packaging plasmid pCMV-dR8.2 dvpr (Addgene #8455) and envelop plasmid pCMV-VSVG (Addgene #8454) using Lipofectamine™ 2000. The lentiviral supernatant was used to transduce cells. The transduced cells were treated with 2 µg/ml puromycin to make stable Rap2b knockdown cell lines. Low-passage (less than 10) stably transfected cells were used. Knockdown efficiency was assessed by western blotting and qRT-PCR.
Cell cycle analysis
Two × 105 HCT116 cells were seeded into each well of a 6-well plate or into a 60-mm dish. The next day, cells were untreated or treated with 1 μM adriamycin. After various times as indicated, cells were harvested and subject to propidium iodide (PI) staining.56 Briefly, cells were fixed and permeablized with 70% ethanol overnight at −20°. Cells were then washed with PBS and treated with 10 μg/ml of RNase at room temperature for 1 h. After RNase treatment, cells were incubated with 0.1 mg/ml of PI at room temperature for at least 10 min. The stained cells were loaded into a BD FACSCalibur flow cytometer, and the data were collected for cell cycle analysis by FlowJo v7.5.5. The experiments were repeated at least 3× to calculate the average percentage of each cell cycle phase.
Ectopic expression of Rap2b
The Rap2b gene was cloned into the pENTR/D-TOPO vector (Invitrogen), and the resulting TOPO vector was then transferred into the pLenti6/V5-DEST vector by LR recombination using the Gateway LR Clonase II enzyme mix. The resulting expression vector pLenti6/V5-Rap2b was used to make lentivirus to transduce NIH3T3, HCT116 or MCF10A cells. Five μg/ml blasticidin was used to select stably transfected cells. The Rap2b overexpressed cells were harvested for western blotting and anchorage-independent growth assays.
Anchorage-independent growth assay
0.5% agar in 1× growth medium was poured into a 6-well plate at 1 ml/well. Rap2b overexpressed MCF10A cells were counted and mixed with 0.35% agarose (final) in 1× growth medium, and 1.5 ml mixture was added into three wells with 2,500 cells/well. The plates were incubated at 37°C in a humidified incubator for 28 d, and the cells were fed every other day. The colonies were counted under a microscope each week to calculate the efficiency of colony formation. H-RasV12 and luciferase overexpressed MCF10A cells were used as positive and negative controls, respectively.
Supplementary Material
Acknowledgments
We thank Stuart Yuspa and Peter Blumberg for critically reading the manuscript and their useful comments. We also thank Chi-Ping Day for technical advice on anchorage-dependent assay.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
J.H.’s laboratory was funded by the intramural research program of the Center for Cancer Research (CCR), National Cancer Institute and National Institutes of Health, USA. This research was also supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases and National Institute of Health.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24364
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24364
References
- 1.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rozan LM, El-Deiry WS. p53 downstream target genes and tumor suppression: a classical view in evolution. Cell Death Differ. 2007;14:3–9. doi: 10.1038/sj.cdd.4402058. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, Huang J. Integrative genome-wide approaches in embryonic stem cell research. Integr Biol (Camb) 2010;2:510–6. doi: 10.1039/c0ib00068j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlereth K, Charles JP, Bretz AC, Stiewe T. Life or death: p53-induced apoptosis requires DNA binding cooperativity. Cell Cycle. 2010;9:4068–76. doi: 10.4161/cc.9.20.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helton ES, Chen X. p53 modulation of the DNA damage response. J Cell Biochem. 2007;100:883–96. doi: 10.1002/jcb.21091. [DOI] [PubMed] [Google Scholar]
- 7.Aylon Y, Oren M. Living with p53, dying of p53. Cell. 2007;130:597–600. doi: 10.1016/j.cell.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Jänicke RU, Sohn D, Schulze-Osthoff K. The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ. 2008;15:959–76. doi: 10.1038/cdd.2008.33. [DOI] [PubMed] [Google Scholar]
- 9.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–39. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 10.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–51. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das S, Raj L, Zhao B, Kimura Y, Bernstein A, Aaronson SA, et al. Hzf Determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell. 2007;130:624–37. doi: 10.1016/j.cell.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka T, Ohkubo S, Tatsuno I, Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638–50. doi: 10.1016/j.cell.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 2005;123:641–53. doi: 10.1016/j.cell.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 14.Caratozzolo MF, Micale L, Turturo MG, Cornacchia S, Fusco C, Marzano F, et al. TRIM8 modulates p53 activity to dictate cell cycle arrest. Cell Cycle. 2012;11:511–23. doi: 10.4161/cc.11.3.19008. [DOI] [PubMed] [Google Scholar]
- 15.Xu Y, Chen X. Glyoxalase II, a detoxifying enzyme of glycolysis byproduct methylglyoxal and a target of p63 and p73, is a pro-survival factor of the p53 family. J Biol Chem. 2006;281:26702–13. doi: 10.1074/jbc.M604758200. [DOI] [PubMed] [Google Scholar]
- 16.Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010;9:4323–7. doi: 10.4161/cc.9.21.13584. [DOI] [PubMed] [Google Scholar]
- 17.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–3. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 18.Reifenberger G, Liu L, Ichimura K, Schmidt EE, Collins VP. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993;53:2736–9. [PubMed] [Google Scholar]
- 19.Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012;3:228–35. doi: 10.18632/oncotarget.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature. 2009;457:51–6. doi: 10.1038/nature07618. [DOI] [PubMed] [Google Scholar]
- 21.Gasparini G, Longo R, Sarmiento R, Morabito A. Inhibitors of cyclo-oxygenase 2: a new class of anticancer agents? Lancet Oncol. 2003;4:605–15. doi: 10.1016/S1470-2045(03)01220-8. [DOI] [PubMed] [Google Scholar]
- 22.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 23.Li M, He Y, Feng X, Huang J. Genome-wide studies of the transcriptional regulation by p53. Biochim Biophys Acta. 2012;1819:684–7. doi: 10.1016/j.bbagrm.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 25.Li M, He Y, Dubois W, Wu X, Shi J, Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell. 2012;46:30–42. doi: 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 27.Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, et al. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36:3639–54. doi: 10.1093/nar/gkn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KH, Li M, Michalowski AM, Zhang X, Liao H, Chen L, et al. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc Natl Acad Sci USA. 2010;107:69–74. doi: 10.1073/pnas.0909734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi YJ, Lin CP, Ho JJ, He X, Okada N, Bu P, et al. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol. 2011;13:1353–60. doi: 10.1038/ncb2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006;10:105–16. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brosh R, Sarig R, Natan EB, Molchadsky A, Madar S, Bornstein C, et al. p53-dependent transcriptional regulation of EDA2R and its involvement in chemotherapy-induced hair loss. FEBS Lett. 2010;584:2473–7. doi: 10.1016/j.febslet.2010.04.058. [DOI] [PubMed] [Google Scholar]
- 33.Rouault JP, Falette N, Guéhenneux F, Guillot C, Rimokh R, Wang Q, et al. Identification of BTG2, an antiproliferative p53-dependent component of the DNA damage cellular response pathway. Nat Genet. 1996;14:482–6. doi: 10.1038/ng1296-482. [DOI] [PubMed] [Google Scholar]
- 34.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–8. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang G, Zhang G, Pittelkow MR, Ramoni M, Tsao H. Expression profiling of UVB response in melanocytes identifies a set of p53-target genes. J Invest Dermatol. 2006;126:2490–506. doi: 10.1038/sj.jid.5700470. [DOI] [PubMed] [Google Scholar]
- 36.el-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–74. [PubMed] [Google Scholar]
- 37.Wu GS, Burns TF, McDonald ER, 3rd, Jiang W, Meng R, Krantz ID, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–3. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 38.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–94. doi: 10.1016/S1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 39.Catalano A, Rodilossi S, Caprari P, Coppola V, Procopio A. 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. EMBO J. 2005;24:170–9. doi: 10.1038/sj.emboj.7600502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong Y, Stambrook PJ. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc Natl Acad Sci USA. 2004;101:14443–8. doi: 10.1073/pnas.0401346101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu G, Chen X. Regulation of the p53 transcriptional activity. J Cell Biochem. 2006;97:448–58. doi: 10.1002/jcb.20700. [DOI] [PubMed] [Google Scholar]
- 42.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 43.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–9. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 44.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–9. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 46.Han JA, Kim JI, Ongusaha PP, Hwang DH, Ballou LR, Mahale A, et al. P53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. EMBO J. 2002;21:5635–44. doi: 10.1093/emboj/cdf591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koeppel M, van Heeringen SJ, Smeenk L, Navis AC, Janssen-Megens EM, Lohrum M. The novel p53 target gene IRF2BP2 participates in cell survival during the p53 stress response. Nucleic Acids Res. 2009;37:322–35. doi: 10.1093/nar/gkn940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seoane J, Le HV, Massagué J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–34. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- 49.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 2006;13:951–61. doi: 10.1038/sj.cdd.4401916. [DOI] [PubMed] [Google Scholar]
- 51.Wilkinson DS, Tsai WW, Schumacher MA, Barton MC. Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-beta-mediated transcription repression. Mol Cell Biol. 2008;28:1988–98. doi: 10.1128/MCB.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy M, Ahn J, Walker KK, Hoffman WH, Evans RM, Levine AJ, et al. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999;13:2490–501. doi: 10.1101/gad.13.19.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai WW, Nguyen TT, Shi Y, Barton MC. p53-targeted LSD1 functions in repression of chromatin structure and transcription in vivo. Mol Cell Biol. 2008;28:5139–46. doi: 10.1128/MCB.00287-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carvajal LA, Hamard PJ, Tonnessen C, Manfredi JJ. E2F7, a novel target, is up-regulated by p53 and mediates DNA damage-dependent transcriptional repression. Genes Dev. 2012;26:1533–45. doi: 10.1101/gad.184911.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 56.Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1:1458–61. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.