

Abstract
Rationale
The neuroprotective agent riluzole has antidepressant-like properties in humans, but its mechanisms of action are unclear. Despite the increasing utility of transgenic and knockout mice in addressing such issues, previous studies aimed at characterizing biochemical mechanisms have been conducted in rats.
Objectives
We sought to optimize an oral riluzole administration protocol with antidepressant-like consequences in C57BL/6 mice, a common background strain in genetically-modified mice.
Methods
Riluzole (6–60 μg/ml) was dissolved in tap water and replaced regular drinking water for up to 3 weeks; sensitivity to tail suspension, forced swimming, and the locomotor response to extinction training in a model of “incentive disengagement” were tested. Peripheral and central effects of long-term 60 μg/ml treatment were also evaluated.
Results
Riluzole had dose-dependent antidepressant-like effects in the forced swim test and like chronic fluoxetine, exerted antidepressant-like actions in an adaptation of the “incentive disengagement” model at the highest concentration tested. This 60 μg/ml concentration also restored hippocampal Brain-derived Neuroptrophic Factor (BDNF) expression after chronic corticosteroid exposure and increased glutamate glial transporter 1 (GLT-1, or EAAT2) expression without significantly affecting baseline locomotor activity, thymus and adrenal gland weights, or blood serum corticosterone. The lowest 6 μg/ml concentration increased locomotor activity, consistent with an anxiolytic-like effect.
Conclusions
Riluzole’s therapeutic potential for treating mood disorders may involve GLT-1 and BDNF, and we suggest this protocol could be used to further characterize its precise long-term biochemical mechanisms of action in animal models of depression.
Keywords: riluzole, depression, glutamate, antidepressant, incentive, BDNF, extinction
Introduction
Riluzole is a neuroprotective agent approved by the FDA for use in slowing the progression of amyotrophic lateral sclerosis. Riluzole inhibits presynaptic glutamate release and increases glial cell glutamate uptake (Azbill et al. 2000; Sung et al. 2003; Frizzo et al. 2004; Fumagalli et al. 2008), and it also has antidepressant-like properties in humans (Coric et al. 2003; Singh et al. 2004; Zarate et al. 2004, 2005; Sanacora et al. 2004, 2007; Pittenger et al. 2008; but see Mathew et al. 2010). Recent work in rats indicates riluzole increases glutamate clearance from the synaptic cleft concomitant with the amelioration of stress-induced depressive-like behavior (Banasr et al. 2010). This work importantly identified an antidepressant-like dosing protocol for riluzole in rats, but comparable doses for mice have not been established. This is despite the increasing utility of transgenic and knockout mice in identifying genetic and biochemical mechanisms of antidepressant sensitivity and mood regulation. Given the increasing recognition of glutamatergic and glial involvement in the psychopathology of depression (Rajkowska and Miguel-Hidalgo 2007), a better understanding of how riluzole ameliorates depressed mood may shed light onto the neurobiological mechanisms of depression itself.
The present studies were aimed at establishing a robust, easily-replicable riluzole administration protocol with antidepressant-like consequences in C57BL/6 mice, as this strain is commonly used in knockout and transgenic mouse models. A primary goal was to minimize the stress associated with chronic drug administration, so the protocol relies on dissolving riluzole in the drinking water, rather than administering daily injections; similar approaches have been taken with tricyclic (Caldarone et al. 2003) and SSRI (Gourley et al. 2008a; Gourley and Taylor 2009) antidepressants.
In rats, riluzole appears to have antidepressant-like qualities only when administered chronically, even in tests sensitive to acute treatment with traditional antidepressants such as the Porsolt forced swim test (Banasr et al. 2010). Although this finding may be confounded by sedation associated with acute administration, we nonetheless exclusively tested the effects of chronic riluzole treatment on immobility during forced swimming and tail suspension. We also adapted the “incentive disengagement” model of acute depressive-like behavior in rats (Klinger et al. 1974) for use in evaluating antidepressant-like compounds in mice. This model is based on the observation that when rats are trained to obtain food reward and are then deprived of the expected appetitive reinforcement, subsequent locomotor activity acutely declines. The model anticipates that an antidepressant-like compound will, by contrast, normalize or increase locomotor activity, reflecting its efficacy in preventing an acute putatively depressive-like state resulting from “disengagement” from the incentive value of the outcome (Klinger et al. 1974; reviewed Willner 1984).
Expression of Brain-derived Neurotrophic Factor (BDNF) and more recently, glutamate glial transporter 1 (GLT-1; also referred to as EAAT2), are implicated in antidepressant action (Banasr and Duman 2007), while prolonged corticosteroid exposure is widely acknowledged to increase the likelihood of depressive-like behavior in both rodents and humans. For this reason, we also investigated the effects of riluzole on BDNF and GLT-1 in the hippocampus of normal and corticosteroid-exposed mice, as restoration or promotion of factors thought to protect against stress-related neurodegenerative events may aid in alleviating depressed mood.
Materials and methods
Riluzole administration
Experimentally naïve group-housed male C57BL/6 mice 9–12 weeks of age at the start of the experiments (Charles River Laboratories, Kingston, NY) were used throughout. Mice received riluzole (Sigma Aldrich, St. Louis, MO) + 2% w/v saccharin in the drinking water in 6 (n=6), 30 (n=8), or 60 (n=8) μg/ml concentrations, translating to ~1.2, 5.5, or 13.2 mg/kg/day p.o., respectively. Riluzole was dissolved by stirring the compound in room temperature tap water overnight; solutions were changed every 48 hours, and control animals consumed saccharin alone. All animals received food ad libitum unless otherwise noted in a humidity- and temperature-controlled colony (lights on at 0700 hours), and procedures were Yale University IACUC-approved.
Locomotor monitoring
Locomotor activity was periodically monitored (at 1, 7, and 22 days of treatment) in animals used for behavioral studies. Mice were individually monitored in clean cages normally used to house rats (41×20×20 cm) with clean corncob bedding for 30 min using the Omnitech Digiscan Micromonitor system (Columbus, OH) equipped with 16 photocells. Photobeam breaks were analyzed by 2-factor (dose x time point) analysis of variance (ANOVA) with repeated measures and Tukey’s post-hoc tests.
Tail suspension test
In an initial group of mice treated with the highest riluzole dose (60 μg/ml, n=8/group), immobility in the tail suspension test was quantified at 10 days of treatment. Animals were individually suspended by taping the tail 1/2 cm from the tip to flexible plastic tubing secured to the edge of a table in a quiet room. Immobility time during the 6 min test was scored by a single blinded observer. When mice climbed the plastic tubing, they were gently returned to the hanging position; no animal climbed more than 3 times. Immobility scores were compared by unpaired t-test.
Forced swim test
At 2 weeks of treatment, all mice were tested in the Porsolt forced swim task (Porsolt et al. 1977). Here, mice were placed in a glass cylinder (24 cm × 15.5 cm diameter) filled to 15 cm with 23°C water for 8 min. Immobility, defined as the absence of movements other than those required to keep the head above water, was scored by a single rater blinded to the treatment. One animal’s scores were two standard deviations outside of the mean and excluded. Otherwise, immobility scores were compared by 1-factor (dose) ANOVA with Tukey’s post-hoc tests.
Incentive disengagement test
The “incentive disengagement” test of acute depressive-like behavior is based on the observation that locomotor activity in rats declines when animals trained to obtain reward are deprived of the expected appetitive reinforcement (Klinger et al. 1974). This occurs even when rats are placed in a different—rather than the training—environment for locomotor monitoring, and the reduction in locomotor activity has been interpreted as reflecting a sense of loss, i.e., incentive disengagement from the appetitive outcome. To adapt this test to mice, 8 adult male drug-naïve mice were food-restricted to ~93% of their free-feeding body weight. Animals were then trained to nose poke for 20 mg grain-based food reinforcers (Bio-Serv, Frenchtown, NJ) using standard operant conditioning chambers (Med-Associates, Georgia, VT). Daily sessions ran 2 hours in duration for 4 consecutive days, at which point mice responded at a stable rate. During training, every 1–3 responses, as randomly determined by the operating computer was reinforced (i.e., variable ratio 2 schedule of reinforcement). When animals completed the response requirement, a 2-second 2.9 kHz tone signaled the availability of reinforcement, and the house light was exterminated for 2 seconds. Immediately after the last 2 instrumental training sessions, locomotor activity was monitored in 15-min time bins for 1 hour in clean cages otherwise used to house rats (41×20×20 cm) with clean corncob bedding. Mice had been previously habituated to the locomotor monitoring room and a clean cage of the same dimensions for 1 hour.
The following day, mice were subjected to a 1-hour extinction session during which no food reinforcement was delivered in the operant conditioning chambers. Locomotor activity was again monitored immediately after this session. To confirm changes in locomotor activity patterns could not be attributed to testing order effects, instrumental responding was reinstated the following day in a 2-hour reinforced instrumental test session identical to that used for instrumental training. Locomotor activity was again monitored for 1 hour in a clean cage after the session ended. Photobeam breaks were analyzed by 2-factor (time bin x instrumental event) ANOVA with repeated measures and Tukey’s post-hoc tests, and with linear regression analyses against instrumental responses made prior to locomotor monitoring.
In riluzole-treated mice, we allowed mice 3 days to recover from forced swim testing, then trained mice to nose poke for food as described. Animals that did not acquire the task within the experimental time frame (1 2-hour session/day for 4 days) were excluded. Photobeam counts during the 15 min immediately after extinction were normalized to the last 15 min of the previous test day when reinforcement had been available and compared between groups by 1-factor (dose) ANOVA with Tukey’s post-hoc tests. This test day occurred 3 weeks into riluzole administration. The overall test design (tail suspension, then forced swimming, followed by operant conditioning; see table 1) was chosen in an attempt to introduce the least “stressful” tasks first and the more persistently “stressful” task (that requiring food restriction) last.
Table 1. Experimental events in riluzole-treated mice.
Riluzole-treated mice were tested in several behavioral tasks (results shown in figures 1 and 2). The timing of experimental events relative to the duration of riluzole administration is indicated. The events highlighted in gray constitute “incentive disengagement” training and testing.
| Day | Experimental event | Duration |
|---|---|---|
| 1 | Locomotor monitoring | 30 min. |
| 7 | Locomotor monitoring | 30 min. |
| 10 | Tail suspension test (high dose only) | 6 min. |
| 14 | Forced swim test | 8 min. |
| 17 | Instrumental training | 120 min. |
| 18 | Instrumental training | 120 min. |
| 19 | Instrumental training | 120 min. |
| 20 | Instrumental training followed by locomotor activity monitoring | 120 min. + 60 min. |
| 21 | Extinction training followed by locomotor monitoring | 120 min. + 15 min. |
| 22 | Locomotor monitoring; peripheral measures collected | 30 min. |
Fluoxetine treatment
As a positive control, a separate group of mice was treated with fluoxetine hydrochloride (Spectrum Chemical, Garderia, CA; 160 μg/ml + 2% saccharin in the drinking water, as in Gourley et al. 2008a) or saccharin alone for 3 weeks (n=11 and 12). This concentration translated to 27.0 mg/kg/day on average. Mice were tested in the “incentive disengagement” model as described above with the test day falling on day 21 of treatment. Normalized photobeam counts were compared by unpaired t-test.
Gland extraction and blood serum analysis
Mice administered the highest (60 μg/ml) concentration of riluzole were euthanized at 22 days of administration by rapid decapitation (n=8/group). Trunk blood, adrenal glands, and thymus glands were collected. Glands were weighed in pairs, and values were normalized to body weight. Trunk blood was chilled, spun, and serum was extracted for corticosterone (CORT) analysis by enzyme-linked immunosorbent assay (ELISA) conducted in accordance with the manufacturer’s instructions (Assay Designs, Inc., Ann Arbor, MI) and our previous reports (Gourley et al., 2008b). Due to equipment failure, only a subset of samples was analyzed (n=6 control; n=5 riluzole); these were run in duplicate. Measures were compared by unpaired t-tests.
Corticosteroid exposure
We investigated the effects of 3 weeks of 60 μg/ml riluzole treatment on putatively antidepressant-sensitive protein targets in the hippocampus of adult male mice, half of which were first exposed to chronic oral CORT (25 μg/ml free base; 4-pregnen-11β 21-DIOL-3 20-DIONE 21-hemisuccinate; Steraloids, Inc., Newport, RI) to evaluate the effects of chronic riluzole in the “normal” brain and in a model of depression in mice (Gourley et al., 2008a,b). CORT was dissolved in tap water and neutralized to a pH of 7.0–7.4 with HCl. Mice were presented with CORT in place of normal drinking water for 14 days, resulting in a dose of ~6.5 mg/kg/day in the experiments shown here. Animals were weaned with 3 days of 12.5 μg/ml CORT, then 3 days with 6.25 μg/ml, to allow for gradual recovery of endogenous CORT secretion. After CORT, half of the mice were then administered riluzole dissolved in saccharin exactly as described above for 3 weeks. This 2×2 experimental design resulted in 4 groups: water + saccharin, CORT + saccharin, water + riluzole, and CORT + riluzole.
Brain tissue dissection
At the end of the 3-week riluzole administration period, mice were euthanized by rapid decapitation. Brains were immediately frozen on dry ice and later cut into 1 mm coronal slices. Bilateral hippocampal samples were collected with tissue punches (1.2 mm diameter) placed immediately lateral to the third ventricle and ventral to the CA1 cell body layer, the goal being to enrich samples with dentate gyrus tissue. “CA1/CA3-rich” samples were collected from the tissue slice caudal to this initial slice by targeting the tissue core to the prominent ventral and lateral CA1/CA3 subregions. This approach resulted here in the anatomically unambiguous dissection of 14 control (water + saccharin), 14 CORT (CORT + saccharin), 7 CORT + riluzole, and 9 riluzole-only tissue samples and has previously allowed us to identify differential effects of antidepressant treatment on other targets within these major hippocampal subregions (Gourley et al., 2008a).
Immunoblotting and BDNF ELISA
Tissue samples were frozen at −80°C and later sonicated in lysis buffer [137 mM NaCl, 20 mM tris-Hcl (pH=8), 1% igepal, 10% glycerol] and restored at −80°C. Protein concentrations were determined using a Bradford colorimetric assay (Pierce, Rockland, IL). For immunoblotting, 20 μg/sample of the CA1/CA3-rich tissue homogenate were added to 10 μl Laemmli buffer (20% glycerol, 2% SDS, Bromphenol blue) and boiled for 10 min. Samples were separated by SDS-PAGE on 8–16% gradient tris-glycine gels (Invitrogen, Carlsbad, CA). Primary antibodies were anti-GAPDH (Ms; 1:20K; Advanced Immunochemical Inc., Long Beach, CA) and anti-GLT-1 (Ms; 1:500; Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were incubated overnight and then incubated with IRDye 700 Dx Anti-Rb IgG and IRDye 800 Dx Anti-Ms IgG for 1 hour (1:5000; Rockland Immunochemicals, Gilbertsville, PA). Bands were quantified using fluorescence densitometry analysis (Licor Odyssey Imaging System).
Individual GLT-1 fluorescence values were normalized to the corresponding GAPDH loading controls; these ratios were then converted to a percentage of the control mean from the same membrane to control for fluorescence variance between gels. Variability in the control group was generated by converting each individual value to a percentage of its own group mean.
BDNF quantification in dentate gyrus-rich samples was conducted using a 2-site BDNF ELISA kit in accordance with the manufacturer’s instructions (Promega, Madison, WI), except tissue was diluted 1:4 in blocking buffer, and the extraction procedure was excluded. Samples were run in duplicate on a single plate, and BDNF concentration values were normalized to the total protein content in each sample. BDNF is expressed as pg/ml wet tissue.
For both GLT-1 and BDNF, group means were compared by 2-factor ANOVA with CORT and riluzole as factors. Post-hoc comparisons were Tukey’s t-tests. Values were square-root- (for BDNF concentrations) or arcsin- (for GLT-1 percentages) transformed in order to ensure normal variance (Ferguson 1978), and values >2 standard deviations outside of the mean were excluded. p≤0.05 was considered significant; p>0.05 but ≤0.1 was considered a trend.
Results
Effects of riluzole on locomotor activity and in models of antidepressant efficacy
In the first series of experiments, mice were treated with riluzole for a total of 22 days to allow for multiple tests and recovery time between them (table 1). During this period, locomotor activity was periodically monitored. Chronic treatment at these concentrations did not appear to be sedative; by contrast, the lowest 6 μg/ml concentration increased ambulatory activity [main effect of concentration F(3,33)=6.7, p<0.001; post-hoc ps≤0.01 relative to all other groups] (fig. 1a). Mice administered 30 or 60 μg/ml did not differ from control mice (ps>0.8).
Figure 1. Riluzole has antidepressant-like properties in mice.
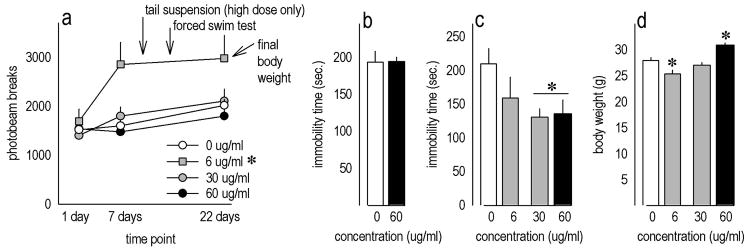
(a) Locomotor activity was periodically monitored in mice chronically treated with riluzole. Oral riluzole did not appear to be sedative at the concentrations tested, and the 6 μg/ml concentration actually increased photobeam counts; the timing of experimental events relative to the time at which the animals were monitored is indicated. See also table 1. (b) Several mice administered 60 μg/ml riluzole were tested in the tail suspension test at 10 days of treatment, but no drug effects were identified. (c) In the forced swim test, however, riluzole dose-dependently decreased immobility time. (d) At the end of the 22-day treatment protocol, 6 μg/ml treatment had decreased body weights, while 60 μg/ml increased body weights. Bars and symbols represent means (+SEM) per treatment group (*p<0.05).
An initial cohort of mice administered the highest 60 μg/ml concentration showed no response in the tail suspension test of antidepressant efficacy at 10 days of treatment (t14=−0.02, p>0.9) (fig. 1b); because this concentration had no effect, lower concentrations were not tested. Despite this finding, 30 and 60 μg/ml reduced immobility in the forced swim test [F(3,32)=3, p=0.05; post-hoc ps<0.04 relative to control] when measured at 14 days of treatment (fig. 1c).
By the end of the 22-day treatment period, mice treated with the lowest concentration (6 μg/ml) were lighter than control animals [F(3,33)=12.4, p<0.001; post-hoc p=0.03], while 60 μg/ml increased body weights (ps≤0.004 relative to all other groups) (fig. 1d).
Incentive disengagement
In view of differing effects in the tail suspension and forced swim tests of antidepressant efficacy, we adapted a model of depressive-like behavior termed “incentive disengagement.” We first confirmed that when expected reinforcement was withheld in drug-naïve instrumentally-trained mice, locomotor activity acutely declined, as in previous reports in rats: Indeed, in drug-naïve mice, photobeams broken in the 15 min after extinction training were reduced relative to counts generated in the first 15 min after reinforced test sessions [time bin x condition F(9,63)=8.1, p<0.001; post-hoc ps<0.003] (fig. 2a), and photobeam counts generated during these initial 15 minutes after extinction training were predicted by the number of nonreinforced instrumental responses the mice had performed (r2=0.56, p=0.03) (fig. 2b), pointing to a relationship between instrumental responding and subsequent locomotor activity. Importantly, reinstatement of food-reinforced responding also reinstated robust locomotor activity equivalent to that prior to extinction training (p=0.003 relative to post-extinction; ps>0.9 relative to reinforced sessions) (fig. 2a), indicating the decline in activity was not simply a consequence of testing order.
Figure 2. Validation and utility of the incentive disengagement model of acute depressive-like behavior.

(a) The incentive disengagement model was adapted for use in mice: Animals learned to earn food pellets in an operant conditioning chamber. After stable responding was established, locomotor activity was monitored immediately after the session; two sessions are shown with reinforcements earned by squares and locomotor activity counts represented by connected circles. When reinforcement was withheld (“non-reinforced” condition), locomotor activity declined. Responding for food was reinstated the following day, and locomotor activity was restored. (b) As added evidence that locomotor activity relates to an animal’s recent instrumental experience, photobeam breaks in the first 15 min after the nonreinforced test session correlated with nonreinforced responses made prior to locomotor monitoring. Symbols represent individual mice. (c) When mice were reinforced, locomotor activity in the first 15 min was approximately 166% of the last 15 min the previous day. By contrast, if reinforcement was withheld, locomotor activity was only approximately 119% of this habituated baseline. Bars represent group means in a within-subjects comparison. (d) Chronic fluoxetine occluded the expected decline in locomotor activity relative to saccharin control treatment, and (e) 60 μg/ml riluzole also increased activity, suggestive of an antidepressant-like effect, while mice treated with 6 μg/ml showed decreased activity. Bars represent group means +SEMs in between-subjects comparisons (*ps≤0.05).
When mice were monitored after a reinforced operant conditioning session, locomotor activity in the first 15 min was increased to approximately 166% of the last 15 min the previous day. By contrast, if reinforcement was withheld, locomotor activity was only approximately 119% of the same baseline (fig. 2c); antidepressant treatments would be expected to occlude this decline. Consistent with this model, mice administered chronic fluoxetine generated more locomotor counts than control mice after extinction training (t21=−2.2, p=0.04) (fig. 2d). Mice treated with 60 μg/ml riluzole were also more active than control and lower-dose groups [F(3,28)=12.1, p≤0.001; post-hoc ps<0.006 relative to all other groups] (fig. 2e) consistent with an antidepressant-like response. Unexpectedly, the lowest dose—6 μg/ml—decreased normalized locomotor scores after extinction training (p=0.009 relative to control). Statistical analyses here refer to normalized scores, but raw photobeam counts are also provided in table 2.
Table 2. Photobeam counts before and after instrumental extinction training.
Mean photobeam breaks (+/−SEM) gathered after the final day of instrumental training and immediately after extinction training the following day in riluzole- and fluoxetine-treated mice are shown. These values represent the raw data used to generate normalized counts represented in figures 2d and e. Control mice are collapsed into a single group. Note that only 60 μg/ml riluzole and fluoxetine appreciably increase photobeam counts after extinction training.
| Group | Post-training photobeam counts (min. 45–60) | Post-extinction photobeam counts (min. 0–15) |
|---|---|---|
| saccharin control | 1504.8 +/− 138.0 | 1591.1 +/− 256.0 |
| 6 μg/ml riluzole | 1876.7 +/−291.2 | 998.5 +/− 304.8 |
| 30 μg/ml riluzole | 1330.6 +/− 130.8 | 854.6 +/− 99.7 |
| 60 μg/ml riluzole | 1282.0 +/− 108.3 | 1630.3 +/− 209.6 |
| 160 μg/ml fluoxetine | 2017.3 +/− 179.1 | 3472.3 +/− 130.3 |
Peripheral measures
We collected several peripheral measures from mice chronically (22 days) treated with riluzole at an antidepressant-like concentration (60 μg/ml): Thymus gland weights did not significantly differ between groups (t14=1.6, p=0.13) (fig. 3a). Adrenal gland weights did not differ (t14=0.4, p=0.4) (fig. 3b); consistent with this finding, blood serum CORT also did not differ between groups (t9=1.1, p=0.3) (fig. 3c).
Figure 3. Chronic riluzole (60 μg/ml) has no effects on thymus and adrenal gland weights or blood serum CORT.
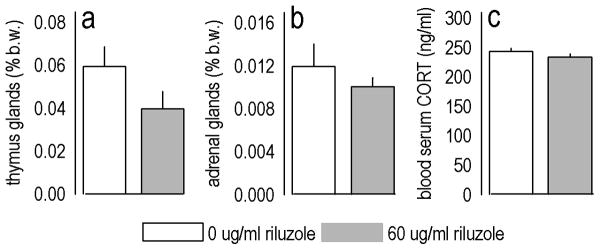
(a) After 22 days of riluzole treatment, thymus gland weights were statistically unchanged [expressed as a percent total body weight (b.w.)]. (b) Adrenal gland weights were also unchanged, and (c) riluzole did not appear to affect circulating CORT. Bars represent group means (+SEM).
Central effects of riluzole in a chronic depression model
We next analyzed the central actions of riluzole (60 μg/ml for 3 weeks) in a glucocorticoid-based model of depression. When we quantified BDNF in dentate gyrus-rich tissue samples, a CORT x riluzole interaction was identified [F(1,39)=8.6, p=0.005] (fig. 4a). Post-hoc tests indicated prior CORT exposure decreased BDNF as expected (p=0.02), while subsequent riluzole treatment restored BDNF levels in CORT-exposed mice (p=0.01 relative to CORT alone). Surprisingly, however, post-hoc tests also indicated mice treated with riluzole alone did not differ from CORT-exposed mice, and levels were reduced at a trend level of significance relative to control values (p=0.1).
Figure 4. Biochemical effects of riluzole (60 μg/ml) after corticosteriod exposure.

(a) In glucocorticoid-exposed mice, riluzole treatment restored BDNF expression in dentate gyrus-rich tissue samples, but riluzole in the absence of prior CORT reduced BDNF at a trend level of significance relative to control. (b) In CA1/CA3-rich samples, prior CORT also reduced GLT-1 expression, which was restored with riluzole treatment. Representative blots loaded in the same order as the bar graph below are shown; GAPDH (37 kD) served as a loading control. Bars represent means (+SEM) per treatment group (*p<0.05; †p=0.1).
We also evaluated the expression of GLT-1 in CA1/CA3-rich tissue samples. Again, a CORT x riluzole interaction was identified [F(1,39)=5.3, p=0.03] (fig. 4b), with post-hoc tests indicating prior CORT exposure decreased GLT-1 expression (p=0.02), and levels were restored by subsequent riluzole treatment (relative to CORT p=0.004). A main effect of riluzole was also identified (p=0.02).
Discussion
Riluzole is a neuroprotective agent that both inhibits glutamate release by acting as a Na+-channel blocker and increases glutamate turnover; it may also have therapeutic utility in treating depression (Mathew et al. 2005), but precise molecular mechanisms of antidepressant-like action are still being established. Transgenic and knockout mice may aid in this process, but an antidepressant-mimicking dosing protocol for mice has not, to our knowledge, been established. For this reason, we optimized an oral riluzole administration protocol for use in C57BL/6 mice and found that chronic administration of a 30 or 60, but not 6, μg/ml concentration of riluzole in the drinking water had antidepressant-like effects in the forced swim test. The highest concentration tested (60 μg/ml) also had antidepressant-like effects in an adaptation of the “incentive disengagement” test, recapitulating the effects of chronic fluoxetine treatment. Moreover, riluzole upregulated hippocampal GLT-1 expression, consistent with antidepressant-like actions.
Use of “behavioral despair” and “incentive disengagement” behavioral models
Using two “behavioral despair” models of antidepressant efficacy, the forced swim test and the tail suspension test, we found dissimilar effects of chronic 60 μg/ml riluzole treatment. In the former, riluzole produced a robust antidepressant-like response, while in the latter, the compound had no effects. While it is possible that testing order played a role in our findings (tail suspension preceded forced swimming), we think it more likely that C57BL/6 mice are not ideal for use in the tail suspension test due to high activity levels, as has been previously suggested (Mayorga and Lucki 2001; Crowley et al. 2005). To resolve our disparate findings, we adapted a model of “transient malaise” termed incentive disengagement first described in a report by Klinger et al. 1974. This model is based on the observation that locomotor activity in rats gradually declines when instrumentally-trained animals are deprived of expected appetitive reinforcement, even when rats are placed in a neutral—rather than the training—environment. This reduction in locomotor activity has been proposed to reflect a sense of loss and was recapitulated here in adult drug-naïve C57 mice (fig.2). An antidepressant-like compound would be expected to occlude the decline in activity, as was indeed observed with 60 μg/ml riluzole and fluoxetine here. Notably, this concentration of riluzole did not otherwise affect locomotor activity.
Although the incentive disengagement model is significantly less well-characterized than the forced swim and tail suspension tests, it has the advantage that animals’ behavioral responses derive from a violation of reward expectancies as opposed to exposure to a threatening situation, as is the case in both tests of “behavioral despair.” Thus incentive disengagement models provide an alternative modality by which to test putatively antidepressant-like compounds. Addition of this test to the repertoire of assays used to quantify antidepressant (or pro-depressive) manipulations in animals may be timely, as behavioral despair models have garnered criticism for failing to reflect depressive-like symptoms (e.g., Holmes 2003; O’Neil and Moore 2003).
Molecular correlates of riluzole efficacy
In an effort to identify molecular correlates of riluzole efficacy, a separate group of mice were chronically exposed to CORT using a protocol that mimics the circadian rhythmicity of CORT hyper-secretion in stressed rats (Pekary et al., 2008), induces multiple depressive-like behaviors that persist for a significant duration of the animal’s lifespan (Gourley et al. 2008a,b,c; Gourley and Taylor 2009), diminishes hippocampal neurogenesis (David et al. 2009), and disrupts hypothalamic-pituitary-adrenal axis negative feedback (Gourley et al. 2009). Consistent with neurotrophic hypotheses of depression and antidepressant efficacy (Duman et al. 1997), the corticosteroid protocol used here also reduces hippocampal BDNF and phosphorylation of downstream targets including cAMP-response Element Binding Protein (CREB) (fig.4; Gourley et al. 2008b). Subsequent riluzole treatment restored BDNF to homeostatic levels, thus reflecting the actions of classical antidepressants (Nibuya et al. 1995; reviewed Groves 2007), but BDNF levels in mice treated with riluzole alone (i.e., in the absence of prior CORT exposure) differed from neither the control nor CORT-exposed group, and qualitatively more closely resembled the suppressed CORT group. This unexpected discrepancy may in part be an issue of dose, as previous reports that identified riluzole-induced BDNF over-expression (Katoh-Semba et al. 2002; Fumagalli et al. 2006) tested higher doses than those achieved by even the highest concentration here.
A potentially more pertinent issue remains: Despite comparable effects on BDNF, two manipulations tested here—prior chronic CORT exposure and chronic riluzole treatment in the absence of prior CORT exposure—had opposite effects in the forced swim test, with riluzole increasing mobility (fig.1), and prior chronic CORT decreasing mobility (Gourley et al. 2008b). Together these findings suggest BDNF over-expression is not necessary for an antidepressant-like response in this test. Conti and colleagues (2002) drew the same conclusion after demonstrating that CREB-deficient mice are unable to generate a BDNF response to desipramine but still show forced swim sensitivity. Thus, while hippocampal BDNF over-expression or restoration may be sufficient to increase mobility (Shirayama et al. 2002; Gourley et al. 2008b), it may not be necessary. Similarly, selective dentate gyrus bdnf knockdown increases immobility under some, but not all, circumstances (Adachi et al. 2008; Taliaz et al. 2010).
Riluzole may alternatively act by facilitating glutamate clearance from the synaptic cleft (Mathew et al. 2005). GLT-1 (also referred to as EAAT2) is the most predominantly expressed glial glutamate transporter in the rodent hippocampus, and increased prefrontal cortical GLT-1 mRNA expression was recently associated with riluzole’s antidepressant-like efficacy in both chronically stressed and non-stressed rats (Banasr et al. 2010). Ceftriaxone, which also increases glutamate turnover and glial transporter expression (Rothstein et al. 2005), also has antidepressant-like effects in rodents (Mineur et al. 2007), while prefrontal gliotoxin infusions conversely induce depressive-like behavior (Banasr and Duman 2008). With regards to stressor or stress hormone exposure, GLT-1 mRNA or protein is increased immediately after stressor exposure in a glucocorticoid-dependent manner in the hippocampus (Reagan et al. 2004; Wood et al. 2004; Autry et al. 2006). This rapid over-expression has been interpreted as a response to increased glutamate efflux (Lowy et al. 1993, 1995) and decreased glutamate clearance (Yang et al. 2005) after stressor exposure. Our findings indicate corticosteroid-induced GLT-1 up-regulation is not persistent, however, since GLT-1 expression was diminished 3 weeks after chronic exposure. As in the rat prefrontal cortex (Banasr et al. 2010), riluzole non-specifically (i.e., in both CORT-exposed and control mice) increased GLT-1. Together with previous work, these data suggest a model in which GLT-1 gradually declines in response to prolonged corticosteroid exposure, and riluzole promotes GLT-1 expression and thereby engenders behavioral resilience.
Effects of 6 μg/ml riluzole
We have primarily focused our discussion on the antidepressant-like consequences of chronic 60 μg/ml riluzole treatment. While this concentration did not affect general locomotor activity, it decreased immobility in the Porsolt forced swim test and increased ambulation in the “incentive disengagement” model of acute depressive-like behavior, consistent with an antidepressant-like effect. By contrast, the lowest concentration tested (6 μg/ml) increased locomotor activity in an open field and reduced activity in the “incentive disengagement” model. Riluzole is generally considered a sedative compound based on studies of higher doses, but it also has separable anxiolytic-like properties in rats (Mirza et al. 2005; Munro et al. 2007). Increased activity here may reflect an anxiolytic-like response to the open field used for locomotor monitoring that biased “incentive disengagement” scores towards diminished sensitivity, since this test was conducted in clean fields of the same dimensions and in the same location in the laboratory. This interpretation implies that the doses of riluzole with anxioloytic-like vs. antidepressant-like consequences are dissociable, in which case anxiolytic properties would be more strongly associated with riluzole’s primary actions as a Na+-channel inhibitor (Mirza et al. 2005), and its antidepressant-like properties with off-target effects, but further studies specifically testing this hypothesis are necessary.
Conclusions
To date, published data on riluzole’s use as an antidepressant come in the form of open-label, uncontrolled clinical trials or case studies (Pittenger et al. 2008). Because research in the field of affective disorders has not developed fundamentally new antidepressants with distinct mechanisms of action in several decades, these findings offer promise for increasing the diversity of well-tolerated antidepressant treatment options for depressed patients, though controlled clinical studies are necessary, and the mechanisms of antidepressant-like action are still not entirely known. Use of a riluzole administration protocol with antidepressant-like consequences in genetically-modified mouse models may aid this process.
Acknowledgments
This work was supported by NIH MH079680 (SLG), NIH MH081211 (GS), the Interdisciplinary Research Consortium on Stress, Self-control and Addiction [UL1-DE19586 and the NIH Roadmap for Medical Research/Common Fund, AA017537 (JRT)], and the Connecticut Department of Mental Health and Addiction Services (JRT, GS).
Dr. Sanacora has received consulting fees from AstraZeneca, Bristol-Myers Squibb, Evotec, Eli Lilly & Co., Johnson & Johnson, Roche, Novartis and Sepracor Inc. He has also received additional grant support from AstraZeneca, Bristol-Myers Squibb, Merck & Co., Roche, and Sepracor Inc. In addition, he is a co-inventor on a filed patent application by Yale University (PCTWO06108055A1) concerning the use of glutamate modulating drugs as antidepressants and anxiolytics.
The authors thank Drs. Mounira Banasr and Mary Torregrossa for advice and feedback.
Works Cited
- Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol Psychiatry. 2008;63:642–629. doi: 10.1016/j.biopsych.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Grillo CA, Piroli GG, Rothstein JD, McEwen BS, Reagan LP. Glucocorticoid reulgation of GLT-1 glutamate transporter isoform expression in rat hippocampus. Neuroendocrinology. 2006;83:371–379. doi: 10.1159/000096092. [DOI] [PubMed] [Google Scholar]
- Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- Banasr M, Duman RS. Regulation of neurogenesis and gliogenesis by stress and antidepressant treatment. CNS Neurol Disord Drug Targets. 2007;6:311–320. doi: 10.2174/187152707783220929. [DOI] [PubMed] [Google Scholar]
- Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psych. 2008;64:863–870. doi: 10.1016/j.biopsych.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, Behar KL, Sanacora G. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psych. 2010;15:501–511. doi: 10.1038/mp.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldarone BJ, Karthigeyan K, Harrist A, Hunsberger JG, Witmack E, King SL, et al. Sex differences in response to oral amitriptyline in three animal models of depression in C57BL/6J mice. Psychopharmacology. 2003;170:94–101. doi: 10.1007/s00213-003-1518-7. [DOI] [PubMed] [Google Scholar]
- Conti AC, Cryan JF, Dalvi A, Lucki I, Blendy JA. cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. J Neurosci. 2002;22:3262–3268. doi: 10.1523/JNEUROSCI.22-08-03262.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coric V, Milanovic S, Wasylink S, Patel P, Malison R, Krystal JH. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology. 2003;167:219–220. doi: 10.1007/s00213-003-1396-z. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Blendy JA, Lucki I. Strain-dependent antidepressant-like effects of citalopram in the mouse tail suspension test. Psychopharmacology. 2005;183:257–264. doi: 10.1007/s00213-005-0166-5. [DOI] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- Ferguson GA. Statistical analysis in psychology and education. McGraw Hill; New York: 1978. [Google Scholar]
- Frizzo ME, Dall’Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24:123–128. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli E, Bigini P, Barbera S, De Paola M, Mennini T. Riluzole, unlike the AMPA antagonist RPR119990, reduces motor impairment and partially prevents motoneuron death in the wobbler mouse, a model of neurodegenerative disease. Exp Neurol. 2006;198:114–128. doi: 10.1016/j.expneurol.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–176. doi: 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Wu FJ, Kiraly DD, Ploski JE, Kedves AT, Duman RS, Taylor JR. Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol Psychiatry. 2008a;63:353–359. doi: 10.1016/j.biopsych.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley SL, Kiraly DD, Howell JL, Olausson P, Taylor JR. Acute hippocampal BDNF restores motivational and forced swim performance after corticosterone. Biol Psychiatry. 2008b;64:884–890. doi: 10.1016/j.biopsych.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley SL, Wu FJ, Taylor JR. Corticosterone regulates pERK1/2 in a chronic depression model. Ann N Y Acad Sci. 2008c;1148:509–514. doi: 10.1196/annals.1410.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley SL, Taylor JR. Regulation and reversal of a persistent depression-like syndrome in rodent. Current Protocols in Neuroscience. 2009;Chapter 9(Unit 9.32) doi: 10.1002/0471142301.ns0932s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley SL, Kedves AT, Olausson P, Taylor JR. A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology. 2009;34:707–716. doi: 10.1038/npp.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves JO. Is it time to reassess the BDNF hypothesis of depression? Mol Psychiatry. 2007;12:1079–1088. doi: 10.1038/sj.mp.4002075. [DOI] [PubMed] [Google Scholar]
- Holmes PV. Rodent models of depression: Reexamining validity without anthropomorphic inference. Crit Rev Neurobiol. 2003;15:143–174. doi: 10.1615/critrevneurobiol.v15.i2.30. [DOI] [PubMed] [Google Scholar]
- Katoh-Semba R, Asano T, Euda H, Morishita R, Takeuchi IK, Inaguma Y, Kato K. Riluzole enhances expression of brain-derived neurotrophic factor with consequent proliferation of granule precursor cells in the rat hippocampus. FASEB J. 2002;16:1328–1330. doi: 10.1096/fj.02-0143fje. [DOI] [PubMed] [Google Scholar]
- Klinger E, Barta SG, Kemble ED. Cyclic activity changes during extinction in rats: A potential animal model of depression. Animal Learn Mem. 1974;2:313–316. [Google Scholar]
- Lowy MT, Gault L, Yamamoto BK. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61:1957–1960. doi: 10.1111/j.1471-4159.1993.tb09839.x. [DOI] [PubMed] [Google Scholar]
- Lowy MT, Wittenberg L, Yamamoto BK. Effects of acute stress on hippocampal glutamate levels and spectrin proteolysis in young and aged rats. J Neurochem. 1995;65:268–274. doi: 10.1046/j.1471-4159.1995.65010268.x. [DOI] [PubMed] [Google Scholar]
- Mathew SJ, Keegan K, Smith L. Glutamate modulators as novel interventions for mood disorders. Rev Bras Psiquiatr. 2005;27:243–248. doi: 10.1590/s1516-44462005000300016. [DOI] [PubMed] [Google Scholar]
- Mathew SJ, Murrough JW, aan het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placebo-controlled continuation trial. Int J Neuropyschopharmacol. 2010;13:71–82. doi: 10.1017/S1461145709000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayorga AJ, Lucki I. Limitations on the use of the C57BL/6 mouse in the tail suspension test. Psychopharmacology. 2001;155:110–112. doi: 10.1007/s002130100687. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psych. 2007;61:250–252. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- Mirza NR, Bright JL, Stanhope KJ, Wyatt A, Harrington NR. Lamotrigine has an anxiolytic-like profile in the rat conditioned emotional response test of anxiety: a potential role for sodium channels? Psychopharmacology. 2005;180:159–168. doi: 10.1007/s00213-005-2146-1. [DOI] [PubMed] [Google Scholar]
- Munro G, Erichsen HK, Mirza NR. Pharmacological comparison of anticonvulsant drugs in animal models of persistent pain and anxiety. Neuropsychopharmacology. 2007;53:609–618. doi: 10.1016/j.neuropharm.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil MF, Moore NA. Animal models of depression: are there any? Hum Psychopharmacol. 2003;18:239–254. doi: 10.1002/hup.496. [DOI] [PubMed] [Google Scholar]
- Pekary AE, Sattin A, Blood J, Furst S. TRH and TRH-like peptide expression in rat following episodic or continuous corticosterone. Psychoneuroendocrinology. 2008;2008:1183–1197. doi: 10.1016/j.psyneuen.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Coric V, Banasr M, Bloch M, Krystal JH, Sanacora G. Riluzole in the treatment of mood and anxiety disorders. CNS Drugs. 2008;22:761–786. doi: 10.2165/00023210-200822090-00004. [DOI] [PubMed] [Google Scholar]
- Porsolt R, Le Pichon M, Jalfe M. Depression: a new animal model sensitive to antidepressant treatment. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ. Gliogenesis and glial pathology in depression. CNS Neurol Disord Drug Targets. 2007;6:219–233. doi: 10.2174/187152707780619326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan LP, Rosell DR, Wood GE, Spedding M, Muñoz C, Rothstein J, McEwen BS. Chronic restraint stress up-regulates GLT-1 mRNA and protein expression in the rat hippocampus: reversal by tianeptine. Proc Natl Acad Sci U S A. 2004;101:2179–2184. doi: 10.1073/pnas.0307294101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Kendell SF, Fenton L, Coric V, Krystal JH. Riluzole augmentation for treatment-resistant depression. Am J Psychiatry. 2004;161:2132. doi: 10.1176/appi.ajp.161.11.2132. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V, Krystal JH. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry. 2007;61:822–825. doi: 10.1016/j.biopsych.2006.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y, Chen AC-H, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Zarate CA, Krystal AD. Case report: successful riluzole augmentation therapy in treatment-resistant bipolar depression following the development of rash with lamotrigine. Psychopharmacology. 2004;173:227–228. doi: 10.1007/s00213-003-1756-8. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaz D, Stall N, Dar DE, Zangen A. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry. 2010;15:80–92. doi: 10.1038/mp.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P. The validity of animal models of depression. Psychopharmacology. 1984;83:1–16. doi: 10.1007/BF00427414. [DOI] [PubMed] [Google Scholar]
- Wood GE, Young LT, Reagan LP, Chen B, McEwen BS. Stress-induced structural remodeling in hippocampus: Prevention by lithium treatment. Proc Natl Acad Sci. 2004;101:3973–3978. doi: 10.1073/pnas.0400208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CH, Huang CC, Hsu KS. Behavioral stress enhances hippocampal CA1 long-term depression through the blockade of the glutamate uptake. J Neurosci. 2005;25:4288–4293. doi: 10.1523/JNEUROSCI.0406-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D, Charney DS, Manji HK. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161:171–174. doi: 10.1176/appi.ajp.161.1.171. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Quiroz JA, Singh JB, Denicoff KD, De Jesus G, Luckenbaugh DA, Charney DS, Manji HK. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57:430–432. doi: 10.1016/j.biopsych.2004.11.023. [DOI] [PubMed] [Google Scholar]