

Abstract
Molecular therapy using a small interfering RNA (siRNA) has shown promise in the development of novel therapeutics. Various formulations have been used for in vivo delivery of siRNAs. However, the stability of short double-stranded RNA molecules in the blood and efficiency of siRNA delivery into target organs or tissues following systemic administration have been the major issues that limit applications of siRNA in human patients. In this study, multifunctional siRNA delivery nanoparticles are developed that combine imaging capability of nanoparticles with urokinase plasminogen activator receptor-targeted delivery of siRNA expressing DNA nanocassettes. This theranostic nanoparticle platform consists of a nanoparticle conjugated with targeting ligands and double-stranded DNA nanocassettes containing a U6 promoter and a shRNA gene for in vivo siRNA expression. Targeted delivery and gene silencing efficiency of firefly luciferase siRNA nanogenerators are demonstrated in tumor cells and in animal tumor models. Delivery of survivin siRNA expressing nanocassettes into tumor cells induces apoptotic cell death and sensitizes cells to chemotherapy drugs. The ability of expression of siRNAs from multiple nanocassettes conjugated to a single nanoparticle following receptor-mediated internalization should enhance the therapeutic effect of the siRNA-mediated cancer therapy.
1. Introduction
Recent advances in the areas of RNA interference (RNAi) and nanotechnology offer a great opportunity to develop molecular targeted therapies to sensitize drug-resistant cancer cells to therapeutic agents. [1–8] RNAi enables sequence specific gene silencing by promoting degradation of specific mRNAs. [9] At present, clinical trials using liposome or polymer formulated RNA-based small interfering RNA (siRNA) delivery systems are ongoing in cancer patients. [1,10] siRNAs are chemically synthesized double-stranded RNA molecules typically with 21 to 23 nucleotides (nt). [9] The major challenges for applying short double-stranded RNA molecules for cancer therapy include the stability of siRNAs in the blood circulation and tissues, the low delivery efficiency of siRNAs, and the high cost of producing RNA-based siRNAs. Unprotected siRNAs are degraded by ribonucleases in serum within 5 min after intravenous delivery. Chemical modification of siRNAs can only extend half-lives to 30 min or a few hours. [11] One approach to protect RNA-based siRNAs from rapid degradation after systemic delivery is using nanoparticle delivery systems. [12–22] For example, a transferrin receptor targeted cyclodextrin polymer nanoparticle carrying a subunit of ribonucleotide reductase siRNAs has been developed and used in a clinical trial for cancer therapy. [1,23] Magnetic iron oxide nanoparticles (IONPs) conjugated with siRNAs for an anti-apoptotic survivin gene have also been shown to inhibit survivin gene expression and monitor the delivery of siRNA-nanoparticles into the tumor by magnetic resonance imaging (MRI). [2] However, only limited numbers of siRNA molecules can be delivered into tumor cells after systemic delivery. Extensive studies have been conducted to develop a RNA nanoparticle-based siRNA delivery system. [8,22,24] Chemically modified and folate receptor-targeted RNA nanoparticles that are based on bacteriophage phi29 DNA packaging motors (pRNAs) have been developed to carry single or multiple siRNAs in their scaffold domains. This pRNA nanoparticle siRNA delivery carrier showed high in vivo stability with the blood half-life of 5 to 10 h and could retain in tumor tissues for over 8 h. Tumor targeted delivery and efficacy of gene silencing have also been demonstrated in animal tumor models. [8,22,24]
Since siRNA is expressed from a RNA polymerase III (U6 or H1) promoter, a short hairpin siRNA (shRNA) gene has been cloned into expression vectors containing a polymerase III promoter to produce shRNAs from plasmids or viral vectors following transfecting into cells. [25–28] Those shRNAs are further processed into siRNAs by a cellular endoribonuclease. [9] Although gene silencing using viral vectors has been demonstrated in vivo in animal models, potential inflammatory and immunogenic effects may prevent their repeated administration. Additionally, most viral vectors have similar sizes to nanoparticles but can only carry one copy of shRNA gene in each vector and, therefore, have relatively low efficiency for delivering the shRNA gene and generating siRNAs. Liposome and polymeric nanoparticles have been used to encapsulate non-viral plasmid vectors expressing shRNA genes. The shRNA expressing plasmid vectors have ~5 Kb nucleotides with particle sizes ranging from 20 to 160 nm, depending on condensing conditions. [29–31] It is likely that limited copies of the plasmids can be encapsulated or conjugated to a single nanoparticle with a size < 100 nm, which is an optimal size for intratumoral delivery. [32] Previous studies have provided proof of concept demonstrations that shRNA expressing DNA cassettes containing a U6 promoter and a shRNA gene can be synthesized by a two step PCR amplification protocol. [33,34] However, such an approach has low efficiency and high incidence of errors in PCR amplification of the DNA cassettes due to the presence of a complementary sequence in the shRNA gene and multi-step PCR using long primers. Therefore, it may not be suitable for the development of therapeutics. New approaches for targeted delivery of siRNAs for enhanced gene silencing efficiency and the ability to monitor siRNA delivery using non-invasive imaging are needed to accelerate the clinical translation of siRNA cancer therapies.
In this study, we have developed a multifunctional siRNA delivery nanoparticle platform that combines the imaging capability of the nanoparticles with receptor-mediated delivery of siRNA-expressing DNA cassettes based on the concept of gene therapy. This new theranostic nanoparticle consists of an amphiphilic polymer-coated nanoparticle core, either fluorescent quantum dot (QD) or MRI contrast-enhancing magnetic IONP, conjugated with 10 to 20 DNA nanocassettes that contain a U6 promoter and a shRNA gene for in vivo siRNA gene expression following intracellular delivery. The nanoparticle is conjugated to the amino terminal fragment (ATF) of the urokinase plasminogen activator (uPA), which targets its cellular receptor, uPAR (Figure 1A). [35] This receptor is highly expressed in tumor, angiogenic endothelial, and stromal cells in many types of human cancers. [36–38] Previously, we demonstrated target specificity and sensitivity of uPAR-targeted nanoparticles in optical and MR imaging of breast and pancreatic cancers in animal tumor models. [39,40] Since the binding of ATF conjugated nanoparticles to uPAR leads to the internalization of the nanoparticles, we proposed that endocytosis of the nanoparticle-DNA-nanocassettes into the endosomes results in the cleavage of the amide bond between nanoparticles and DNA cassettes and release of DNA-cassettes into cytoplasm. After DNA cassettes enter into the cell nucleus, interactions of cellular transcriptional factors with the U6 promoter of the DNA-nanocassettes activate transcription of shRNA genes, which are then processed into double-stranded siRNAs for targeted gene silencing (Figure 1B).
Figure 1.

Schematic illustration of the design of siRNA-nanogenerators and mechanism of targeted delivery and production of siRNAs in cells A) Production of uPAR-targeted nanoparticles for delivery of siRNA-expressing DNA nanocassettes. B) Proposed mechanism of internalization of the nanoparticles and expression of siRNAs from the nanocassettes.
2. Results
2.1. Development and Characterization of Receptor-Targeted Nanoparticles Carrying siRNA-Expressing DNA Nanocassettes
To develop siRNAs as therapeutic agents for clinical applications, it is necessary to use simple, consistent, and low cost methods to produce large amounts of siRNAs for in vivo delivery. Our strategy is to use receptor-targeted nanoparticles for delivering DNA nanocassettes containing the U6 promoter and shRNA gene that are able to express siRNAs in cells. To enable the production of large amounts of siRNA expressing DNA cassettes by PCR amplification, we engineered shRNA expression plasmids by cloning chemically synthesized double stranded oligonucleotides containing a shRNA gene into a shRNA expression plasmid (p-Silencer 2.1-U6 Neo plasmid) as PCR templates (Figure 1A). Two pairs of universal PCR primers were used to amplify 550 or 750 bp double stranded DNA fragments containing the 5′-flank region of the plasmid sequence immediately before the U6 promoter, a U6 promoter, a shRNA gene, and 3′-flank region of the plasmid immediately after the shRNA gene (Figure 1A). Our results showed that 550 or 750 bp siRNA expressing DNA nanocassettes were amplified by PCR (Figure 2A). These double stranded DNA fragments have molecular weights ranging around 300 to 400 kDa and sizes of several nanometers.
Figure 2.

Characterization of targeted nanoparticles carrying siRNA expressing DNA nanocassettes. A) Gel electrophoresis analysis of PCR products. The gel picture shows PCR products of Survivin siRNA (750 bp), control siRNA (750 bp), luciferase siRNA (550 bp) and control siRNA (550 bp) expressing DNA cassettes. B) Gel electrophoresis of QD-DNA nanocassettes. Red QD (emission wavelength of 620 nm) and green DNA fluorescent signals were detected on the gel. Left: red QD; Middle: DNA green fluorescence; Right: overlaying QD with DNA–staining image. QDs with DNA nanocassettes showed orange color. C) Nanoparticle size measurements by dynamic light scattering.
uPAR-targeted nanoparticles carrying siRNA expressing DNA cassettes were then produced by conjugating 17 kDa human ATF (hATF) peptides and DNA nanocassettes to amphiphilic polymer coated QDs or magnetic IONPs (Figure 1A). We determined conjugation of hATF and DNA nanocassettes to QDs using gel electrophoresis (Figure 2B). Our results showed that conjugation of DNA nanocassettes to QDs did not significantly affect the movement of the QDs in the gel, which may be due to negatively charged DNAs. QDs conjugated with hATF peptides moved much slower compared with QDs alone or QDs-DNA nanocassettes. However, QDs conjugated with both hATF and DNA nanocassettes moved faster than hATF-QDs but slower than QDs alone or QDs-DNA-nanocassettes (Figure 2B). Next, we examined the size distribution of various nanoparticles using a dynamic light scattering measurement. As shown in Figure 2C, the mean particle size of hATF-QDs is 25 ± 8 nm. Conjugation of ten siRNA DNA nanocassettes increased the particle size to 56 ± 15 nm.
2.2. Determination of the Ability of Gene Expression from Double-Stranded DNA Gene-Expressing Nanocassettes
First, we determined the feasibility and efficiency of gene expression from the DNA-cassettes after being delivered into cells by transfecting cancer cells with 1.8 kb DNA cassettes containing a cytomegalovirus (CMV) promoter and an enhanced green fluorescent protein (EGFP) gene (Supporting Information). We then examined whether the DNA-cassettes are capable of producing siRNAs to silence gene expression in cells. U6-eGFP siRNA expressing DNA cassettes were co-transfected with p-EGFP-c3 plasmids into human lung cancer H1299 or breast cancer MCF-7 cells for 48 h. We found that co-transfection of the U6-eGFP siRNA cassettes markedly inhibited the level of EGFP gene expression in both cell lines (Figure 3A). This result was further confirmed by using a GFP gene stable MCF-7 cell line, which showed that transfection of U6-eGFP siRNA nanocassettes silenced the expression of an endogenously transduced EGFP gene (Figure 3B).
Figure 3.

Determination of gene silencing efficiency using siRNA gene expressing cassettes. A) Determination of siRNA expression from the U6-siRNA DNA cassettes by co-transfection. Upper two panels: H1299 and MCF-7 cancer cells were transfected with p-EGFP-c3, without or with co-transfection with U6-eGFP siRNA or U6-control siRNA expressing nanocassettes for 48 h. B) MCF-7 GFP gene stable cells were transfected with U6-eGFP siRNA or U6-Control siRNA nanocassettes for 48 h.
2.3. Examination of Targeted Delivery of siRNA-Expressing DNA Nanocassettes and Gene-Silencing Efficiency
To further quantify gene silencing efficiency in tumor cells in vitro and the detection of dynamic changes in the inhibition of gene expression in animal tumor models in vivo using bioluminescence imaging, we amplified U6-luciferase (Luc) siRNA expressing nanocassettes from p-Silencer-firefly luciferase shRNA plasmid. U6-Luc siRNA nanocassettes were then conjugated to QDs with human ATF targeting ligands. First, we found that hATF-QDs-Luc siRNA, but not non-targeted QD-Luc siRNA cassettes, could efficiently enter into MCF-10DCIS human breast cancer cells. It has similar intracellular delivery efficiency compared to the SV40-nuclear localization signal (NLS) peptide conjugated-QD-Luc siRNA cassettes (Figure 4A). uPAR-targeted internalization of QD-Luc siRNA led to a decrease in luciferase activity in those cells. The inhibitory effect was enhanced as the copy number of Luc siRNA nanocassettes on each QD increased from 2 to 10 or 20 (Figure 4B). As a positive control, p-Silencer Luc shRNA plasmids were transfected into the cells and inhibition of luciferase activity was detected in those cells (Figure 4B).
Figure 4.

uPAR-targeted delivery and gene silencing efficiency of luciferase siRNA-expressing DNA nanocassettes in human tumor cell lines. A) Targeted delivery of Luc siRNA expressing DNA cassettes into human cancer cells. B) Efficiency of gene silencing in cancer cells. Luciferase activity in firefly luciferase gene stably transfected human breast cancer MCF-10DCIS cell lysates was measured at 24 h following nanoparticle incubation. Luciferase units from the no treatment cell lysate serves as 100%. C) Comparison of gene silencing efficiency of delivery of unconjugated RNA-based siRNAs with targeted nanoparticles carrying siRNA expressing DNA nanocassettes. Cells were cultured in 96-well plates and then incubated with 20 pmol of luciferase siRNA (Invitrogen) or an equal molar DNA concentration of ATF-IONP-Luc siRNA expressing cassettes. Luciferase activity in the wells was measured 48 h following the incubation using the Xenogen IVIS system. Luciferase activity of untreated cells serves as 100%.
Next, we determined whether targeted delivery of multiple copies of siRNA-generating nanocassettes on a single nanoparticle has a higher efficiency of knocking-down gene expression compared with direct delivery of RNA-based siRNAs. Unconjugated double stranded RNA Luc siRNAs or siRNA-expressing DNA cassettes that were conjugated to magnetic IONPs were delivered into luciferase positive MIA PaCa-2 pancreatic cancer cells. Our result showed that the tumor cells incubated with 20 pmol of DNA equivalent concentration of hATF-Luc siRNA-IONPs for 48 h had 65% reduction in the luciferase activity while transfecting 20 pmol of RNA-based Luc siRNAs decreased the luciferase activity by 39% in the cells (Figure 4C).
One of the major obstacles in applying siRNA technology to cancer therapy is the low delivery and gene silencing efficiencies in the tumor following systemic administration. To determine the gene silencing efficiency of uPAR-targeted siRNA expressing nanocassettes, hATF-QD-Luc siRNA nanocassettes were injected via the tail vein into nude mice bearing orthotopic luciferase positive MCF-10DCIS tumors. Non-invasive bioluminescence imaging was used at different time points to determine changes in the luciferase activity in the tumors. 24 h after the injection, we found that the level of luciferase activity decreased by 42% in the mice that received hATF-QD-Luc siRNA nanocassettes. The inhibitory effect was further enhanced at 72 h and lasted over 120 h after the injection. Although luciferase activity returned to the preinjection level at 140 h, it is likely that the inhibitory effect was retained in the tumor since the growth of the tumor volume should have significantly increased the luciferase activity, as was shown in the mouse found with 160% increase in luciferase activity at 140 h after receiving non-targeted QD-Luc siRNA nanocassettes (Figure 5A). The mouse that received non-targeted QD-Luc siRNA nanocassettes showed increases in luciferase activity at all time points following system delivery (Figure 5A).
Figure 5.
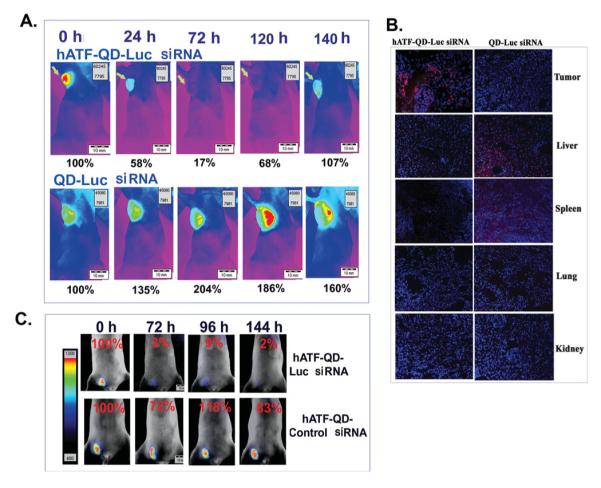
Targeted delivery of QDs carrying Luc siRNA-expressing DNA nanocassettes silenced the gene expression in human breast cancer xenografts in nude mice. A) Luciferase activity in nude mice bearing MCF-10DCIS human breast tumor xenografts that received hATF-QD-Luc siRNA nanocassettes or non-targeted QD-Luc siRNA nanocassettes. Scale bar: upper right. Same scale was used for all images. Arrows: an orthotopic tumor in the mammary fat pad. B) Examination of biodistribution of QDs carrying siRNA expressing cassettes in frozen tissue sections of tumor and normal organs collected from the mice received a tail vein injection of the nanoparticles for 140 h. Red: QD signal (Em 620 nm). Blue: DAPI nuclear counterstaining. C) Validation of specific gene silencing effect after systemic delivery of hATF-QD-Luc siRNA or control scrambled siRNA-nanocassettes. Bioluminescence images were overlaid with bright-field images of the mice. Numbers in the figure show changes in the percentages of luciferase activity compared with the level in the tumor before the nanoparticle injection. Similar results were observed in three repeat mice in each group.
Selective delivery of the targeted nanoparticles into the tumors was further confirmed by histological analysis of the tumor and normal tissues collected from the mice following systemic delivery of 200 pmol of the targeted or non-targeted QDs (Figure 5B) and by ex vivo optical imaging of tumor and normal organs (Supporting Information). In the mice that received uPAR-targeted QDs-carrying Luc siRNA nanocassettes, strong red QD signals were found in the tumors (Figure 5B and Supporting Information). However, QD signal was not found in the mice that received non-targeted QD-Luc siRNAs (Figure 5B). Furthermore, the levels of QD accumulation in the liver and spleen of the mice that received the targeted QD-Luc siRNA cassettes were markedly decreased compared with those of the mice injected with non-targeted QDs (Figure 5B). QD signal was not detected in the lung, kidney and heart in the mice that received either targeted or non-targeted nanoparticles (Figure 5B and Supporting Information).
To determine whether the changes in luciferase activity in the tumor were the result of specific silencing of the luciferase gene, mice bearing MCF-10DCIS tumors in the low abdominal mammary gland received a tail vein injection of either hATF-QD-Luc siRNA or hATF-QD-control siRNA nanocassettes. Consistent with the above observation, a significant decrease in luciferase activity (> 90%) was detected in the tumor at 72 h and the inhibitory effect was still strong at 144 h following the injection (Figure 5C). However, only a moderate decrease in luciferase activity (17 to 28%) was detected in the tumors of mice receiving hATF-QD control siRNA nanocassettes (Figure 5C). Therefore, the effect of downregulation of luciferase gene expression is likely due to the targeted delivery of luciferase siRNA-expressing nanocassettes into the tumor.
2.4. Effect of Targeted Delivery of Survivin siRNA Nanocassettes Using Nanoparticles on Cell Death Induction and Drug Sensitivity in Human Cancer Cells
Taking advantages of the specific gene knock-down function of siRNA-expressing DNA nanocassettes, we produced uPAR-targeted QDs carrying survivin siRNA expressing nanocassettes and found a high efficiency of intracellular nanoparticle delivery in MD-MB-231 breast cancer cells (Figure 6A). Survivin is an anti-cell death gene that confers resistance of cancer cells to therapeutic agents. [41–43] Western blot analysis revealed that the level of survivin proteins was markedly down-regulated in cells transfected with survivin siRNA expressing nanocassettes (Figure 6B). In cultured cells, the targeted gene knock-down effect by hATF-QD Survivin siRNA expressing DNA nanocassettes was similar as that of SV40-NLS-mediated internalization of the QD-Survivin siRNA nanocassettes (Figure 6B). However, SV40-NLS-QD-siRNA nanocassettes could not be used for in vivo delivery due to the lack of specificity. Our result further showed that inhibition of survivin expression led to the activation of the apoptotic cell death since a high level of active caspase 3 (17 KDa fragments) was detected in those cell groups by Western blot analysis (Figure 6B).
Figure 6.

Targeted silencing of survivin gene expression in human tumor cells A) Internalization of hATF-QD-Survivin siRNA expressing DNA cassettes by MDA-MB-231 breast cancer cells. B) Western blot analysis. Following treatment, cell lysates were then collected for Western blot analysis using antibodies against survivin or caspase 3. Activation of caspase 3 is shown as the detection of the cleaved caspase 3 fragments (low molecular weight bands at 17 kDa). C) Effect of nanoparticle delivery of Survivin-siRNA nanocassettes. Left panel: Fluorescent images show internalization of hATF-QD-Survivin siRNA nanocassettes in MIA PaCa-2 pancreatic cancer cells. Red: QD signal. Right panel: Crystal violet cell proliferation assay 2 days following treatment. O.D. value of no treatment group was used as 100%.
We further confirmed targeted internalization of hATF-QD-Survivin siRNA expressing nanocassettes in the MIA PaCa-2 cell line (Figure 6C). Treatment of the pancreatic cancer cells with hATF-QD-Survivin siRNA expressing nanocassettes for 2 days induced cell death and significantly reduced the percentage of viable cells (Figure 6C). However, there was no significant change in the percentage of viable cells that received QDs only, non-targeted QD-Survivin siRNA nanocassettes, or hATF-QD-Control siRNA nanocassettes (Figure 6C).
Finally, we examined the effect of survivin gene silencing using hATF-QD-Survivin siRNA expressing nanocassettes on cell sensitivity to chemotherapy drugs. In this study, MIA PaCa-2 cancer cells were treated concomitantly with a chemotherapy drug, gemcitabine, and hATF-QD-Survivin siRNA or hATF-QD-Control siRNA expressing nanocassettes. Pancreatic cancer cells have a low sensitivity to free gemcitabine treatment and 60% of viable cells were detected by cell proliferation assay 72 h following 100 μ M of gemcitabine treatment (Figure 7A). However, co-treatment of gemcitabine with hATF-QD-Survivin siRNA expressing nanocassettes significantly enhanced the inhibitory effect on tumor cell growth at all drug concentrations (p < 0.0005, student’s-t test). For example, the combination of knocking-down survivin gene with 1 μM of gemcitabine treatment significantly decreased the percentage of viable cells from 94% in free drug treated group to 18% in cells that received combination therapy (Figure 7A). We further demonstrated that the enhanced effect on tumor cell growth inhibition is due to the activation of the apoptotic cell death pathway since a high caspase 3 activity was detected in cells treated with the combination of gemcitabine with hATF-QD-Survivin siRNA expressing nanocassettes (Figure 7B).
Figure 7.
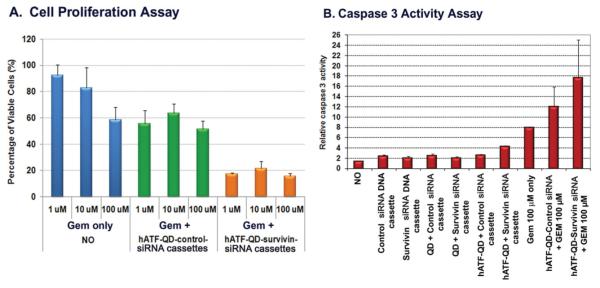
Combination effect of survivin gene silencing and drug treatment A) Cell proliferation assay. The O.D. value of no treatment cells was used as 100%. Student’s t-test: Gem only vs. Gem + hATF-QD-Survivin siRNA expressing cassettes, p < 0.0005 for all three concentrations. B) Caspase 3 activity assay. O.D. value from the non treated cell lysate was used as 1.
3. Discussion
Development of novel approaches for efficient delivery of siRNAs into tumors has been an intensive research area in cancer nanomedicine. However, challenges for applications of this strategy for developing effective cancer therapeutic agents, such as in vivo stability, intra-tumoral delivery and gene silencing efficiencies in the tumor cells, remain to be addressed. The design of the siRNA nanoparticle delivery platform reported in this study has important features to address those issues. First, this is a multifunctional siRNA delivery nanoparticle system with imaging capability and targeted delivery of siRNA-expressing DNA cassettes. End-protected double stranded DNA fragments produced by PCR amplification are nanometer-sized DNA cassettes that have a high stability in the blood and tissues. Nanoparticle conjugated DNA cassettes should have longer half-life in the blood circulation, compared with the delivery of unconjugated siRNAs or nanocassettes alone.
To solve the problem of a low delivery efficiency of siRNAs into cells, we used uPAR-targeted nanoparticles to bring the siRNA expressing nanocassettes into tumors as well as inside tumor cells. Our result showed that the uPAR-targeted nanoparticles play an important role in efficient delivery of siRNA expressing DNA nanocassettes into tumor cells since a very low level QD was found in the cells and tumors from the mice treated with non-targeted nanoparticle-siRNA nanocassettes. In tumor-bearing mice, a marked inhibition of luciferase activity was only detected in the tumors of the mice following systemic delivery of uPAR-targeted nanoparticles carrying Luc siRNA cassettes, but not in the tumors of the mice that received non-targeted nanoparticles-Luc siRNA cassettes. It is likely that an increase in tumor accumulation and receptor-mediated internalization of the nanoparticles into cells contribute to the significant inhibition of luciferase gene expression in the tumor xenografts in nude mice.
Additionally, the ability of delivery of over ten siRNA expressing DNA nanocassettes by a single nanoparticle and expression of multiple copies siRNAs from each DNA cassette further enhanced the efficiency of knocking-down gene expression. Using EGFP siRNA as a model system, we demonstrated the ability and efficient of the DNA nanocassettes in expression of a shRNA gene after being delivered into cells. At present, the precise mechanisms of release of the DNA cassettes from the endosomes and intracellular transportation into the cell nucleus for shRNA gene transcription have yet to be elucidated. It is possible that the DNA nanocassettes can be released from the nanoparticles by cleaving the amide bond between the nanoparticles and DNA cassettes or by degradation of polymer-coating in the endosomes or lysosomes. These small DNA fragments may pass through the endosomal membrane. Additionally, conjugation of the DNA fragments at the 3′-end of the expressing cassettes makes it possible to express the shRNA gene from the nanoparticle-conjugated nanocassettes after endosomal escape of the nanoparticles. Investigations into these mechanisms are currently underway.
The advantage of intracellular expression of siRNAs from the DNA cassettes has also been shown in vitro in tumor cells that received an equal molar concentration of the DNA cassettes delivered by the nanoparticles or RNA-based siRNAs. Furthermore, strong and targeted gene silencing effects following delivery of luciferase siRNA expressing nanocassettes using the receptor-targeted nanoparticles have been shown in a human breast cancer xenograft model in nude mice. Importantly, the combination of targeted delivery and in vivo expression of siRNAs enabled significant inhibition of the level of luciferase gene expression in the tumors for over 6 days. Therefore, targeted delivery of the U6 promoter-siRNA expressing DNA nanocassettes using nanoparticles is a promising approach for the development of siRNA therapeutics with increased delivery efficiency as well as enhanced the effectiveness and duration of gene silencing in cancer cells.
Drug resistance is the major challenge in cancer treatment. Increasing evidence shows that cell survival pathways, especially inhibition of proteins in the apoptotic family, such as survivin, confer apoptosis or drug resistance in cancer cells. [41–43] Survivin is highly expressed in many human cancer types and can interact with other proteins to block apoptosis. [44] Several types of nanoparticles carrying RNA-based survivin siRNAs have been developed and examined for their effects on inhibition of survivin gene expression. [2,45] In this study, we produced survivin siRNA expressing DNA nanocassettes and showed that uPAR-targeted delivery of the nanocassettes inhibited survivin gene expression and led to the activation of apoptotic cell death in human cancer cells.
Since cancer cells develop various mechanisms to resist cell death, the combination of activation of the apoptotic pathway by drug treatment with inhibition of anti-apoptotic factors, such as survivin, using siRNAs may produce more potent anti-tumor effects on drug resistant tumor cells. Results of our study demonstrated that the combination of a chemotherapy drug, gemcitabine, with survivin siRNA nanocassette delivery significantly enhanced the sensitivity of human pancreatic cancer cells to the drug treatment. For future development as cancer therapeutics, it is feasible to produce multifunctional nanoparticles carrying both siRNA expressing nanocassettes and chemotherapy drugs for effective cancer therapy. Although QDs were used as a nanoparticle delivery system to conduct the proof of concept studies in cells and mice, we have also developed magnetic IONPs carrying siRNA expressing nanocassettes that have potential for the development of theranostic siRNA nanoparticles for targeted therapy and non-invasive imaging of the therapeutic response in human patients.
4. Conclusion
In summary, we have developed a new approach for targeted delivery and expression of siRNAs in vivo using DNA-based siRNA-expressing nanocassettes and receptor-targeted nanoparticles. We demonstrated the specificity of delivery and gene silencing efficiency of this siRNA delivery system in cell lines and in an animal tumor model. Our results showed that targeted delivery of survivin siRNA expressing nanocassettes enhanced sensitivity of human cancer cells to chemotherapeutic drugs. The advantages of this DNA-based siRNA generating nanoparticle delivery system include: 1) highly stable and small size DNA fragments containing both the promoter and gene sequences for expressing siRNAs inside cells; 2) the receptor targeted nanoparticle carrier that allows efficient delivery into target tissues as well as intracellular delivery; 3) the nanoparticles are also imaging probes that enable non-invasive imaging of siRNA nanocassette delivery and tumor response to therapy; and 4) the capability of the nanoparticle carrier for simultaneous delivery of therapeutic agents that activate cell death (e.g. chemotherapeutic drugs) and inhibit cell survival pathways (siRNAs) may enhance the effectiveness of cancer therapy.
5. Experimental Section
Human Cancer Cell Lines
H1299 human lung, MCF-7 and MDA-MB-231 human breast, and MIA PaCa-2 human pancreatic cancer cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in the medium as suggested by the ATCC. The MCF-10DCIS (or MCF-10DCIS.com) human breast cancer cell line was obtained from Dr. Fred Miller at the Barbara Ann Karmanos Cancer Institute (Detroit, MI) [46] and cultured in the DMEM/F12 medium supplemented with 5% horse serum. The dual firefly luciferase and enhanced green fluorescence protein (eGFP) gene stable MCF-10DCIS cell line was produced by transducing cells with a lentiviral vector, LV-pUB-Fluc-eGFP. [45] Firefly luciferase gene stable MIA PaCa-2 human pancreatic cancer cell line was kindly provided by Dr. Rosa Hwang, MD Anderson Cancer Center, Houston, TX.
Engineering siRNA Expressing Plasmids
The shRNA expressing plasmids were generated by cloning 69 to 77 nt of chemically synthesized double stranded oligonucleotides (Sigma Aldrich, St. Louis, MO) containing the following structure: 5′-GGATCC (Bam H1)-19 to 23 nt shRNA sense sequences-TTCAAGAGA (Loop sequence)-19 to 20 shRNA antisense sequences-TTTTTTGGAAA (Terminate sequence)-AAGCT (Hind III), into Bam H1-Hind III cloning site of p-Silencer 2.1-U6 Neo plasmid (Ambion/Invitrogen, Grand Island, NY). The following are siRNA sense sequences used for this study: 1). Random control: 5′-AAGAGGCTTGCAACAGTGCA-3′; 2) Survivin: 5′-GAGGCTGGCTTCATCCACTGCCC-3′; 3) Firefly luciferase: 5′-CGGATTACCAGGGATTTCA-3′; and 4) Enhanced green fluorescence protein (EGFP) 5′-CAAGCTGACCCTGAAGTTC-3′. After demonstrating their effects on the downregulation of gene expression in tumor cell lines following transfection, these p-Silencer shRNA plasmids were used as templates for PCR amplification of the DNA cassettes (Figure 1A).
PCR Amplification of siRNA-Expressing DNA Cassettes
Two pairs of universal PCR primers were used to amplify the double stranded DNA cassettes from the p-Silencer shRNA plasmids. The PCR primer pair for the small cassette was: 5′-GATGTGCTGCAAGGCGATTA-3′ (Forward) and 5′-AGTGAGCGCAACGCAATT-3′ (Reverse). The primer pair for the large cassette was: 5′-AACTGTTGGGAAGGGCGA-3′ (Forward) and 5′-AGTGAGCGCAACGCAATT-3′ (Reverse). All PCR primers were made by Integrated DNA Technology, Coralville, Iowa. Both reverse primers were modified at the 5′ end with an amine group for conjugation to a nanoparticle. To protect the DNA cassettes from digestion by deoxyribonucleases, the reverse primers were also modified at the 5′ end with a phosphorothioate linkage (Figure 1A). Using p-Silencer shRNA plasmids as templates and above PCR primer pairs, 550 bp or 750 bp of firefly luciferase, survivin, GFP, and control siRNA expressing DNA cassettes were amplified using the following PCR conditions: 95 °C for 5 min; 94 °C for 30 s, 55 °C for 30 s, 72 °C for 90 s, 30 cycles; and 72 °C, 8 min. PCR samples were then ethanol precipitated and re-suspended in H2O. DNA fragments were purified from the gel using the QIAEX II Agarose Gel Extraction Kit (Qiagen, Valencia, CA).
Production of uPAR-Targeted Nanoparticles Carrying siRNA Expressing DNA Nanocassettes
The recombinant amino terminal fragment (ATF, 135 aa) of human uPA (17 kDa) was produced from E. coli BL21 bacterial expression system using a pET20a plasmid (Invitrogen, Grand Island, NY) containing the ATF cDNA sequence as described previously. [40] Recombinant human ATF (hATF) was purified from bacterial extracts using a Ni2+ NTA-agarose column (Qiagen) and our established protocol. [40]
uPAR-targeted nanoparticles carrying siRNA expressing DNA cassettes were produced by two steps. First, hATF peptides were conjugated to amphiphilic polymer coated quantum dots (QDs, emission wavelength 620 nm, Ocean Nanotech, LLC, Springdale, AR) or magnetic iron oxide nanoparticles (IONPs, 10 nm core size, Ocean Nanotech, LLC) at a molar ratio of 1 nanoparticle to 10 hATF by forming an amide bond between the amine group of hATF and the carboxyl group of the amphiphilic polymer, mediated by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) and sulfo-NHS (Pierce, Rockford, IL). Nanoconjugates were purified using the Nanosep 100 k column (Pall Corp, Ann Arbor, MI). Purified PCR fragments were then conjugated to hATF-nanoparticles at a ratio of 1 nanoparticle:10 or 20 DNA cassettes by EDAC mediated conjugation of the 3′-terminal amine group of the DNA fragments with the carboxyl group of the polymer coating (Figure 1A). Conjugated nanoparticles were purified using a magnet sorter (Ocean Nanotech, LLC) at 4 °C overnight. After conjugation, the nanoparticles were diluted and examined for particle size distribution using a Zetasizer Nano (Malvern Instruments Inc., Southborough, MA). Conjugation of DNA cassettes and ATF peptides to QDs was examined by electrophoresis in 0.8% agarose gel for 1 h. The gel was stained with SYBR green DNA dye, and examined by the Olympus OV-100 imaging system (Olympus America Inc., Central Valley, PA).
Cell Transfection
Cells were co-transfected with pEGFP-c3 plasmids and U6-eGFP siRNA or U6-control siRNA expressing DNA nanocassettes using Lipofectamine 2000 Reagent (Invitrogen). MCF-7 GFP gene stable cells, kindly provided by Dr. Adam Marcus at Emory University, were transfected with U6-eGFP siRNA or U6-Control siRNA expressing cassettes for 48 h and then examined under an inverted fluorescence microscope. 3 μg of DNA or DNA equivalent amount of QDs were used for above studies.
Determination of Target Specificity of hATF-Conjugated Nanoparticles: Cells were plated in 24-well culture plates for 24 h and then incubated with various nanoparticles at an equal molar concentration (15 pmol of QDs) for 24 h. Control unconjugated DNA cassettes or plasmids were transfected into cells. As a delivery efficiency control, siRNA expressing nanocassettes were also conjugated to QDs with the NLS peptides from SV40 virus, CGGGPKKKRKVE. The peptide sequence was provided by Dr. Steven Dowdy at University of California, San Diego. The SV-40-NLS peptides were synthesized by Genscript USA Inc. (Piscataway, NJ). Culture plates were then examined using an inverted Olympus fluorescence microscope.
Luciferase Activity Assay
Following different treatments, cells were collected from culture plates. Cell lysates were examined for luciferase activity using the Single Luciferase Assay System (Promega Corp., Madison, WI). The level of the luciferase activity was measured by a luminometer (Lumistar galaxy, BMG, Winooski, VM) for single samples or using the Xenogen IVIS system (Caliper Life Sciences, Mountain View, CA) for 96-well plates.
Western Blot Analysis
Cells were incubated with various QD-hATF/survivin-siRNA expressing DNA nanocassettes or non-targeted QD-survivin siRNA expressing DNA nanocassettes for 48 h. Control siRNA and survivin siRNA DNA nanocassettes were transfected into MDA-MB-231 cells. After treatment, cells were lysed with cell lysis buffer. [41] A total of 30-50 μg of the proteins were resolved on 12% polyacrylamide-SDS gels and then transferred to PVDF membranes (Bio-Rad laboratories, Hercules, CA). The membranes were blocked with 5% nonfat milk in Tris-buffered saline for 1 h, and incubated overnight with primary antibodies for survivin, caspase-3, and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA). After three washes, the membranes were incubated with anti-goat, anti-rabbit, or anti-mouse secondary antibodies conjugated with horseradish-peroxidase (Santa Cruz Biotechnology) for 1 h. The levels of specific proteins in each lysate were detected by enhanced chemiluminescence using ECL plus (Amersham International, Buckingham, UK) followed by autoradiography.
Cell Proliferation Assay
Crystal violet assay was used to determine the percentage of viable cells. Cells were plated in 96-well culture plates for 24 h and then treated with various nanoparticles or transfected with survivin siRNA expressing DNA cassettes for 48 h. To detect drug sensitivity, MIA PaCa-2 cells were treated with gemcitabine (Eli Lilly Co., Indianapolis, IN, USA) without or with nanoparticles carrying survivin siRNA expressing DNA cassettes for 48 h. Cells were fixed with 4% paraformaldehyde in PBS and then stained with crystal violet. Percentage of viable cells in the experimental groups was determined by measurements O.D. at 590 nm using Spectra Max Plus (Molecular Devices, Sunnyvale, CA).
Caspase 3 Activity Assay
MIA PaCa-2 cells were treated with various nanoparticles in the presence or absence of gemcitabine for 48 h. Cell lysates were analyzed for their levels of caspase 3-like activity, which is generated by caspases 3, 7, and 10, using an Ac-DEVD-AFC substrate (Calbiochem, San Diego, CA). Measurements were made using a fluorescence microplate reader (Spectra Max Gemini xs, Molecular Devices) at an excitation wavelength of 408 nm and an emission wavelength of 500 nm.
Detection of Targeted Delivery and Gene Silencing Effects in an Animal Tumor Model
An orthotopic human breast cancer xenograft model was established by injecting 1 × 107 of dual firefly luciferase and GFP positive MCF-10DCIS cells into the mammary fat pad of the nude mice. The tumor bearing mice then received 200 pmol of QDs carrying luciferase siRNA-expressing DNA cassettes (~2 nmol of DNA cassettes) by tail vein injection. Before and at different time points following the nanoparticle administration, 2 mg/Kg of luciferin substrate was injected intraperitoneally into the mice for 5 min before each bioluminescence imaging procedure using identical imaging conditions and an Olympus OV-100 small animal imaging system (Olympus America Inc.).
Supplementary Material
Acknowledgements
We thank Dr. Fengzhi Li for providing the survivin shRNA sequence, Dr. Steven Dowdy for information on the SV40 nuclear localization sequence, Dr. Rosa Hwang for luciferase gene stable MIA PaCa-2 cell line, and Dr. Adam Marcus for the MCF-7 GFP stable cell line and for his assistance with the Olympus OV-100 imaging system at the Emory Integrated Cell Imaging Core. This research project was supported by the following NIH grants: U01CA151810 (Yang and Mao), R01CA154129A01 (Yang), U54 CA119338 (Nie), and P50CA128613 (Shin). Dr. Young-Seok Cho was supported by a Songeui Scholar Research Grant funded by the Catholic University of Korea. Dr. Lily Yang is the Nancy Panoz Chair of Surgery in Cancer Research.
Footnotes
Supporting Information Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Y.-S. Cho, Emory University School of Medicine The Catholic University of Korea, Atlanta, GA 30322, USA Uijeongbu, 480717, Korea.
Dr. G. Y. Lee, Emory University School of Medicine The Catholic University of Korea, Atlanta, GA 30322, USA Uijeongbu, 480717, Korea.
Dr. H. K. Sajja, Emory University School of Medicine The Catholic University of Korea, Atlanta, GA 30322, USA Uijeongbu, 480717, Korea
Dr. W. Qian, Emory University School of Medicine The Catholic University of Korea, Atlanta, GA 30322, USA Uijeongbu, 480717, Korea
Z. Cao, Emory University School of Medicine The Catholic University of Korea, Atlanta, GA 30322, USA Uijeongbu, 480717, Korea
Dr. W. He, First Affiliated Hospital Sun Yat-Sen University Guangzhou, Guangdong 510180, PR China
Dr. P. Karna, Emory University School of Medicine The Catholic University of Korea, Atlanta, GA 30322, USA Uijeongbu, 480717, Korea
Dr. X. Chen, National Institute of Biomedical Imaging and Bioengineering Bethesda, MD 20892, USA
Dr. H. Mao, Emory University School of Medicine The Catholic University of Korea, Atlanta, GA 30322, USA Uijeongbu, 480717, Korea
Dr. Y. A. Wang, Ocean Nanotech, LLC Springdale, AR 72764, USA
Dr. L. Yang, Department of Surgery National Institute of Biomedical Imaging Emory University School of Medicine and Bioengineering Clinic C, Room C-4088, 1365 C Clifton Road, Bethesda, MD 20892, USA NE, Atlanta, GA 30322, USA.
References
- 1.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Medarova Z, Pham W, Farrar C, Petkova V, Moore A. Nat. Med. 2007;13:372–377. doi: 10.1038/nm1486. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg MS, Xing D, Ren Y, Orsulic S, Bhatia SN, Sharp PA. Proc. Natl. Acad. Sci. USA. 2011;108:745–750. doi: 10.1073/pnas.1016538108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pirollo KF, Chang EH. Cancer Res. 2008;68:1247–1250. doi: 10.1158/0008-5472.CAN-07-5810. [DOI] [PubMed] [Google Scholar]
- 5.Ren Y, Cheung HW, von Maltzhan G, Agrawal A, Cowley GS, Weir BA, Boehm JS, Tamayo P, Karst AM, Liu JF, Hirsch MS, Mesirov JP, Drapkin R, Root DE, Lo J, Fogal V, Ruoslahti E, Hahn WC, Bhatia SN. Sci. Transl. Med. 2012;4:147ra112. doi: 10.1126/scitranslmed.3003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, Li J, Liu F, Huang L. Mol. Ther. 2012;20:609–615. doi: 10.1038/mt.2011.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T, He H. Small. 2009;5:2673–2677. doi: 10.1002/smll.200900621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shu D, Shu Y, Haque F, Abdelmawla S, Guo P. Nat. Nanotechnol. 2011;6:658–667. doi: 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hannon GJ. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 10.Burnett JC, Rossi JJ, Tiemann K. Biotechnol. J. 2011;6:1130–1146. doi: 10.1002/biot.201100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao S, Dagnaes-Hansen F, Nielsen EJ, Wengel J, Besenbacher F, Howard KA, Kjems J. Mol. Ther. 2009;17:1225–1233. doi: 10.1038/mt.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartlett DW, Davis ME. Biotechnol. Bioeng. 2007;97:909–921. doi: 10.1002/bit.21285. [DOI] [PubMed] [Google Scholar]
- 13.Yezhelyev MV, Qi L, O’Regan RM, Nie S, Gao X. J. Am. Chem. Soc. 2008;130:9006–9012. doi: 10.1021/ja800086u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel PC, Hao L, Yeung WS, Mirkin CA. Mol. Pharm. 2011;8:1285–1291. doi: 10.1021/mp200084y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elmen J, Thonberg H, Ljungberg K, Frieden M, Westergaard M, Xu Y, Wahren B, Liang Z, Orum H, Koch T, Wahlestedt C. Nucleic Acids Res. 2005;33:439–447. doi: 10.1093/nar/gki193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang XL, Xu R, Wu X, Gillespie D, Jensen R, Lu ZR. Mol. Pharm. 2009;6:738–746. doi: 10.1021/mp800192d. [DOI] [PubMed] [Google Scholar]
- 17.Hasan W, Chu K, Gullapalli A, Dunn SS, Enlow EM, Luft JC, Tian S, Napier ME, Pohlhaus PD, Rolland JP, DeSimone JM. Nano Lett. 2012;12:287–292. doi: 10.1021/nl2035354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tassa C, Shaw SY, Weissleder R. Acc. Chem. Res. 2011;44:842–852. doi: 10.1021/ar200084x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwok A, Hart SL. Nanomedicine. 2011;7:210–219. doi: 10.1016/j.nano.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Gao K, Huang L. Mol. Pharm. 2009;6:651–658. doi: 10.1021/mp800134q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu G, Choi KY, Bhirde A, Swierczewska M, Yin J, Lee SW, Park JH, Hong JI, Xie J, Niu G, Kiesewetter DO, Lee S, Chen X. Angew. Chem. Int. Ed. 2012;51:445–449. doi: 10.1002/anie.201105565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haque F, Shu D, Shu Y, Shlyakhtenko LS, Rychahou PG, Evers BM, Guo P. Nano Today. 2012;7:245–257. doi: 10.1016/j.nantod.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Proc. Natl. Acad. Sci. USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelmawla S, Guo S, Zhang L, Pulukuri SM, Patankar P, Conley P, Trebley J, Guo P, Li QX. Mol. Ther. 2011;19:1312–1322. doi: 10.1038/mt.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 26.Miyagishi M, Taira K. Nat. Biotechnol. 2002;20:497–500. doi: 10.1038/nbt0502-497. [DOI] [PubMed] [Google Scholar]
- 27.McAnuff MA, Rettig GR, Rice KG. J. Pharm. Sci. 2007;96:2922–2930. doi: 10.1002/jps.20968. [DOI] [PubMed] [Google Scholar]
- 28.Bot I, Guo J, Van Eck M, Van Santbrink PJ, Groot PH, Hildebrand RB, Seppen J, Van Berkel TJ, Biessen EA. Blood. 2005;106:1147–1153. doi: 10.1182/blood-2004-12-4839. [DOI] [PubMed] [Google Scholar]
- 29.Dunlap DD, Maggi A, Soria MR, Monaco L. Nucleic Acids Res. 1997;25:3095–3101. doi: 10.1093/nar/25.15.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fink TL, Klepcyk PJ, Oette SM, Gedeon CR, Hyatt SL, Kowalczyk TH, Moen RC, Cooper MJ. Gene Ther. 2006;13:1048–1051. doi: 10.1038/sj.gt.3302761. [DOI] [PubMed] [Google Scholar]
- 31.Blessing T, Remy JS, Behr JP. Proc. Natl. Acad. Sci. USA. 1998;95:1427–1431. doi: 10.1073/pnas.95.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W, Popovic Z, Jain RK, Bawendi MG, Fukumura D. Proc. Natl. Acad. Sci. USA. 2011;108:2426–2431. doi: 10.1073/pnas.1018382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castanotto D, Li H, Rossi JJ. RNA. 2002;8:1454–1460. doi: 10.1017/s1355838202021362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gou D, Jin N, Liu L. FEBS Lett. 2003;548:113–118. doi: 10.1016/s0014-5793(03)00630-6. [DOI] [PubMed] [Google Scholar]
- 35.Appella E, Robinson EA, Ullrich SJ, Stoppelli MP, Corti A, Cassani G, Blasi F. J. Biol. Chem. 1987;262:4437–4440. [PubMed] [Google Scholar]
- 36.Nielsen BS, Rank F, Illemann M, Lund LR, Dano K. Int. J. Cancer. 2007;120:2086–2095. doi: 10.1002/ijc.22340. [DOI] [PubMed] [Google Scholar]
- 37.Blasi F, Carmeliet P. Nat. Rev. Mol. Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 38.Pyke C, Graem N, Ralfkiaer E, Ronne E, Hoyerhansen G, Brunner N, Dano K. Cancer Res. 1993;53:1911–1915. [PubMed] [Google Scholar]
- 39.Yang L, Mao H, Cao ZH, Wang YA, Peng XH, Wang XX, Sajja HK, Wang LY, Duan HW, Ni CC, Staley CA, Wood WC, Gao XH, Nie SM. Gastroenterology. 2009;136:1514–1525. doi: 10.1053/j.gastro.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang L, Peng XH, Wang YA, Wang X, Cao Z, Ni C, Karna P, Zhang X, Wood WC, Gao X, Nie S, Mao H. Clinical Cancer Res. 2009;15:4722–4732. doi: 10.1158/1078-0432.CCR-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng XH, Karna P, Cao Z, Jiang BH, Zhou M, Yang L. J. Biol. Chem. 2006;281:25903–25914. doi: 10.1074/jbc.M603414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altieri DC. Nat. Rev. Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 43.Yang L, Cao Z, Yan H, Wood WC. Cancer Res. 2003;63:6815–6824. [PubMed] [Google Scholar]
- 44.Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, Marusawa H, Zou H, Armstrong R, Matsuzawa S, Salvesen GS, Reed JC, Altieri DC. J. Biol. Chem. 2004;279:34087–34090. doi: 10.1074/jbc.C400236200. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Zhu X, Zhang X, Liu B, Huang L. Mol. Ther. 2010;18:1650–1656. doi: 10.1038/mt.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller FR, Santner SJ, Tait L, Dawson PJ. J. Natl. Cancer Inst. 2000;92:1185–1186. doi: 10.1093/jnci/92.14.1185a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.