

Abstract
STAT3 is involved in protection of the heart provided by ischemic preconditioning. However, the role of this transcription factor in the heart in chronic stresses such as hypertension has not been defined. We assessed whether STAT3 is important in hypertension-induced cardiac remodeling using mice with reduced STAT3 activity due to a S727A mutation (SA/SA). Wild type (WT) and SA/SA mice received angiotensin (ANG) II or saline for 17 days. ANG II increased mean arterial and systolic pressure in SA/SA and WT mice, but cardiac levels of cytokines associated with heart failure were increased less in SA/SA mice. Unlike WT mice, hearts of SA/SA mice showed signs of developing systolic dysfunction as evidenced by reduction in ejection fraction and fractional shortening. In the left ventricle of both WT and SA/SA mice, ANG II induced fibrosis. However, fibrosis in SA/SA mice appeared more extensive and was associated with loss of myocytes. Cardiac hypertrophy as indexed by heart to body weight ratio and left ventricular anterior wall dimension during diastole was greater in WT mice. In WT+ANG II mice there was an increase in the mass of individual myofibrils. In contrast, cardiac myocytes of SA/SA+ANG II mice showed a loss in myofibrils and myofibrillar mass density was decreased during ANG II infusion. Our findings reveal that STAT3 transcriptional activity is important for normal cardiac myocyte myofibril morphology. Loss of STAT3 may impair cardiac function in the hypertensive heart due to defective myofibrillar structure and remodeling that may lead to heart failure.
Keywords: Cardiac hypertrophy, cardiac remodeling, heart failure, myofibrils, fibrosis, cytokines
1. Introduction
In response to hypertension, the left ventricle of the heart undergoes hypertrophy, which is initially beneficial in maintaining cardiac function.1 However, with sustained hypertension, cardiac hypertrophy invariably proves detrimental with heart failure the result.1 How cardiac myocytes initially adapt to increased wall stress is poorly understood, as are events responsible for transitioning to heart failure. Conceivably, loss or reduction in the initial adaptive response may contribute to progression of cardiac failure.
We tested the hypothesis that the transcription factor signal transducer and activator of transcription 3 (STAT3) is important in the adaptive response of the heart during development of increased blood pressure in a mouse model of angiotensin II (ANG II)-induced hypertension. Upon activation, STAT3 is phosphorylated on Y705 and undergoes dimerization and translocation to the nucleus where it induces expression of certain genes. Multiple studies have shown STAT3 to be protective to the heart in the acute setting of ischemia, being implicated in both early and delayed phases of preconditioning through up regulation of genes for anti-oxidant and anti-apoptotic proteins (metallothioneins, SOD2, Bcl-xL, c-FLIPs, and Mcl-1), angiogenic factors (VEGF), and inducers of biological mediators (COX2, NOS2, HO-1).2–7 However, the importance of STAT3 as a protective factor to cardiac myocytes in a chronic stress setting has not been elucidated.
Through association with hypertrophic agonists observed on cultured cardiac myocytes and overexpression studies in transgenic mice, STAT3 was initially implicated in cardiac myocyte hypertrophy, but the actual contribution of this transcription factor to cardiac muscle growth in vivo is unsettled.8 Two studies suggest STAT3 may be important for long-term cardiac myocyte viability. STAT3 was recently shown to play a protective role in CVB3-induced myocarditis by preventing excessive fibrosis and progression to dilated cardiomyopathy.9 In addition, with advancing age, mice with cardiac myocyte-restricted postnatal loss of STAT3 develop heart dysfunction characterized by increased left ventricular end diastolic diameter, severe fibrosis, and decreased fractional shortening.10 In our study, we employed mice homozygous for STAT3 S727A (SA/SA), thereby precluding phosphorylation of this residue necessary for full transcriptional activity of STAT3.11 These mice appear phenotypically normal, although IL-6-induced STAT3 transcriptional activity was shown to be reduced to 50% in embryonic fibroblasts of mice homozygous for the S727A mutation.12 The knockout/knockin transgenic model avoids unintended alterations in miRNA homeostasis that theoretically may arise due to loss of a mRNA transcript with the gene knockout approach.13
2. Methods and materials
2.1. Materials
Angiotensin II acetate was from Bachem (Torrance, CA, USA). Cell Signaling Technology (Danvers, MA, USA) was the source for antibodies against STAT3 (#9139), phospho-STAT3 Y707 (#9131), and Bcl-xL (#2764). Antibodies for phosphor-STAT3 S727 (sc-8001-R), SOD2 (sc-137254), and GAPDH (sc-166545/sc-25778) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Secondary antibodies were from LI-COR Biosciences (Lincoln, NE, USA). Cytokines were measured with Elisa kits from R&D Systems (Minneapolis, MN).
2.2. Animals
Surgical procedures and protocols were approved by the Institutional Animal Care and Use Committee of The University of Mississippi Medical Center and comply with NIH Guidelines for Care and Use of Laboratory Animals. The STAT3 S727A knock-in mouse line on the C57 Black 6 background was obtained from Drs. Shen and Levy (The Rockefeller University, NY). The colony was maintained by breeding mice heterozygous for the STAT3 S727A mutation. Female adult (5 – 7 months old) mice homozygous for STAT3 S727A (SA/SA) and wild type (WT) littermates (controls) were used for the study. Animals were maintained on 8640 Teklad 22/5 rodent diet (Harlan Laboratories, Indianapolis, Indiana).
2.3. Experimental Protocol
There were 4 groups of mice: WT+Saline (n = 3), WT+ANG II (1000 ng/kg/min; n = 7), SA/SA+Saline (n = 3), SA/SA+ANG II (1000 ng/kg/min; n = 5). Saline (0.9%) or ANG II was delivered via Alzet miniosmotic pumps (Model 1002) implanted subcutaneously. Animals were treated with ibuprofen (0.1 mg/25g IP) as analgesic. At completion of the study animals were anesthetized with isoflurane. Mice were then perfused with PBS via a peristaltic pump and catheter through the left ventricle. Draining was allowed by a small cut in the liver. Once blood was cleared, the heart was collected and weighed. A small piece of left ventricle was snap-frozen to determine IL-6, TGF-β1, MCP-1, and VEGF. The other part was placed in 10% buffered formalin for histology. A piece of tail was taken to confirm genotype.
2.4. Blood Pressure
Mice were anesthetized with 1.5% isoflurane and given caprofen (1mg/kg IP) as analgesic and atropine sulfate (0.1mg/kg IP) to reduce airway secretions. A telemetric pressure transmitter device (Model TA11PAC-10) from Data Science International (St. Paul, MN, USA) was implanted using sterile techniques. The catheter was inserted through the left carotid artery into the aortic arc and the transmitter secured into a subcutaneous pocket. Mice were allowed to recover in shoebox cages for 24 hours on heating pads and then a total of 10 days prior to baseline recordings. Mean arterial pressure (MAP), systolic blood pressure (SBP), diastolic blood pressure (DBP) and heart rate (HR) were measured 24-hours per day using computerized methods for data collection as described.14 Daily MAP and HR were obtained from the average of 24 hours of recording using a sampling rate of 500 Hz with duration of 10 seconds for every 10–minute period. Following 3 days of stable recordings of SBP, MAP, and HR, mice entered into the study. The pattern of blood pressure change for SA/SA and WT mice was essentially the same between the nighttime and 24 hour record and thus we present our data as the mean of 24 hours. Mice received food and water throughout the study and were placed in a quiet room on a 12-hour light cycle. In a preliminary study, two age-matched mice of each group (WT and SA/SA) were infused with saline for the same period of time (17 days) and no difference in blood pressure was seen by telemetry; thus ruling out an impact of age or strain on blood pressure.
2.5. Echocardiography
Cardiac function was assessed using a VEVO 770 high resolution in vivo imaging system from VisualSonics (Toronto, ON, Canada). Echocardiography was performed twice -prior to pump implantation and one day prior to study completion. Measurements were made with a 707B RMV scanhead with a center frequency of 25 MHz and frequency band ranging from 12.5 to 37.5 MHz. Mice were anesthetized with 1.5% isoflurane and placed on prewarmed EKG transducer pad with heart rate, body temperature, and EKG monitored. Two-dimensional B-mode parasternal long axis views were obtained first to visualize aortic and mitral valves. The transducer was then rotated clockwise 90° to obtain the parasternal short axis view. Ejection fraction (EF), fractional shortening (FS), and left ventricular anterior wall dimension during diastole (LVAWd) were all determined from the M-mode images.
2.6. Histology
Fixed hearts were cut laterally through the mid region to cross sections of both the left and right ventricles. The heart material was embedded in paraffin, sectioned (5 μm thickness), and stained with Masson’s Trichrome stain for myocardial morphology and collagen. Regions of the left and right ventricle were imaged with a Nikon E-600 and images collected with a 40X (NA = 0.75) objective. Sequential, adjacent images (15–20 images per section) were recorded from each section to eliminate morphology selection bias. Images were collected as TIFF files and morphometric analysis carried out using MetaMorph 6.1 image analysis software.
2.7. Myocardial Tissue and Collagen Analysis
The area of the total image measured was 257240.65 μm2. Area of myocardial tissue and area occupied by collagen in an image were determined using the HSI (hue, saturation, intensity) color model of the set color threshold subroutine in MetaMorph. Comparison of the amount of collagen in myocardium was performed by determination of the ratio of area occupied by collagen to area occupied by myocardium expressed as a percent. A minimum of 10 images were analyzed from each heart section of each animal in the control and experimental groups. Three to five mice were analyzed for each group.
Analysis of cardiac myocyte morphology and myofibrillar morphology was determined in myocytes that satisfied the following: in the trichrome images, the myocytes had to be cut in cross section with both border of cell and nucleus visible. Myocytes not meeting these criteria were excluded. The trichrome color images were converted to 8-bit gray scale images and the red channel image used for analysis of myocyte and myofibrillar morphologies. Individual myocytes were outlined as regions of interest (ROI’s) and the region bounded by ROI measured as the total cross sectional area of the myocyte. The myofibrils contained within each myocyte ROI were isolated by adjusting the threshold scale to include only the stained myofibrils and area occupied by myofibrils (myofibril area, myofA), area not occupied by myofibrils (hole area, HA) and integrated optical density (IOD) of the myofibrils measured using the integrated morphometrics subroutine of MetaMorph. The loss of myofibrils and myofibrillar integrity (MI) in each myocyte is expressed as a percent of the ratio of the hole area to total area of the myocyte (MI% = HA/TMA × 100). The normalized apparent mass of the myobrils in each myocyte is expressed by the ratio of the IOD of myofibrils to area of the myofibrils in the each myocyte (myofibillar mass = IOD/myofA). The IOD of the cytoplasmic myofibrillar region labeled by trichrome stain delineated by the threshold boundaries was considered to be representative of the cellular cross sectional “mass” of the region and a measurement of the total amount of labeled material in the region.15 The use of IOD to quantify the relative amount of structures has been applied to the determination of morphological changes in the actin cytoskeleton16,17 and microvasculature.18 Values of IOD were calculated directly from the integrated morphometry subroutine of MetaMorph image analysis software. Using the software’s optical calipers, the measurements were refined by setting specific boundary conditions for area and IOD for acceptance of the signal from myofibrillar content of the selected individual cardiac myocytes. The relative myofibrillar content of each cardiac myocytes measured was normalized by dividing the IOD value by the myofibrillar area in each cardiac myocyte.
2.8. Statistical Analysis
Values are reported as mean ± SEM for n number of independent observations. Statistical significance was determined by Student’s t-test for single comparisons, one-way ANOVA and post hoc test as identified in the figure legends, or two-way ANOVA. P ≤ 0.05 was taken as significant.
3. Results
3.1. Increase in Blood Pressure
ANG II increased MAP in WT and SA/SA mice. In both, a modest increase was noted initially, with the increase being slightly greater in WT mice. However, by day 11, MAP was slightly more elevated in SA/SA mice (Fig. 1A). SBP was increased comparably by ANG II in both WT and SA/SA mice (Fig. 1B), while DBP was more elevated in SA/SA mice (Fig. 1C). Consequently, while pulse pressure was increased above 40 mmHg in both WT and SA/SA, the increase was more dramatic in WT mice (Fig. 1D). No difference was observed between the two groups in HR (data not shown).
Figure 1.
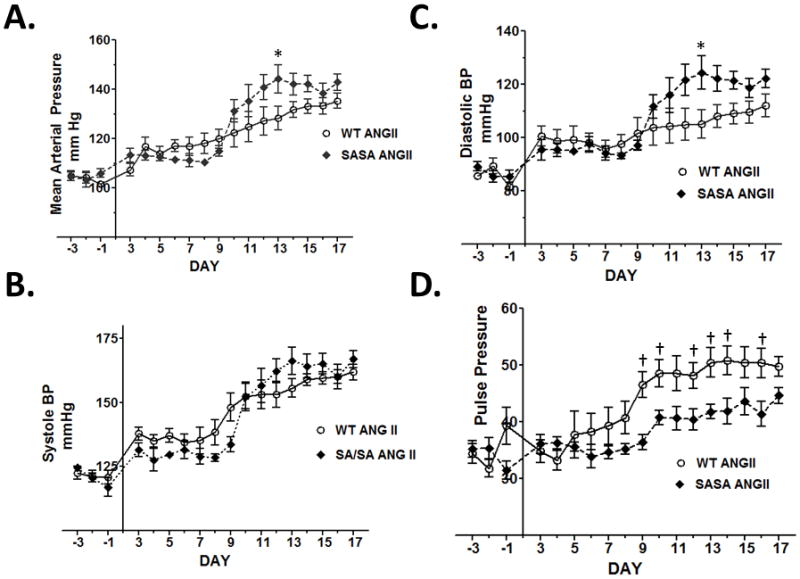
ANG II-induced hypertension in WT and SA/SA mice. Mice were implanted with a telemetry device to record blood pressure. Ten days later (Day −3) baseline measurements were made. Mini-osmotic pumps were implanted on Day 0 to deliver saline or ANG II (1000 ng/kg/min). Blood pressure data were analyzed by 2-way ANOVA. Source of variation: (A) mean arterial pressure: genotype (P < 0.01), ANG II (P< 0.001), and interaction (P < 0.05).(B) Systolic blood pressure: genotype (not significant), ANG II (P < 0.001), interaction (not significant); (C) Diasotic blood pressure: genotype (P < 0.01), ANG II (P < 0.001), interaction (P < 0.05); (D) Pulse pressure: genotype (P < 0.001), ANG II (P < 0.001), interaction (not significant). P < 0.05 WT vs SA/SA same day (*Bonferroni’s test or †t-test); n = 6 WT and 4 SA/SA.
3.2. Cardiac Hypertrophy and Inflammatory Cytokines
Both WT and SA/SA exhibited cardiac hypertrophy with ANG II (Table 1). However, the increase in heart to body weight ratio was greater in WT (32.9%) than in SA/SA (19.5%) mice. We also examined cytokines in the left ventricle linked to STAT3 and cardiac remodeling: VEGF,7 MCP-1,19 TGF-β1,20 and IL-6.21 Ventricular levels of VEGF tended to be higher in WT mice treated with ANG II, although not reaching statistical significance (Table 1). Cardiac MCP-1, TGF-β1, and IL-6 were significantly increased by ANG II in both WT and SA/SA; however, for all the increase was not as pronounced in SA/SA (Table 1). With ANG II, ventricular levels (pg/μg) were significantly higher in WT vs. SA/SA mice for MCP-1, TGF-β1, and IL-6. After ANG II treatment, cardiac STAT3 phosphorylated on S727 was detected in WT, but not SA/SA mice; STAT3 phosphorylated on Y705 was not observed in ventricles of either WT or SA/SA mice (Fig. 2A). No difference was seen between WT and SA/SA mice in cardiac levels of SOD2 or Bcl-xL (Fig. 2B).
Table 1.
Hemodynamics and cardiac parameters
| WT Saline | SA/SA Saline | WT ANG II | SA/SA ANG II | ||
|---|---|---|---|---|---|
| SBP | Before | - | - | 122.5 ± 2.6 (6) | 124.5 ± 0.7 (4) |
| After | - | - | 161.8 ± 3.0 (6)† | 166.9 ± 3.4 (4)† | |
| DBP | Before | - | - | 85.8 ± 1.0 (6) | 89.2 ± 1.7 (4) |
| After | - | - | 112.0 ± 4.3 (6)† | 122.2 ± 3.5 (4)† | |
| MAP | Before | - | - | 105.0 ± 2.0 (6) | 104.8 ± 1.8 (4) |
| After | - | - | 135.4 ± 3.0 (6)† | 142.9 ± 3.3 (4)† | |
| HR | Before | - | - | 592.5 ± 20.6 (6) | 594.3 ± 10.5 (4) |
| After | - | - | 552.7 ± 16.6 (6) | 568.7 ± 14.2 (4) | |
| EF | Before | 63.0 ± 2.0 (3) | 61.0 ± 2.2 (3) | 72.6 ± 10.0 (5) | 63.7 ± 7.1 (5) |
| After | 62.6 ± 1.8 (3) | 58.7 ± 1.8 (3) | 71.5 ± 0.8 (5) | 51.7 ± 6.9 (5)* | |
| FS | Before | 33.0 ± 1.7 (3) | 32.2 ± 1.5 (3) | 35.5 ± 4.4 (5) | 34.3 ± 4.9 (5) |
| After | 33.4 ± 0.7 (3) | 31.3 ± 0.8 (3) | 40.0 ± 0.6 (5) | 26.4 ± 4.3 (5)* | |
| HW/BW (mg/g) | 5.50 ± 0.23 (4) | 5.22 ± 0.30 (3) | 7.31 ± 0.23 (6)φ | 6.24 ± 0.19 (5)**,‡ | |
| VEGF (pg/μg) | 47.47 ± 3.03 (3) | 44.20 ± 4.47(3) | 64.13 ± 6.24 (7) | 46.18 ± 4.44 (4) | |
| MCP-1 (pg/μg) | 1.50 ± 0.79 (3) | 1.10 ± 0.21 (3) | 35.14 ± 2.58 (7) φ | 22.28 ± 1.56 (4)**,§ | |
| TGF-β1 (pg/μg) | 8.30 ± 0.91(3) | 6.13 ± 0.43 (3) | 56.40 ± 4.38 (7) φ | 38.00 ± 2.86 (4)**,§ | |
| IL-6 (pg/μg) | 3.93 ± 0.65 (3) | 3.00 ± 0.06 (3) | 13.59 ± 1.41 (7) φ | 9.05 ± 0.56 (4)*,‡ | |
P < 0.001 vs. respective Before value;
P < 0.05,
P < 0.01 vs. WT ANG II;
P < 0.001 vs. WT Saline;
P < 0.05,
P < 0.001 vs. SA/SA Saline. Numbers in parentheses represent the number of mice in each group. Data were analyzed by Student’s t test (Before vs. After) or one-way ANOVA with Newman-Keuls multiple comparison test. SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure; HR, heart rate; EF, ejection fraction; FS, fractional shortening; WT, wild-type; SA/SA, mice with S727A mutation; ANG II, angiotensin II; HW, heart weight; BW, body weight.
Figure 2.
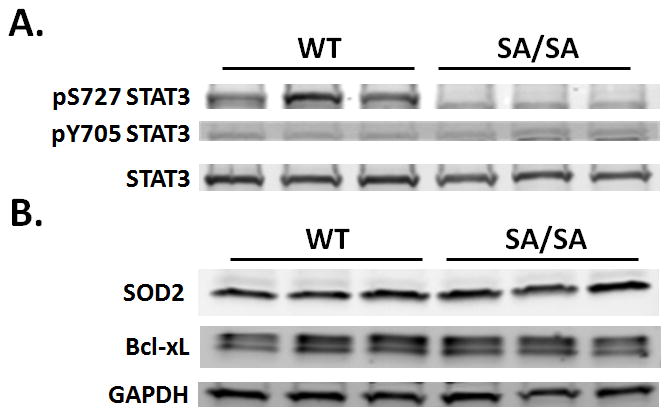
Effect of ANG II on STAT3 activation and expression of protective proteins. WT and SA/SA mice were treated with saline or ANG II for 17 days. Ventricles of the heart were processed for Western analysis. (A) Activation of STAT3. Homogenates containing equal protein levels were probed for STAT3 S727 and Y705 phosphorylation and total STAT3. (B) Ventricles were assayed for expresion SOD2 and Bcl-xL. GAPDH was probed for to ensure equal loading. Each lane represents an individual mouse heart.
3.3. Cardiac Function
No significant differences were noted in SBP, HR, EF, or FS between WT and SA/SA mice before ANG II (Table 1). Unlike WT mice, hearts of SA/SA mice exhibited lower EF and FS with ANG II (Table 1). Unlike WT mice, individual SA/SA mice exhibited a decrease in EF and FS over the course of ANG II treatment (Fig. 3A & B). For individual mice, there was an increase in left ventricular anterior wall diameter in diastole (LVAWd) in WT mice with ANG II that was not observed in SA/SA mice, indicative of greater cardiac hypertrophy in WT mice (Fig. 3C).
Figure 3.
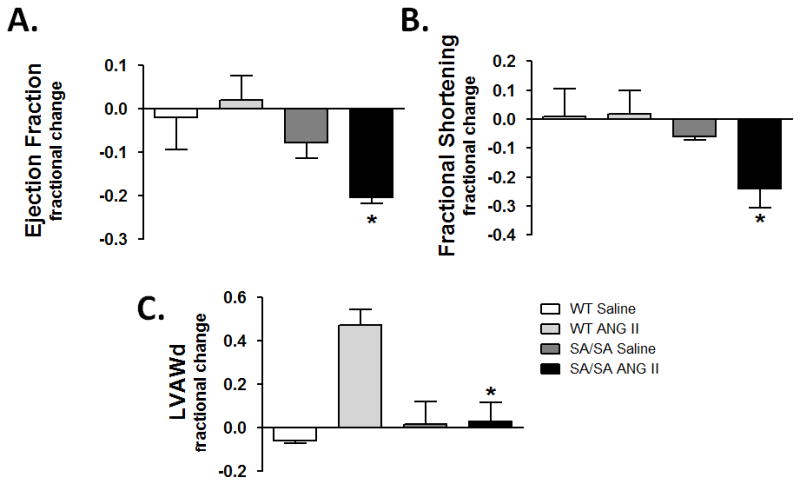
Changes in ventricular function and wall thickness in individual mice. Echocardiography was performed on WT and SA/SA mice before and after treatment with saline or ANG II. Change in (A) ejection fraction (EF), (B) fractional shortening (FS), and (C) LVAWd between days −1 and 16 were calculated for each mouse. *P < 0.05 SA/SA+ANG II vs. WT+ANG II (One-way ANOVA with Bonferroni’s multiple comparison test).
3.4. Myocardial Collagen and Fibrosis
Myocardial tissue from WT+Saline and SA/SA+Saline had the same morphology, distribution and content of collagen. Collagen was present in normal appearing perimysium and endomysium connective tissue supporting myocytes and bundles of myocytes (Figs. 4A, panels A & B). Treatment of WT or SA/SA mice with ANG II resulted in formation of foci of fibrosis (Figs. 4A, panels C & D). These foci were characterized by loss of cardiac myocytes and increase in collagen deposition in regions in which myocytes were lost. These fibrotic regions appeared predominately in the left ventricle. Fibrosis in the right ventricle was seen in close proximity with the intraventricular septum. Although the magnitude of formation of these collagen foci was essentially the same in WT+ANG II and SA/SA+ANG II hearts (Fig. 4B) their morphology was different. In the WT+ANG II myocardium, these fibrotic lesions consisted of several smaller adjacent regions (Fig. 4A, panel C) or appeared more like a fibrotic network extending from a focus and intercalating around the myocytes (Fig. 5A, panel A). Larger more extensive regions of fibrosis were present in the SA/SA+ANG II myocardium. In these regions of fibrosis, many myocytes were lost and replaced by connective tissue (Fig. 5A, panel B). Affected myocytes in the fibrotic lesion exhibited smaller cross-sections, but still retained nuclei and some myofibrils. Some myocytes associated with these fibrotic regions exhibited nuclear damage and apparent nuclear hypertrophy, suggesting progressive loss of myocytes in the fibrotic regions was due to myocyte necrosis. There was an absence of mast cells and macrophages in these fibrotic lesions indicating inflammation was not involved in loss of cardiac myocytes and collagen deposition in the myocardial tissue of WT+ANG II and SA/SA+ANG II animals.
Figure 4.
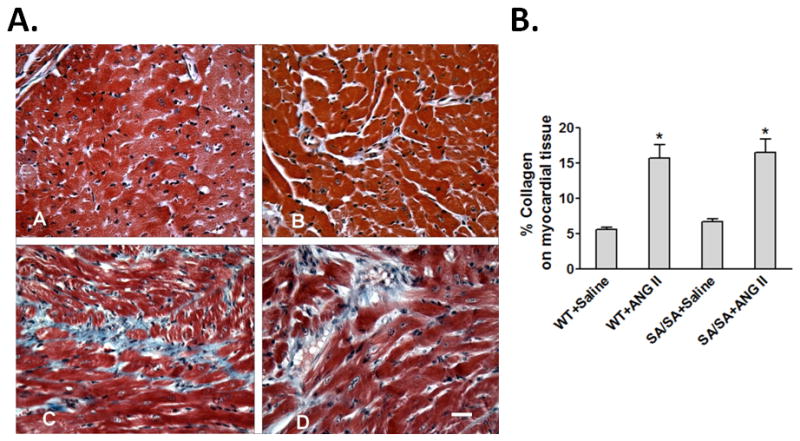
Hypertension-induced cardiac collagen. (A) Trichrome staining of myocardium. Panel A, WT+Saline: Typical cross-sectional morphology of myocardium in the left ventricle of WT mice. Panel B, SA/SA+Saline: Typical cross-sectional morphology of myocardium in left ventricle of SA/SA mice. Panel C, WT+ANG II: An increase in collagen (blue stain) is seen as small foci of fibrosis scattered in the left ventricular myocardium. Panel D, SA/SA+ANG II: The collagen fibrosis comprises larger regions and appears more extensive in SA/SA left ventricular myocardium. Scale bar = 50 μm. Bar Graph: Percent change in collagen among WT and SA/SA mice treated with saline or ANG II. *P < 0.05 SA/SA+ANG II and WT+ANG II vs. both saline controls; Kruskal-Wallis ANOVA followed by Dunn’s post hoc test.
Figure 5.

Hearts of SA/SA mice showed differences in collagen and myocyte morphology. (A) Examples of variable and extensive collagen fibrosis in WT and SA/SA left ventricular myocardium. Panel A, WT+ANG II: Left ventricle. Collagen fibrosis appears as a focal region with increase in thickness of endomysium associated with adjacent cardiac myocytes. There appears to be minimal destruction or loss of myocytes associated with regions of fibrosis. Panel B, SA/SA+ANG II. Left ventricle. A typical large region of myocardial damage is characterized by extensive collagen fibrosis and loss and destruction of cardiac myocytes. In the central region of the fibrotic lesion, cardiac myocytes have been completely lost and undergoing destruction. The cardiac myocytes in the fibrotic lesion are reduced in size, but may still retain their nucleus (arrow c). Other cardiac myocytes associated with collagen lesion exhibit hypertrophied nuclei and condensed chromatin (arrow n). Scale Bar = 50 μm. (B) Changes in cardiac myocyte morphology. Trichrome stained sections of the left ventricles of WT and SA/SA mice treated with saline or ANG II were analyzed for loss of myofibrils and mass of myofibrils. Ventricles of SA/SA mice treated with ANG II showed significant loss of myofibrils compared to other groups (upper). Mass of the myofibrils was increased in WT, but decreased in SA/SA mice treated with ANG II (lower). Myofibrils of SA/SA mice exhibited a greater density than wild type mice. *P < 0.05; Kruskal-Wallis ANOVA followed by Dunn’s post hoc test.
3.5. Cardiac Myocyte Size and Myofibril Mass
No significant difference in size of cardiac myocytes measured as cross-sectional area was seen among groups (data not shown). There was no difference in myofibrillar content between WT+Saline and SA/SA+Saline control groups (Fig. 5B). Treatment of WT animals with ANG II resulted in a trend towards an increase in loss of myofibrillar content; however, this loss was not significantly greater than seen in WT+Saline controls. SA/SA+ANG II animals exhibited a significant (P<0.05) loss in myofibrillar content when compared to SA/SA+Saline, WT+Saline, and WT+ANG II groups (Fig. 5B). This suggests that cardiac myocytes of SA/SA are more susceptible to adverse effects of ANG II resulting in greater loss of myofibrils and thus decreased cardiac function.
Treatment of WT mice with ANG II showed an increase in mass density of individual myofibrils (Fig. 5B). This suggests ANG II may have caused some enlargement or hypertrophy of myofibrils. This possibility would be consistent with the trend towards a potential loss of myofibrils in WT+ANG II hearts. With myofibrils being lost and remaining mass of individual myofibrils being larger would mean surviving myofibrils had become enlarged or were undergoing hypertrophy. The effect of ANG II on mass and distribution of myofibrils in SA/SA animals contrasts with that seen in WT mice (Fig. 5B). The mass of individual myofibrils in SA/SA+ANG II hearts was significantly less than seen in SA/SA+Saline hearts. In SA/SA myocardium, ANG II induced a loss of myofibrils accompanied by a loss of individual myofibrillar mass density. In SA/SA myocytes ANG II treatment resulted in fewer and smaller myofibrils compared to SA/SA+Saline control mice. Since myofibrillar mass of SA/SA+Saline controls is significantly greater for WT+Saline controls, this would suggest that myofibrillar morphologies for WT and SA/SA animals are uniquely different and myofibrils in the SA/SA myocytes are larger than those in the WT myocytes.
4. Discussion
We report that reduced STAT3 transcriptional activity adversely affected the response of the heart to ANG II hypertension at an early stage. Hearts of mice expressing a STAT3 S727A mutation exhibited less cardiac hypertrophy, associated with a diminution in EF and FS. The reduction in cardiac function may have resulted from enhanced loss of myofibrils as well as reduction in individual myofibrillar mass in hearts of SA/SA mice treated with ANG II. Cardiac hypertrophy with ANG II at this early stage was likely due to an increase in fibrosis (not individual myocyte size) in WT and SA/SA hearts, as well as increased myofibrillar mass in WT mice. However, total left ventricular collagen will need to be measured before a definitive conclusion can be made. A loss of myofibrils (i.e., reduction in their volume density) has been reported for both the hibernating myocardium and failing heart.22,23 Whether they are associated as well with a reduction in mass density of individual myofibrils as observed here in SA/SA mice treated with ANG II has not been previously assessed to the best of our knowledge.
Notably, the worsened response of hearts of STAT3 S727A mice to increased blood pressure occurred in a situation of lower expression of inflammatory cytokines associated with maladaptive cardiac remodeling. Hearts of WT and SA/SA mice treated with ANG II exhibited elevated levels of MCP-1, IL-6, and TGF-β1. Increased expression of each of these, MCP-1,24,25 IL-6,26 and TGF-β1,27 has been linked to adverse cardiac remodeling (especially fibrosis) and dysfunction in pressure overload/hypertension, as well as transition of compensatory hypertrophy to heart failure. The increase in inflammatory cytokines was less in SA/SA mice, likely due to decreased STAT3 transcriptional activity. Mouse promoters (TRED ID) for MCP-1 (126767), IL-6 (129719), and TGF-β1 (130404) contain multiple potential STAT3 binding elements (TT(N)4-6AA).28 Alternatively, we cannot discount the possibility that the different pattern of cardiac cytokine expression that we observed was secondarily related to differences in the magnitude or phenotype of the adaptive response of the heart to increased blood pressure between wild type and SA/SA mice.29,30 In any case, the finding in the present study of worsened cardiac function in the face of reduced inflammatory cytokine expression suggests these factors have beneficial actions on cardiac myocytes during the onset of hypertension-induced cardiac remodeling.
ANG II induced hypertension in WT and SA/SA mice. Others had shown that ANG II-induced hypertension in the mouse is dependent upon IL-6,31 a major activator of STAT3, although IL-6 infusion alone does not cause hypertension. IL-6 knockout mice failed to develop hypertension upon chronic treatment with ANG II and the failure was associated with loss of JAK2 and STAT3 activation in the kidney.31 Also, ANG II-induced hypertension is blocked by JAK2 inhibitor AG490.32 No difference was observed between WT and IL-6 knockout mice in renal blood flow with ANG II infusion; however, ANG II decreased sodium excretion and evidence was found that renal JAK-STAT signaling may contribute to sodium retention.31,32 Our finding would suggest that IL-6 affects renal sodium absorption via a JAK-dependent, but STAT3-independent mechanism.
DBP, which has been implicated in end organ damage, was slightly more elevated by ANG II in SA/SA mice than WT mice, perhaps due to reduced cardiac function. Consequently, the increase in pulse pressure, which is also associated with damage to the heart, was not as marked in SA/SA mice. In any case, these differences in blood pressure over the short time frame involved are unlikely to explain the differences observed in heart function between SA/SA and WT mice. However, we cannot rule out the possibility that the SA/SA genotype sensitized the heart to these slight changes in blood pressure. Thus, rather than reflecting solely an impaired response of the heart to chronic stress due to STAT3 deficiency, our findings may reveal a fundamental role for STAT3 in the normal workings of the heart. The greater increase in diastolic blood pressure observed in the SA/SA hearts under hypertension may have been due to inadequate cardiac performance that may have further contributed to deterioration in cardiac structure and performance. Theoretically, greater left ventricular end systolic wall stress in the SA/SA hearts due to an inadequate adaptive response may have led to greater local production of ANG II and oxidative stress that accelerated the deterioration in cardiac function and structure.33
STAT3 has been previously linked to a preconditioning response protecting the heart against oxidative injury acutely.34 Enhanced STAT3 signaling was also recently shown to limit infarct expansion and facilitate cardiac repair 2 weeks after infarction in the mouse heart.35 This finding and ours suggest that STAT3 activation is linked to a coordinated adaptive response of cardiac myocytes to chronic stress. The protective effects of STAT3 against ischemia/reperfusion have been attributed to upregulation of anti-apoptotic and anti-oxidant genes, such as Bcl-xL and SOD2.34 However, no difference in expression of these proteins was seen between WT and SA/SA mice (Fig. 2B). We did observe a difference between the two in myofibrillar density before hypertension and a decrease in myofibrillar density in SA/SA mice, but not WT mice, with hypertension (Fig. 5B). One interpretation of this observation is that STAT3 transcriptional activity is important in packaging or assembly of myofilaments. Alternatively, nongenomic actions of STAT3 may be involved in the protective actions of STAT3 in hypertension-induced cardiac hypertrophy. Recent studies have shown that STAT3 is found in the mitochondria of different cell types, including cardiac myocytes, and regulates respiration.36 How STAT3 regulates mitochondrial function is not known; however, S727 phosphorylation may be involved as impaired respiration of mitochondria from STAT3−/− pro-B cells could be restored by a mimetic of constitutively S727 phosphorylated STAT3, while a mimetic of Y705 phosphorylation was ineffective.37
Conclusions and Perspectives
We show that STAT3 is important in early adaptation of the heart to increased blood pressure. Attenuated STAT3 activity resulted in alterations in cardiac remodeling as evidenced by changes in the pattern of fibrosis deposition and reduced cardiac hypertrophy. Reduction in STAT3 activity was associated with development of cardiac dysfunction marked by a combined loss of myofibrillar content of cardiac myocytes and a reduction in the mass of contractile filaments. These changes occurred even though the expression of inflammatory cytokines was reduced. Due to small sample sizes and the limited statistical power, the failure to detect certain group differences in this study should be interpreted with caution. Future work will need to establish whether STAT3 directly impacts on protein stability and turnover or acts indirectly, such as through the generation of reactive oxygen species.
Acknowledgments
This work was supported by grants to GWB from NHLBI (5R01HL088101-06) and to MK from The Lebanese University (MK-02-2011), The Lebanese National Council for Scientific Research (CNRS;05-10-09), and The COMSTECH-TWAS (09-122 RG/PHA/AF/AC_C).
Footnotes
Disclosure: None of the authors have any conflicts of interest to declare.
References
- 1.Booz GW. Left ventricular physiology in hypertension. In: Lip YH, Hall JE, editors. Comprehensive Hypertension. Mosby; Philadelphia: 2007. pp. 113–121. [Google Scholar]
- 2.Bolli R, Stein AB, Guo Y, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu Y, Xuan YT. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol. 2011;50:589–597. doi: 10.1016/j.yjmcc.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Oshima Y, Fujio Y, Nakanishi T, Itoh N, Yamamoto Y, Negoro S, Tanaka K, Kishimoto T, Kawase I, Azuma J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc Res. 2005;65:428–435. doi: 10.1016/j.cardiores.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 4.Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, Wu WJ, Tan W, Bolli R. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res. 2004;64:61–71. doi: 10.1016/j.cardiores.2004.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xuan YT, Guo Y, Zhu Y, Han H, Langenbach R, Dawn B, Bolli R. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol. 2003;35:525–537. doi: 10.1016/s0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, Osugi T, Izumi M, Oshima Y, Nakaoka Y, Hirota H, Kishimoto T, Yamauchi-Takihara K. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104:979–981. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 7.Osugi T, Oshima Y, Fujio Y, Funamoto M, Yamashita A, Negoro S, Kunisada K, Izumi M, Nakaoka Y, Hirota H, Okabe M, Yamauchi-Takihara K, Kawase I, Kishimoto T. Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J Biol Chem. 2002;277:6676–6681. doi: 10.1074/jbc.M108246200. [DOI] [PubMed] [Google Scholar]
- 8.Haghikia A, Stapel B, Hoch M, Hilfiker-Kleiner D. STAT3 and cardiac remodeling. Heart Fail Rev. 2011;16:35–47. doi: 10.1007/s10741-010-9170-x. [DOI] [PubMed] [Google Scholar]
- 9.Lindner D, Hilbrandt M, Marggraf K, Becher PM, Hilfiker-Kleiner D, Klingel K, Pauschinger M, Schultheiss HP, Tschöpe C, Westermann D. Protective Function of STAT3 in CVB3-Induced Myocarditis. Cardiol Res Pract. 2012;2012:437623. doi: 10.1155/2012/437623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA. 2003;100:12929–12934. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zgheib C, Zouein FA, Kurdi M, Booz GW. Differential STAT3 Signaling in the Heart: Impact of Concurrent Signals and Oxidative Stress. JAK-STAT. 2012;1:102–111. doi: 10.4161/jkst.19776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen Y, Schlessinger K, Zhu X, Meffre E, Quimby F, Levy DE, Darnell JE., Jr Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol. 2004;24:407–419. doi: 10.1128/MCB.24.1.407-419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tallam LS, da Silva AA, Hall JE. Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension. 2006;48:58–64. doi: 10.1161/01.HYP.0000227966.36744.d9. [DOI] [PubMed] [Google Scholar]
- 15.Walter RJ, Jr, Berns MW. Digital Image Processing and Analysis. In: Inoue S, editor. Video Microscopy. Plenum Press; New York and London: 1986. pp. 327–392. [Google Scholar]
- 16.Fuseler JW, Valarmathi MT. Modulation of the migration and differentiation potential of adult bone marrow stromal stem cells by nitric oxide. Biomaterials. 2012;33:1032–1043. doi: 10.1016/j.biomaterials.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 17.Fuseler JW, Millette CF, Davis JM, Wayne Carver W. Fractal and image analysis of morphological changes in the actin cytoskeleton of neonatal cardiac fibroblasts in response to mechanical stretch. Microsc Microanal. 2007;13:128–132. doi: 10.1017/S1431927607070225. [DOI] [PubMed] [Google Scholar]
- 18.Fuseler JW, Bedenbaugh A, Krishna Y, Baudino TA. Fractal and Image Analysis of the microvasculature in normal intestinal submucosa and intestinal polyps in ApcMin/+ Mice. Microsc Microanal. 2010;16:73–79. doi: 10.1017/S143192760999119X. [DOI] [PubMed] [Google Scholar]
- 19.Morimoto H, Takahashi M, Izawa A, Ise H, Hongo M, Kolattukudy PE, Ikeda U. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ Res. 2006;99:891–899. doi: 10.1161/01.RES.0000246113.82111.2d. [DOI] [PubMed] [Google Scholar]
- 20.Mascareno E, Galatioto J, Rozenberg I, Salciccioli L, Kamran H, Lazar JM, Liu F, Pedrazzini T, Siddiqui MA. Cardiac lineage protein-1 (CLP-1) regulates cardiac remodeling via transcriptional modulation of diverse hypertrophic and fibrotic responses and angiotensin II-transforming growth factor β (TGF-β1) signaling axis. J Biol Chem. 2012;287:13084–13093. doi: 10.1074/jbc.M111.288944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sano M, Fukuda K, Kodama H, Takahashi T, Kato T, Hakuno D, Sato T, Manabe T, Tahara S, Ogawa S. Autocrine/Paracrine secretion of IL-6 family cytokines causes angiotensin II-induced delayed STAT3 activation. Biochem Biophys Res Commun. 2000;269:798–802. doi: 10.1006/bbrc.2000.2364. [DOI] [PubMed] [Google Scholar]
- 22.Hamdani N, Borbély A, Veenstra SP, Kooij V, Vrydag W, Zaremba R, Dos Remedios C, Niessen HW, Michel MC, Paulus WJ, Stienen GJ, van der Velden J. More severe cellular phenotype in human idiopathic dilated cardiomyopathy compared to ischemic heart disease. J Muscle Res Cell Motil. 2010;31:289–301. doi: 10.1007/s10974-010-9231-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bito V, van der Velden J, Claus P, Dommke C, Van Lommel A, Mortelmans L, Verbeken E, Bijnens B, Stienen G, Sipido KR. Reduced force generating capacity in myocytes from chronically ischemic, hibernating myocardium. Circ Res. 2007;100:229–237. doi: 10.1161/01.RES.0000257829.07721.57. [DOI] [PubMed] [Google Scholar]
- 24.Iwasaki J, Nakamura K, Matsubara H, Nakamura Y, Nishii N, Banba K, Murakami M, Ohta-Ogo K, Kimura H, Toh N, Nagase S, Oka T, Morita H, Kusano KF, Ohe T. Relationship between circulating levels of monocyte chemoattractant protein-1 and systolic dysfunction in patients with hypertrophic cardiomyopathy. Cardiovasc Pathol. 2009;18:317–322. doi: 10.1016/j.carpath.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Kai H, Mori T, Tokuda K, Takayama N, Tahara N, Takemiya K, Kudo H, Sugi Y, Fukui D, Yasukawa H, Kuwahara F, Imaizumi T. Pressure overload-induced transient oxidative stress mediates perivascular inflammation and cardiac fibrosis through angiotensin II. Hypertens Res. 2006;29:711–718. doi: 10.1291/hypres.29.711. [DOI] [PubMed] [Google Scholar]
- 26.Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenzel S, Taimor G, Piper HM, Schlüter KD. Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-β expression in adult ventricular cardiomyocytes. FASEB J. 2001;15:2291–2293. doi: 10.1096/fj.00-0827fje. [DOI] [PubMed] [Google Scholar]
- 28.Zhao F, Xuan Z, Liu L, Zhang MQ. TRED: a Transcriptional Regulatory Element Database and a platform for in silico gene regulation studies. Nucleic Acids Res. 2005;33:D103–D1037. doi: 10.1093/nar/gki004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Modesti PA, Vanni S, Bertolozzi I, Cecioni I, Polidori G, Paniccia R, Bandinelli B, Perna A, Liguori P, Boddi M, Galanti G, Serneri GG. Early sequence of cardiac adaptations and growth factor formation in pressure- and volume-overload hypertrophy. Am J Physiol Heart Circ Physiol. 2000;279:H976–H985. doi: 10.1152/ajpheart.2000.279.3.H976. [DOI] [PubMed] [Google Scholar]
- 30.Cambi GE, Lucchese G, Djeokeng MM, Modesti A, Fiaschi T, Faggian G, Sani G, Modesti PA. Impaired JAK2-induced activation of STAT3 in failing human myocytes. Mol Biosyst. 2012;8:2351–2359. doi: 10.1039/c2mb25120e. [DOI] [PubMed] [Google Scholar]
- 31.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension. 2010;56:879–884. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banes-Berceli AK, Al-Azawi H, Proctor D, Qu H, Femminineo D, Hill-Pyror C, Webb RC, Brands MW. Angiotensin II utilizes Janus kinase 2 in hypertension, but not in the physiological control of blood pressure, during low-salt intake. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1169–R1176. doi: 10.1152/ajpregu.00071.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serneri GG, Boddi M, Cecioni I, Vanni S, Coppo M, Papa ML, Bandinelli B, Bertolozzi I, Polidori G, Toscano T, Maccherini M, Modesti PA. Cardiac angiotensin II formation in the clinical course of heart failure and its relationship with left ventricular function. Circ Res. 2001;88:961–968. doi: 10.1161/hh0901.089882. [DOI] [PubMed] [Google Scholar]
- 34.Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J Cardiovasc Pharmacol. 2007;50:126–41. doi: 10.1097/FJC.0b013e318068dd49. [DOI] [PubMed] [Google Scholar]
- 35.Oba T, Yasukawa H, Hoshijima M, Sasaki K, Futamata N, Fukui D, Mawatari K, Nagata T, Kyogoku S, Ohshima H, Minami T, Nakamura K, Kang D, Yajima T, Knowlton KU, Imaizumi T. Cardiac-specific deletion of SOCS-3 prevents development of left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. 2012;59:838–852. doi: 10.1016/j.jacc.2011.10.887. [DOI] [PubMed] [Google Scholar]
- 36.Szczepanek K, Chen Q, Larner AC, Lesnefsky EJ. Cytoprotection by the modulation of mitochondrial electron transport chain: The emerging role of mitochondrial STAT3. Mitochondrion. 2012;12:180–189. doi: 10.1016/j.mito.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]