

Abstract
Background
The prognostic and therapeutic implications of the spectrum of KRAS oncogene substitutions in lung cancer remain poorly understood. The objective of this study was to determine if KRAS oncogene substitutions differed with regard to prognosis or predictive value in lung adenocarcinoma.
Methods
KRAS oncogene substitutions and mutant-allele specific imbalance (MASI) were determined in patients with lung adenocarcinoma and associations with overall survival (OS) and recurrence free survival (RFS), and chemotherapy interactions were assessed.
Results
KRAS mutational analysis was performed on 988 lung adenocarcinomas, and 318 KRAS mutations were identified. In this predominantly early stage cohort (78.6% stage I–III), OS and RFS did not differ by the type of KRAS substitution (OS, p=0.612; RFS P=0.089). There was a trend toward better OS in the subset of patients with KRAS codon 13 mutations (p=0.052), which was not significant in multivariate analysis (p=0.076). RFS did not differ by codon type in univariate analysis (p=0.322). There was a marked difference in RFS based on the presence of MASI in univariate (p=0.004) and multivariate analysis (p=0.009). A test for interaction was performed in order to determine if the effect of chemotherapy on OS and RFS differed based on the type of KRAS substitution, codon type or the presence of MASI. There were no differences in the effects of chemotherapy for any of variables examined.
Conclusions
KRAS codon 13 mutations and MASI are candidate biomarkers for prognosis that may be useful to incorporate in prospective studies evaluating novel therapies in KRAS mutant lung adenocarcinoma.
Keywords: KRAS, lung adenocarcinoma, mutant allele-specific imbalance, prognosis, prediction
Introduction
Lung cancer is the leading cause of cancer-related mortality in the United States.1 Non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancers, while small-cell lung cancer accounts for about 15%.2 Historically treated as a single disease entity, the identification of driver mutations and the development of molecularly targeted agents against the epidermal growth factor receptor (EGFR) and the anaplastic lymphoma kinase (ALK) fusion oncogene have permanently shifted the landscape of NSCLC therapy toward a personalized approach.
Data from the Lung Cancer Mutation Consortium indicate that of 1,000 tumors from patients with lung adenocarcinoma, mutations in the v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) were the most prevalent of the driver mutations, found in 25% of all cases.3 The presence of a KRAS mutation is thought to have prognostic implications with regard to survival; in a meta-analysis of studies assessing RAS mutations in lung adenocarcinomas, the presence of a RAS mutation was associated in a 50% relative increase in the risk of death.4 However, the prognostic implications of RAS mutations have not been validated in a prospective fashion.
The predictive properties of KRAS mutations were explored in a molecular analysis of the patients included in the JBR.10 clinical trial which evaluated the role of cisplatin and vinorelbine in the adjuvant setting.5 In this analysis, KRAS mutations were neither prognostic of survival nor predictive of a differential benefit from adjuvant chemotherapy. Retrospective studies in the metastatic setting have failed to demonstrate a differential chemotherapy effect based on the presence or absence of a KRAS mutation.6,7 The predictive value of individual KRAS oncogene substitutions was explored a pooled analysis of trials evaluating the addition of cetuximab to chemotherapy in the treatment of metastatic colorectal cancer.8 In this study, patients with a G13D KRAS oncogene substitution derived greater benefit from cetuximab-based chemotherapy compared to patients with G12D and other KRAS oncogene substitutions, suggesting clinical heterogeneity amongst subtypes of KRAS mutations.
Furthermore, the significance of KRAS gene copy number change is uncertain. It has been observed that KRAS mutations may be associated with a higher KRAS gene copy number.9–11 The combination of mutations and copy number gain may result in an imbalance between the wild-type allele (W) and the mutant allele (M), in which may result in M being predominant over the W, a scenario defined as mutant allele specific imbalance (MASI).12 The incomplete dominance of M over W is most frequently a result of selective amplification of the M, but may also be due to the presence only of M in the absence of the W, as in acquired uniparental disomy, which frequently leads to complete MASI.13 Recent reports suggest that the combination of a KRAS mutation and copy number gain may be associated with adverse outcome in lung and colon adenocarcinoma patients.12,14,15
The aim of the present study was to assess if the spectrum of KRAS oncogene substitutions differed with regard to their clinical behavior, looking specifically at the type of KRAS amino acid (AA) substitution present, the presence of a codon 12 versus 13 mutation or the presence or absence of MASI.
Material and Methods
This was a retrospective analysis of banked tumor specimens collected from patients with newly diagnosed lung adenocarcinoma at the University of Pittsburgh Medical Center (UPMC) between 2005 and 2011. All formalin-fixed, paraffin-embedded specimens remaining after complete pathologic signout of the case were considered for inclusion in the study and were selected on the basis of meeting a minimum tissue requirement of 300 cells/sample. Guided by hematoxylin and eosin (H&E) stained slides, tumor targets containing more than 70% tumor cells were manually microdissected from the 4-μm unstained histologic sections. KRAS codons 12 and 13 mutational analysis was then performed as previously described.16 Briefly, DNA was isolated from each target using the DNeasy tissue kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. PCR products were sequenced in both the sense and antisense directions using the BigDye Terminator v3.1 cycle sequencing kit on an ABI 3130 automated sequencer (Applied Biosystems, Inc., Foster City, CA, USA) according to the manufacturer’s instructions. The sequences were analyzed using Mutation Surveyor software (SoftGenetics, LLC, State College, PA, USA). Cases were classified as mutated or wild type for KRAS based on the sequencing results. MASI was determined in a semi-quantitative fashion on sequencing electropherograms and was defined as a KRAS mutant peak greater than wild-type peak on (M>W) or a mutant peak equal to wild-type peak (M=W), and no MASI was defined as a mutant peak less than a wild-type peak (M<W) (Figure 2a).
Figure 2.


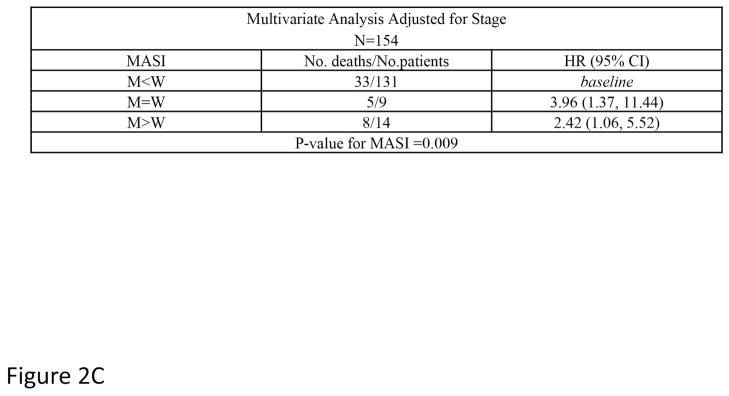
Assessment of recurrence free survival and mutant allele-specific imbalance (MASI) (A) defined on sequencing electropherograms as either a KRAS mutant peak equal to the wild-type peak (M=W) or a KRAS mutant peak greater than the wild-type peak (M>W). (B) Kaplan-Meier survival curve for recurrence free survival and KRAS MASI and (C) a multivariate analysis of recurrence free survival and MASI adjusting for stage.
Baseline demographics, staging and smoking history were obtained through a review of the medical record in the UPMC Medical Archival System. First-line treatment data, follow-up data with regard to first recurrences and survival were collected through the UPMC Network Cancer Registry. Recurrence free survival (RFS) and OS were calculated from the date of surgery (in stage I to III patients) or the date of a diagnostic biopsy (in stage IV patients) to the date of recurrence or death, respectively. Patients with less than 30 days of follow-up were excluded from the survival analyses. Those patients that did not experience the event of interest were censored at the date of their last follow-up. Survival probabilities were estimated using the Kaplan Meier Method. Cox proportional hazard models were used to examine the effects of the KRAS mutations and MASI on OS and RFS, while controlling for age, gender, race, smoking history, stage and use of chemotherapy. Only significant factors were left in the final model with the type of KRAS mutation or MASI. A test for interaction was used to determine if the effect of the KRAS mutation or MASI was modified by the use of chemotherapy both in resected Stage I to III disease (either neoadjuvant or adjuvant chemotherapy) and in the metastatic setting. This study was conducted under an exemption approved by the University of Pittsburgh Institutional Review Board.
Results
Using the inclusion criteria of 300 cells/sample, 988 tumor specimens collected between 2005 and 2011 were sequenced for KRAS mutations. Of those tumors, 318 (32.2%) harbored KRAS mutations, the majority of which were of codon 12 (298, 93.7%). GLY→CYS AA substitutions were the most common codon 12 (139, 46.6%) and codon 13 (10, 58.8%) oncogene substitutions (Table 1). This was a predominantly early stage cohort, consisting of 143 (45.0%) stage I patients, 52 (16.4%) stage II patients, 55 (17.3%) stage III patients, and 64 (20.1%) stage IV patients (Table 1). The median follow-up time was 24.3 months (range 0–6.5 years).
Table 1.
Clinical and pathologic features of patients with KRAS mutant lung adenocarcinoma
| Age (median, range) | 67 (39–92) |
| Male | 129 (40.6%) |
| Female | 189 (59.4%) |
| White | 291 (91.5%) |
| Black | 21 (6.6%) |
| Smoking status | |
| Current Smoker | 102 (32.1%) |
| Former Smoker | 143 (45.0%) |
| Never smoker | 18 (5.7%) |
| Unknown | 55 (17.3%) |
| Stage | |
| I | 143 (45.0%) |
| II | 52 (16.3%) |
| III | 55 (17.3%) |
| IV | 64 (20.9%) |
| KRAS mutation | |
| Codon 12 | 298 (93.7%) |
| Codon 13 | 17 (5.3 %) |
| Codon 12 | |
| GLY→CYS | 139 (46.6%) |
| GLY→VAL | 59 (19.8%) |
| GLY→ASP | 46 (15.4%) |
| Other | 40 (13.4%) |
| Unknown | 14 (4.7%) |
| Codon 13 | |
| GLY→CYS | 10 (58.8%) |
| GLY→ASP | 6 (35.3%) |
| GLY→VAL | 1 (5.9%) |
| MASI | |
| M>W | 19 (6.0%) |
| M=W | 17 (5.3%) |
| M<W | 180 (56.6%) |
| Unknown | 102 (32.1%) |
| Stage I–III* | |
| Chemotherapy | 72 (22.6%) |
| No chemotherapy | 160 (50.3%) |
| Stage IV – 1st line therapy** | |
| Chemotherapy | 56 (17.6%) |
| No chemotherapy | 12 (3.8%) |
All patients with stage I to III disease underwent a surgical resection, with the exception of 7 (2.2%) patients who received concurrent chemoradiotherapy and who were not included in interaction models for chemotherapy benefit. Chemotherapy for stage I–III disease includes both adjuvant and neoadjuvant chemotherapy. An additional 11 (3.5%) patients had an unknown treatment history.
All stage IV patients treated with chemotherapy in the 1st line setting received conventional chemotherapy with the exception of one patient who received erlotinib.
OS did not differ by the type of KRAS AA substitution either in univariate (p=0.612; Figure 1a) or multivariate analysis adjusting for age and stage (p=0.287). Similarly, RFS did not differ by the type of KRAS AA substitution either in univariate (p=0.089; Figure 1b) or multivariate analysis adjusting for stage (p=0.126). Interestingly, there was a trend toward better OS in the subset of patients with KRAS codon 13 mutations (p=0.052; Figure 1c), however this was not statistically significant in multivariate analysis adjusting for age and stage (p=0.076). In addition, RFS did not differ by codon type either in univariate (p=0.322; Figure 1d) or multivariate analysis adjusting for stage (p=0.318). However, RFS differed significantly by MASI both in univariate (p=0.004; Figure 2b) and multivariate analysis controlling for stage (p=0.009). Controlling for stage, MASI as defined by M>W (hazard ratio 2.42; 95% confidence interval, 1.06–5.52) or M=W (hazard ratio 3.96; 95% confidence interval, 1.37–11.44) was associated with a greater risk of recurrence compared with no MASI (M<W) (Figure 2c).
Figure 1.




Kaplan-Meier survival curves for patients with KRAS mutations: (A) overall survival by KRAS amino acid substitution, (B) recurrence free survival by KRAS amino acid substitution, (C) overall survival by KRAS codon type and (D) recurrence free survival by KRAS codon type.
We next examined whether KRAS mutation subtypes differed with regard to predictive value for chemotherapy benefit. A test for interaction was performed in order to determine if the effect of chemotherapy on OS differed based on the type of KRAS AA substitution present, the codon type or the presence or absence of MASI. Adjusting for age and stage, there were no differences in the effects of chemotherapy for any of variables examined (AA substitution, p=0.795; codon type, p=0.438, and MASI, p=0.598) on OS (Table 2). A similar test for interaction was performed in patients with resected stage I to stage III disease for RFS to explore if differences existed with regard to the effects of chemotherapy in the neoadjuvant and adjuvant setting. In this analysis, there was no interaction between the AA substitution (p=0.357), the codon type (p=0.790) or MASI (p=0.392) and chemotherapy benefit (Table 3).
Table 2.
The effect of chemotherapy on OS by KRAS oncogene substitution and MASI adjusting for age and stage
| CT (No. deaths/No. patients) | No CT (No. deaths/No. patients) | Hazard Ratio | 95% Confidence Interval | |
|---|---|---|---|---|
| GLY→CYS | 32/63 | 24/71 | 0.53 | (0.27, 1.05) |
| GLY→VAL | 8/19 | 10/34 | 0.62 | (0.23, 1.72) |
| GLY→ASP | 11/24 | 5/22 | 0.64 | (0.21, 2.00) |
| GLY→ALA | 8/10 | 2/14 | 1.56 | (0.30, 8.25) |
| Other | 4/6 | 4/8 | 0.75 | (0.17, 3.26) |
| Test for interaction AA substitution X treatment, P=0.795 | ||||
| Codon 12 | 65/120 | 48/147 | 0.58 | (0.33, 1.02) |
| Codon 13 | 1/6 | 1/11 | 1.69 | (0.10, 28.03) |
| Test for interaction codon X treatment, P=0.438 | ||||
| M<W | 32/70 | 16/94 | 0.83 | (0.35,1.97) |
| M=W | 5/6 | 4/8 | 0.66 | (0.15, 2.82) |
| M>W | 5/8 | 5/10 | 0.41 | (0.10, 1.63) |
| Test for interaction MASI X treatment, P=0.598 | ||||
Abbreviation: CT, chemotherapy; AA, amino acid; MASI, mutant allele-specific imbalance.
Table 3.
The effect of chemotherapy on RFS by KRAS oncogene substitution and MASI in patients with resected stage I to III disease, adjusted for stage
| CT (No. recurrences/No. patients) | No CT (No. recurrences/No. patients) | Hazard Ratio | 95% Confidence Interval | |
|---|---|---|---|---|
| GLY→CYS | 9/27 | 18/64 | 0.47 | (0.17, 1.30) |
| GLY→VAL | 5/10 | 7/32 | 1.06 | (0.28, 3.94) |
| GLY→ASP | 3/12 | 2/22 | 1.00 | (0.16, 7.60) |
| GLY→ALA | 1/1 | 1/14 | 7.65 | (0.45, 130.19) |
| Other | 3/5 | 3/7 | 1.19 | (0.23, 6.24) |
| Test for interaction AA substitution X treatment, P=0.357 | ||||
| Codon 12 | 23/56 | 33/141 | 0.90 | (0.41, 1.97) |
| Codon 13 | 1/3 | 1/8 | 1.33 | (0.08, 22.18) |
| Test for interaction codon X treatment, P=0.790 | ||||
| M<W | 17/36 | 16/91 | 1.17 | (0.46, 2.96) |
| M=W | 0/0 | 4/8 | 0.53 | (0.10, 2.95) |
| M>W | 2/3 | 6/10 | 0.53 | (0.10, 2.95) |
| Test for interaction MASI X treatment, P=0.392 | ||||
Abbreviation: CT, chemotherapy; AA, amino acid; MASI, mutant allele-specific imbalance.
Discussion
In the largest published series of KRAS mutant lung adenocarcinomas to date, we report no differences in prognosis based on the type of KRAS AA substitution present, a trend toward better survival amongst patients with KRAS codon 13 mutations compared with codon 12 mutations and a markedly negative prognosis associated with the presence of KRAS mutant allele-specific imbalance. It is important to note that this was a largely early stage cohort which is likely a reflection of the minimum tissue requirement needed for inclusion in this analysis.
In cell line models, KRAS codon 12 mutations appear to be a more potent oncogenic driver compared with codon 13 mutations, inducing a higher level of resistance to apoptosis and a predisposition to anchorage-independent growth.17 Remarkably, in the small number of patients with codon 13 mutations in this study, there was a trend toward better OS in univariate analysis, which did not remain significant in multivariate analysis adjusting for age and stage. In the molecular analysis of 1,532 patients included in the LACE meta-analysis, a total of 300 KRAS mutations were identified. KRAS mutations neither considered as a whole nor considered as subsets of AA substitutions or codon types were found to have prognostic value in the resected early stage setting.18,19 Ultimately, KRAS codon 13 mutations are infrequent in number (24 codon 13 mutations in the LACE meta-analysis and 17 in the present study). As such, a robust analysis to further delineate the true prognostic value of KRAS codon 13 in lung adenocarcinoma will require pooling of codon 13 mutations across institutions in order to obtain an adequate number of cases.
In the relapsed metastatic setting, an analysis of KRAS AA substitutions in patients treated on the Biomarker-integrated Approach of Targeted Therapy for Lung Cancer Elimination (BATTLE) trial demonstrated 48 KRAS mutations amongst 268 tumors profiled. The presence of a GLY→CYS or GLY→VAL substitution at codon 12 was associated with significantly worse progression free survival (PFS) compared to the other KRAS AA substitutions or wild-type KRAS.20 Sixty-four patients with KRAS mutant stage IV disease were included in the present analysis, and we were unable to demonstrate an association between KRAS AA substitution subtypes and survival (both OS and RFS) in a multivariate analysis controlling for stage. It should be noted that the BATTLE clinical trial reflects a refractory NSCLC population which is distinct from the present population which represents a predominantly early stage and first line metastatic cohort. Cell line data indicate that codon 12 GLY→CYS and GLY→VAL mutations are associated with activated Ral signaling and decreased factor-dependent Akt activation.20 The difference in the prognostic significance of individual AA substitutions between the present study and the BATTLE analysis may be a reflection of a distinct biology between the relapsed metastatic setting and the adjuvant or first line metastatic setting.
Soh et al. and others previously demonstrated that direct sequencing is a valid method for quantifying MASI using several techniques, including subcloning, plasmid mixture experiments, and restriction fragment length polymorphism combined with band intensity measurement on gel electrophoresis.11,12,15,21,22 Stromal contamination is of concern in the determination of MASI in tumor specimens; however review of H&E slides and manual microdissection limits the proportion of non-tumor DNA in a given sample. The presence of remaining non-tumor DNA would randomly affect all specimens, and non-tumor DNA increases the wild-type peak and decreases the mutant to wild-type peak ratio thereby decreasing the ability to detect MASI on sequencing electropherograms.
We demonstrated that the presence of KRAS mutant MASI was associated with a markedly inferior RFS compared to the absence of MASI. In prior studies, we demonstrated that the presence of MASI was also associated with worse OS, mirroring the adverse RFS with MASI seen in the present study.21 The presence of MASI is frequently associated with KRAS amplification as demonstrated by FISH studies.21 This suggests that the KRAS mutant/wild-type peak ratio seen on sequencing electropherograms is representative of the actual KRAS mutant allele/wild-type allele ratio within the tumor. This allelic imbalance is a reflection both of the post-transcriptional dosage of the KRAS allele and its concomitant kinase activity. While amplification may account for KRAS MASI, other potential mechanisms may include uniparental disomy, chromosome 12 hyperdiploidy, or KRAS homozygous mutation. The implications of KRAS MASI will require prospective validation as a biomarker of prognosis.
In the present study, there was no suggestion of a predictive value for chemotherapy benefit amongst the subtypes of KRAS AA substitutions, the subtypes of KRAS codons or amongst the subpopulations with or without MASI. In both the JBR.10 clinical trial and the LACE meta-analysis, KRAS mutations considered as a whole were not associated with differential chemotherapy effectiveness.5,18 In an analysis of the patients included in the LACE meta-analysis by KRAS mutation subtypes, the subset of patients with KRAS codon 13 mutations fared worse with adjuvant chemotherapy relative to the observation arm.19 In drawing a comparison to the LACE meta-analysis, KRAS codon 13 mutations in the present study were associated with a non-statistically significant trend toward worse OS and RFS with chemotherapy compared with no chemotherapy. However, the number of patients in these sub-analyses is small and the resultant confidence intervals wide.
The major limitation of these predictive analyses (which also applies to the prognostic analyses) is that this represents a biomarker analysis which was not conducted in the context of a prospective randomized clinical trial. Comparisons in this study were made between KRAS mutation subtypes and not relative to a wild-type population, limiting our ability to draw conclusions from our predictive analyses. However, the aims of this study were to ascertain if there were differences in prognosis and predictive benefit between different types of KRAS mutations and not necessarily between KRAS-mutant and KRAS wild-type lung adenocarcinomas. The KRAS wild-type population is in and of itself a molecularly heterogeneous population, that is, there are a significant proportion of patients who are KRAS wild-type, but whose tumors harbor other oncogenic drivers, such as EGFR mutations or ALK translocations amongst others.3 Therefore, we chose to focus the present analyses on a purely KRAS-mutant cohort. The present study is meant to be hypothesis generating, and incorporation of these candidate biomarkers in a prospective fashion in clinical trial design will be necessary to define the true predictive value of KRAS mutation subtypes.
Acknowledgments
Source Funding: M.A.S. is a consultant for Celgene and Teva, and currently receives research funding from Genentech, GSK, Merrimack and Synta.
Grant Support: The study was supported by the NIH/NCI SPORE in Lung Cancer Grant (P50-CA090440) and the NIH/NCI Cancer Center Support Grant (5 P30 CA047904).
Footnotes
Conflict of Interest: For the remaining authors none were declared.
References
- 1.Howlader N, Noone AM, Krapcho M, et al. SEER Stat Fact Sheets: Lung and Bronchus. 2010 http://seer.cancer.gov/csr/1975_2008/
- 2.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kris MG, Johnson BE, Kwiatkowski DJ, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI’s Lung Cancer Mutation Consortium (LCMC) ASCO Meeting Abstracts. 2011;29:CRA7506. [Google Scholar]
- 4.Mascaux C, Iannino N, Martin B, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92:131–139. doi: 10.1038/sj.bjc.6602258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsao MS, Aviel-Ronen S, Ding K, et al. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. J Clin Oncol. 2007;25:5240–5247. doi: 10.1200/JCO.2007.12.6953. [DOI] [PubMed] [Google Scholar]
- 6.Camps C, Jantus-Lewintre E, Cabrera A, et al. The identification of KRAS mutations at codon 12 in plasma DNA is not a prognostic factor in advanced non-small cell lung cancer patients. Lung Cancer. 2011;72:365–369. doi: 10.1016/j.lungcan.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Kalikaki A, Koutsopoulos A, Hatzidaki D, et al. Clinical outcome of patients with non-small cell lung cancer receiving front-line chemotherapy according to EGFR and K-RAS mutation status. Lung Cancer. 2010;69:110–115. doi: 10.1016/j.lungcan.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D Tumor Mutations With Outcome in Patients With Metastatic Colorectal Cancer Treated With First-Line Chemotherapy With or Without Cetuximab. J Clin Oncol. 2012;30:3570–3577. doi: 10.1200/JCO.2012.42.2592. [DOI] [PubMed] [Google Scholar]
- 9.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weir BA, Woo MS, Getz G, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Modrek B, Ge L, Pandita A, et al. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res. 2009;7:1244–1252. doi: 10.1158/1541-7786.MCR-08-0532. [DOI] [PubMed] [Google Scholar]
- 12.Soh J, Okumura N, Lockwood WW, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4:e7464. doi: 10.1371/journal.pone.0007464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet. 1980;6:137–143. doi: 10.1002/ajmg.1320060207. [DOI] [PubMed] [Google Scholar]
- 14.Sasaki H, Hikosaka Y, Kawano O, Moriyama S, Yano M, Fujii Y. Evaluation of Kras gene mutation and copy number gain in non-small cell lung cancer. J Thorac Oncol. 2011;6:15–20. doi: 10.1097/JTO.0b013e31820594f0. [DOI] [PubMed] [Google Scholar]
- 15.Hartman DJ, Davison JM, Foxwell TJ, Nikiforova MN, Chiosea SI. Mutant allele-specific imbalance modulates prognostic impact of KRAS mutations in colorectal adenocarcinoma and is associated with worse overall survival. Int J Cancer. 2012;131:1810–1817. doi: 10.1002/ijc.27461. [DOI] [PubMed] [Google Scholar]
- 16.Chiosea S, Shuai Y, Cieply K, Nikiforova MN, Dacic S. EGFR fluorescence in situ hybridization-positive lung adenocarcinoma: incidence of coexisting KRAS and BRAF mutations. Hum Pathol. 2010;41:1053–1060. doi: 10.1016/j.humpath.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 17.Guerrero S, Casanova I, Farre L, Mazo A, Capella G, Mangues R. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000;60:6750–6756. [PubMed] [Google Scholar]
- 18.Tsao MS, Hainaut P, Bourredjem A, et al. LACE-Bio pooled analysis of the prognostic and predictive value of KRAS mutation in completely resected non-small cell lung cancer (NSCLC) Ann Oncol. 2010;21:Abstract 4218. [Google Scholar]
- 19.Shepherd FA, Bourredjem A, Brambilla E, et al. Prognostic and predictive effects of KRAS mutation subtype in completely resected non-small cell lung cancer (NSCLC): A LACE-Bio study. J Clin Oncol. 2012;30:Abstract 7007. [Google Scholar]
- 20.Ihle NT, Byers LA, Kim ES, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–239. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiosea SI, Sherer CK, Jelic T, Dacic S. KRAS mutant allele-specific imbalance in lung adenocarcinoma. Mod Pathol. 2011;24:1571–1577. doi: 10.1038/modpathol.2011.109. [DOI] [PubMed] [Google Scholar]
- 22.Oakley GJ, 3rd, Chiosea SI. Higher dosage of the epidermal growth factor receptor mutant allele in lung adenocarcinoma correlates with younger age, stage IV at presentation, and poorer survival. J Thorac Oncol. 2011;6:1407–1412. doi: 10.1097/JTO.0b013e31821d41af. [DOI] [PubMed] [Google Scholar]