

Abstract
Deregulation of the mTOR pathway is closely associated with tumorigenesis. Accordingly mTOR inhibitors such as rapamycin and mTOR-selective kinase inhibitors have been tested as cancer therapeutic agents. Inhibition of mTOR results in sensitization to DNA damaging agents, however the molecular mechanism is not well understood. We found that an mTOR-selective kinase inhibitor, AZD8055, significantly enhanced sensitivity of a pediatric rhabdomyosarcoma xenograft toradiotherapy and sensitized rhabdomyosarcoma cells to interstrand crosslinker (ICL) melphalan. Sensitization correlated with drug-induced downregulation of a key component of the Fanconi anemia (FA) pathway, FANCD2 through mTOR regulation of FANCD2 gene transcripts via mTORC1-S6K1. Importantly, we show that FANCD2 is required for the proper activation of ATM-Chk2 checkpoint in response to ICL and that mTOR signaling promotes ICL-induced ATM-Chk2 checkpoint activation by sustaining FANCD2. In FANCD2 deficient lymphoblasts, FANCD2 is essential to suppress endogenous and induced DNA damage, and FANCD2-deficient cells demonstrated impaired ATM-Chk2 and ATR-Chk1 activation, which was rescued by re-introduction of wild type FANCD2. Pharmacological inhibition of PI3K-mTOR-AKT pathway in Rh30 rhabdomyosarcoma cells attenuated ICL-induced activation of ATM, accompanied with the decrease of FANCD2. These data suggest that the mTOR pathway may promote the repair of DNA double strand breaks by sustaining FANCD2 and provide a novel mechanism of how the FA pathway modulates DNA damage response and repair.
Keywords: mammalian target of rapamycin, DNA damage response, ATM, FANCD2, interstrand crosslink
Introduction
Recent data have demonstrated that cyclin dependent kinases (CDKs) are required for the processing of damaged DNA ends and checkpoint activation (1–3), but the molecular mechanism by which CDKs act is unknown. PI3K-AKT and RAS-MAPK are the predominant growth-promoting signaling pathways, which enhance CDK activity and increase cell cycle progression. Cancer genome projects demonstrated that PI3K-AKT and RAS-MAPK are among the most frequently mutated signaling pathways in cancers (4), and both pathways converge on mammalian target of rapamycin (mTOR) signaling (5). mTOR lies at the hub of intracellular and extracellular signal transduction networks via integrating and processing multiple signals, and dictates the rates of macromolecule synthesis and hence cell growth, proliferation and survival (6–8). Aberrant activity of mTOR signaling has been found in most cancers, making mTOR an important target for cancer drug development (7, 9, 10). We previously found that the TOR pathway promotes cell survival at the cost of elevated mutation rate in response to DNA replication stress and DNA damage in yeast (11). Data also demonstrate that inhibiting the mTOR pathway sensitizes many cancer cells to chemotherapy and radiotherapy, however, the molecular mechanism by which this occurs is largely unclear (12–14).
These observations suggest that mTOR signaling maintains some components of the DNA damage and repair response (DDR). Besides homologous recombination, most if not all DNA repair systems are error prone and the major function of DNA damage checkpoint and repair systems is to maximize cell survival. It was intriguing, therefore, to test whether mTOR signaling modulates DDR and enhances DNA damage repair in cancer cells.
In this study, using pediatric rhabdomyosarcoma models in vitro and in vivo, we have investigated the functional linkage between the mTOR pathway and DDR, and found that mTOR signaling sustains FANCD2 mediated signaling pathways to promote DDR via potentiating the ATM checkpoint activation in response DNA double strand breaks.
Materials and Methods
Drugs
AZD8055, MK2206, and PD0332991 were provided by AstraZeneca, Merck, and Pfizer, respectively. Rapamycin was from the NCI drug repository. Melphalan, cisplatin, mitomycin C, cycloheximide and hydroxyurea were purchased from Sigma (St. Louis, MO).
Solid Tumor Xenografts Studies
CB17SC scid−/− female mice (Taconic Farms, Germantown, NY) were used to propagate subcutaneously implanted rhabdomyosarcoma. All mice were maintained under barrier conditions and experiments were conducted using protocols and conditions approved by the institutional animal care and use committee. AZD8055 was administered P.O. daily at 20 mg/kg per day. Rapamycin was dissolved in DMSO (5% final concentration) and diluted in 5% Tween-80 in water and administered I.P. daily at a dose of 5 mg/kg. Tumors were harvested post treatment on day1 or day 4 at different time points.
Cells, siRNA and Plasmids
Rhabdomyosarcoma Rh30 cells were cultured in RMPI 1640 (GIBCO) supplemented with 10% heat-inactivated FBS (GIBCO). Control and ON-TARGETplusSMARTpool siRNAs of CDK4, CDK6, mTOR and FANCD2 were purchased from Dharmacon (Chicago, IL). Plasmid pcDNA3 Myr-AKT1 was a gift of William Sellers (Addgene plasmid 9008). pcDNA-DEST40 and pcDNA-DEST40-FANCD2 Plasmids were provided by Niall G. Howlett. S6K1 knockout MEF was provided by George Thomas. MEF cells were cultured in DMEM (GIBCO) supplemented with 10% heat-inactivated FBS (GIBCO). Lipofectamine 2000 was from Invitrogen (Carlsbad, CA) and transfection of siRNA or plasmids in Rh30 cells was performed according to the manufacture’s instructions.
Immunoblotting and Immunofluorescence Staining
Cells were lysed on ice in RIPA lysis buffer (Cell Signaling Technology) supplemented with protease inhibitors and phosphatase inhibitor (Roche), and 1mM PMSF (Sigma). Immunoblots were probed with the following antibodies: S6, pS6 (S235/236), AKT, pAKT (S473), ATM, pATM (S1981), ATR, DNA-PKcs, CHK1, pCHK1 (S345), CHK2, pCHK2 (T68), p53, p53-S15, mTOR, S6K1, pS6K1 (T89), GSK3β, pGSK3β (S9), p4E-BP1 (T37/46), β-Actin, Myc, GAPDH, Rb, pRb (S780) (Cell Signaling Technology); pH2AX (S139) (Upstate); PARP1, cleaved PARP1 (Abnova); FANCD2 (Epitomics); FANCC, FANCA (Abcam); RAD51, CDK4, CDK6 (Santa Cruz). For immunofluorescence staining, cells grown on sterile coverslips were fixed in 100% cold methanol, washed with TBS and blocked with 1% BSA in PBS. Cells were incubated with the gamma H2AX antibody (Clone JBW301, UPSTATE) diluted in 1% BSA in TBS for 60 min, washed in 1×TBS. The slides were additionally incubated the FANCD2 antibody diluted in 1% BSA in TBS for 60 min, washed in 1×TBS. Secondary antibodies Alexa Fluor 488 goat anti-mouse IgG and TRITC goat anti-rat IgG were from Invitrogen. Sample was simultaneously stained with DAPI to detect nucleus. The fluorescence was viewed with a Zeiss axioskop 2 microscope equipped with DIC, epi-fluorescence and UV blocking filter sets. Images were acquired with a Micromax CCD camera (Princeton Instruments Inc., Monmouth Junction, NJ) and IP lab software (Scanalytics).
RNA Isolation, cDNA Synthesis and RT-PCR
Total RNA from cultured cells was extracted with mirVana miRNA Isolation Kit (Ambion) according to the total RNA isolation protocol. Reverse transcription were performed using the High Capacity RNA-to-cDNA kit (Applied Biosystems) according to the manufacturer’s instructions. Real-time PCR was performed on the 7900HT Fast Real-Time PCR System using the TaqMan® Universal Mastermix II. Human and mouse FANCD2 expression was quantified in real-time with FANCD2 specific FAM dye-labeled MGB-probes and normalized to GAPDH, MYCN or beta-ACTIN (Applied Biosystems).
Cell Cycle Analysis and Colony Formation Assay
Following treatment, cells were trypsinized, fixed with 70% ethanol and stored at 4°C for subsequent FACS analysis of DNA content. For colony formation assay, 500 cells were plated in 10 cm petri dish. Two weeks following treatment, cells were washed with PBS, fixed with cold methanol for 15 min under −20°C, and stained with 1% Crystal Violet (Fisher) in 25% methanol for 15 min. The dishes were washed with water extensively and the blue colonies were counted.
Results
An mTOR Kinase Inhibitor Sensitizes Cancer Cells to Radiotherapy and Chemotherapy
Several mTOR-selective kinase inhibitors have recently been reported that in comparison to rapalogs, abrogate mTORC1-mediated phosphorylation of 4E-BP1 (15, 16). We previously showed that AZD8055 is a potent and specific mTOR kinase inhibitor (17). To test the involvement of mTOR in DDR, we examined the effect of AZD8055 on ionizing radiation using the Rh30 xenograft model. Mice were irradiated (2 Gy/tumor day) using whole body shielding to a range of total doses (from 10–40 Gy) with or without AZD8055 at various starting volumes. In the radiation alone group, radiation treatment induced tumor volume regressions, but tumors regrew and progressed in 14 of 18 mice (78% failure rate) with a radiation dose per volume of tumor of 60 Gy/cm3. In contrast, only in 4 of 15 mice did tumors regrow in the combination XRT-AZD8055 treatment arm (Table 1) with dose per volume of 27 Gy/cm3; a greater than 50% reduction in radiation dose with a similar reduction in failure rate. In Rh30 xenografts AZD8055 has limited single agent activity, slowing tumor progression, but does not induce either stable disease or tumor regression (17). Thus, this mTOR kinase inhibitor may be a promising radiosensitizer.
Table 1.
The mTOR kinase inhibitor, AZD8055, significantly sensitized the pediatric rhabdomyosarcoma xenograft Rh30 to radiotherapy
| Group | Mean Dose | Failures/Total | Failure Rate |
|---|---|---|---|
| XRT | 60 Gy/CC | 14/18 | 78% |
| XRT +AZD | 27 Gy/CC | 4/15 | 27% |
XRT: X-ray treatment; AZD: AZD8055
We previously reported that rapamycin enhanced the activity of the bifunctional alkylating agent, cyclophosphamide, against several pediatric tumor models (18). To extend this observation in vitro, we checked whether AZD8055 sensitizes cultured Rh30 cells to DNA interstrand cross linker (ICL) melphalan, which produces DNA double strand breaks (DSB). We pretreated Rh30 cells with AZD8055 for 16 hr, and transiently exposed the cells to melphalan for 2 hr. Drugs were washed away, and survival was assayed by colony formation. AZD8055 or transient insult with melphalan resulted in 16% and 18% loss of cell viability, respectively. Pretreatment with AZD8055 followed by transient melphalan exposure led to a significant decrease in colony formation (66%) (Fig. 1A and 1B). Thus, targeting mTOR kinase sensitizes cancer cells to ICL based chemotherapies.
Figure 1. The mTOR Pathway Promotes Cell Survival in Response to Mephalan by Sustaining FANCD2.

A, 500 cells were plated in 10 cm dish for 6 hr, then AZD8055 (2 μM) was applied for 16 hr. Melphalan (0.2 μg/mL) was added to untreated or AZD8055 treated cells for 2 hr. The drugs were washed away. Two weeks later, the colonies were counted. Shown was a representative of three independent experiments. B, quantification of the colonies in Figure 1A. Error bars: Mean ± SD (n=3). C, Rh30 cells were treated with AZD8055 (2 μM) for 16 hr. Total proteins were extracted for immunoblotting. D, lymphoblast PD20 cells derived from a Fanconi anemia D2 patient were stably transfected with empty vector (pMMP-Puro) or wild type FANCD2 plasmid (pMMP-wt-FANCD2). FANCD2 and γH2AX were detected by immunoblotting. (−), pMMP-Puro; (+), pMMP-wt-FANCD2. E, Rh30 cells were transfected with control or FANCD2 siRNA. 48 hr later, total proteins were extracted for immunoblotting. F, Rh30 cells were transfected with vector or FANCD2 plasmid. 24 hr later, AZD8055 (2 μM) was added for additional 24 hr. Total proteins were extracted for immunoblotting. G, Rh30 cells were transfected with control or mTOR siRNA. 48 hr later, total proteins were extracted for immunoblotting. H, Rh30 cells were treated with AZD8055 (AZD, 2 μM) or melphalan (MP, 2 μg/ml) for the time indicated. Total proteins were extracted for immunoblotting. UT, untreated. Upper band of FANCD2 staining showed the monoubiquitination of FANCD2. GAPDH and β-Actin served as loading controls.
The mTOR Pathway Controls FANCD2
The above data suggest that mTOR signaling plays an important role for cancer cells to survive DSB producing agents. This may be not due to the decrease of ATM, ATR and DNA-PKcs, as AZD8055 did not alter the protein levels of ATR, RAD51 and DNA-PKcs but slightly increased ATM (Fig. 1C). Of note, exposure to AZD8055 resulted in marked decrease in FANCD2, but not other components of the FA pathway (FANCC, FANCA). FA-pathway deficient cells display phosphorylation of H2AX, a marker of DNA strand breaks, and defect of DNA damage checkpoint in response to DNA damage (19, 20). FA cells, such as FANCD2 deficient PD20 lymphoblast cells, display high levels of γH2AX, and re-expression of FANCD2 in PD20 cells reduced γH2AX levels (Fig. 1D) (19, 20). Similarly, knockdown of FANCD2 by siRNA led to increased γH2AX in Rh30 cells (Fig. 1E). Interestingly, AZD8055 downregulated FANCD2 and induced γH2AX, whereas ectopic overexpression of FANCD2 partially attenuated AZD8055-induced γH2AX (Fig. 1F). These results led us to hypothesize that mTOR signaling may regulate DDR and hence DSB repair by controlling FANCD2 signaling. AZD8055 treatment reduced the levels of FANCD2 in a time-dependent manner (Fig. S1A). Similarly, knockdown of mTOR by siRNA decreased FANCD2 (Fig. 1G). In contrast to melphalan, that causes ICL, inhibition of mTOR signaling did not result in the monoubiquitination of FANCD2 in Rh30 cells (Fig. 1H). In addition, overexpression of wild type AKT1 (Fig. S1B) or myristoylated AKT1 (myr-AKT1) (Fig. S1C) in Rh30 cells increased FANCD2 and was associated with enhanced activity of the AKT pathway as demonstrated by the elevation of the pGSK3β-S9 signal. These data demonstrated that AKT-mTOR signaling controls the levels of FANCD2. To test this further, we treated cultured rhabdomyosarcoma Rh30 cells with mTORC1 specific inhibitor rapamycin, AZD8055 and AKT kinase inhibitor MK2206 (21). Rapamycin inhibited the phosphorylation of pS6K1 (Thr389), a marker of mTORC1 activity, but increased the phosphorylation of AKT at Ser473 due to the mTORC1-S6K1-IRS negative feedback loop (22). Treatment with either AZD8055 or MK2206 resulted in absence of the phosphorylation of both pS6K1 (Thr389), and pAKT (Ser473), indicating inhibition of both mTORC1 and mTORC2 complexes. Similar to AZD8055, MK2006 reduced FANCD2 although the effect of rapamycin was less pronounced (Fig. S1D), demonstrating that AKT-mTOR pathway controls FANCD2.
Our data suggest that mTOR signaling may be involved in the DNA damage response and repair of DSB by regulating the FA signaling pathway. To test this in vivo, we next checked FANCD2 protein levels of the mouse xenografts of Rh18, Rh30 and Rh10 following treatment with AZD8055. Mice received AZD8055 daily for 4 days, and tumors were harvested 1, 4, 8 and 24 hr after the final dose. AZD8055 efficiently and rapidly inhibited both the mTORC1 and mTORC2 complexes, as demonstrated by the decrease of p4E-BP1-T37/46, pS6-S235/6, and pAKT-S473 signals one hour after treatment (Fig. 2A and 2B). At 24 hr post AZD8055 dosing FANCD2 was markedly reduced in all of three tumor models (Fig. 2C). As AZD8055 inhibits both mTORC1 and mTORC2, we additionally checked the FANCD2 of the mouse tumor xenografts Rh30 and Rh18 treated with rapamycin. Similar to AZD8055, inhibition of mTORC1 by rapamycin resulted in apparent downregulation of FANCD2 in vivo (Fig. 2D). These in vivo and in vitro data suggest that FANCD2 is regulated by the mTORC1 pathway.
Figure 2. FANCD2 is Controlled by mTOR Signaling in Pediatric Rhabdomyosarcoma in vivo.
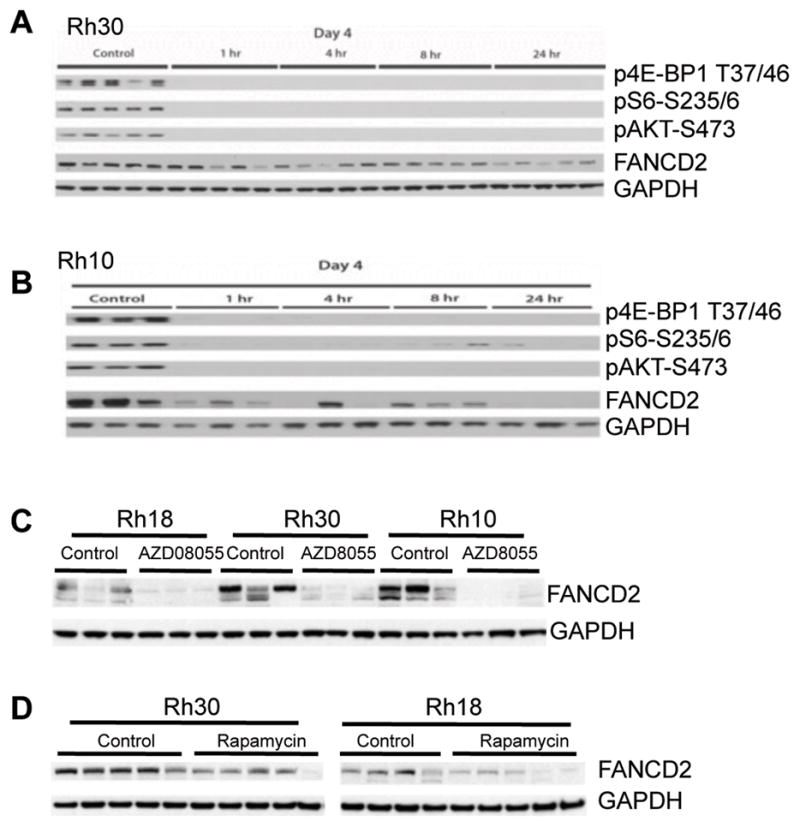
A and B, pediatric rhabdomyosarcoma Rh10 and Rh30 tumor xenograft models were propagated subcutaneously in SCID mice and were treated with mTOR kinase inhibitor AZD8055 at 20 mg/kg/day. Tumors were harvested 1, 4, 8 and 24hr post treatment on day 4 and were pulverized under liquid N2. Total proteins were extracted for immunoblotting. C, mice bearing subcutaneous rhabdomyosarcoma xenografts, Rh18, Rh10 and Rh30 tumor xenografts were treated with mTOR kinase inhibitor AZD8055 (20 mg/kg daily). Tumors were harvested 24 hr after the fourth dose administered. Total proteins were extracted for immunoblotting to detect FANCD2. D, pediatric rhabdomyosarcoma Rh18 and Rh30 tumor xenograft models were propagated subcutaneously in SCID mice and were treated with rapamycin at 5 mg/kg per day. Tumors were harvested 24 hr post treatment on day 1. Total proteins were extracted for immunoblotting.
mTORC1-S6K1 Signaling Controls Transcription of FANCD2
The above results showed that inhibition of mTORC1 led to decreased FANCD2 both in tumor xenografts and cultured cells. As TOR signaling has been implicated in both translation and transcription, we probed the mechanism by which this pathway regulates FANCD2 levels. We determined the mRNA levels of FANCD2 following inhibition of mTOR signaling by real-time RT-PCR. Treatment of Rh30 cells with AZD8055 resulted in a progressive decrease in FANCD2 mRNA (Fig. 3A). Similarly, rapamycin and MK2206 also decreased FANCD2 mRNA (Fig. 3B). Conversely, overexpression of AKT1 increased the mRNA of FANCD2 (Fig. 3C). Thus mTOR signaling appears to regulate either transcription or transcript stability of FANCD2 gene.
Figure 3. The mTOR Pathway Regulates FANCD2 at Transcription Level.

A, Rh30 cells were treated with AZD8055 (2 μM) for the times indicated. The total RNA was extracted to detect FANCD2 mRNA by real-time RT-PCR with GAPDH as internal control. Relative quantity (Ln) of FANCD2 mRNA was plotted. B, Rh30 cells were treated with rapamycin or MK2206 for 12 hr. Total RNA was extracted to detect FANCD2 mRNA by real-time RT-PCR with GAPDH as internal control. Relative quantity (Ln) of FANCD2 mRNA was plotted. C, Rh30 cells were transfected with vector or AKT1-wt plasmid. 48 hr later, the total RNA was extracted to detect FANCD2 mRNA by real-time RT-PCR with GAPDH as internal control. Relative quantity (Ln) of FANCD2 mRNA was plotted. D, Rh30 cells were treated with AZD8055 for 16 hr. Total proteins were extracted for immunoblotting. E, Rh30 cells were treated with rapamycin, AZD8055, MK2206 or PD03332991 (1 μM) for 16 hr. Total proteins were extracted for immunoblotting. F, Rh30 cells were treated with PD03332991 for 12 hr. The total RNA was extracted to detect FANCD2 mRNA by real-time RT-PCR with GAPDH as internal control. Relative quantity (Ln) of FANCD2 mRNA was plotted. G, wild type (MEF WT) and S6K1 KO MEF cells were treated with rapamycin (100 ng/mL) for 24 hr. Total proteins were extracted to detect FANCD2 by immunoblotting with beta actin as loading controls. H, cells were treated as in G, total mRNA was extracted to detect mouse FANCD2 mRNA by real-time RT-PCR with GAPDH as internal control. Relative quantity (Ln) of FANCD2 mRNA was plotted. Error bars: Mean ± SD (n=3).
From yeast to mammalian cells, the TOR pathway promotes cell cycle progression by increasing the activity of cyclin dependent kinases (CDKs) (8). Indeed, AZD8055 robustly reduced both total and pRb-S780 (Fig. 3D). Recently an essential role for CDKs in processing of damaged DNA ends and checkpoint activation has been described (1). Our data demonstrate that mTOR coordinately controls the proteins and mRNAs of FANCD2. These observations suggest that mTOR controls the gene expression of FANCD2 by regulating CDKs. To further test this observation, we treated Rh30 cells with rapamycin, AZD8055 or MK2206 in parallel with a CDK4/6 specific inhibitor PD0332991 (23, 24), and determined FANCD2 by immunoblotting. Similar to AZD8055 and MK2206, PD0332991 significantly reduced FANCD2 (Fig. 3E). We further analyzed the FANCD2 mRNAs of Rh30 cells treated with PD0332991. Consistent with the decreased protein levels, PD0332991 reduced FANCD2 mRNAs (Fig. 3F), demonstrating that the gene transcription/transcript stability of FANCD2 is indirectly controlled by CDKs. We next reduced CDK4 and CDK6 by siRNAs in Rh30 cells. Knockdown of CDK4 led to reduction of both FANCD2 protein (Fig. S2A) and mRNA (Fig. S2B). Interestingly, downregulation of CDK6 by siRNA increased both the protein and mRNA levels of FANCD2, accompanied by an enhanced pRb-S780 signal (Fig. S2A). These data demonstrated that FANCD2 is positively controlled by CDK4, which is consistent with the finding that FANCD2 is regulated by Rb-E2F1 (25).
We further tested whether FANCD2 mRNA is controlled by mTORC1 signaling, by examining whether S6K1 knockout mouse embryonic fibroblast cells (S6K1−/− MEF). S6K1−/− MEFs demonstrated significant downregulation of FANCD2 protein (Fig. 3G) and mRNA (Fig. 3H), and rapamcyin did not further downregulate FANCD2 in S6K1−/− MEFs. Thus, mTOR kinase appears to control FANCD2 principally via the mTORC1-S6K1 pathway.
FANCD2 is Required for the Activation of both ATM-Chk2 and ATR-Chk1 Checkpoints
It was recently reported that FANCD2 mediates the nucleolytic incisions near the ICL and leads to DNA double strand break (DSB) (26, 27), which may result in ATM-Chk2 activation and phosphorylation of H2AX. This model suggests that FANCD2 may be essential for ATM-Chk2 activation in the early steps of FA signaling-mediated repair of ICL-induced DNA lesions. To test this hypothesis, we first compared the status of ATR-Chk1 and ATM-Chk2 activation of the lymphoblast PD20 cells derived from a patient with mutant FANCD2 and the isogenic PD20 cells re-introduced with wild type FANCD2 by immunoblotting. Similar to previous findings (28, 29), FANCD2 deficient PD20 cells demonstrated high levels of γH2AX, an indication of DNA strand breaks while re-introduction of wild type FANCD2 into PD20 cells suppressed γH2AX signal (see Fig. 1D). Reintroduction of wild type FANCD2 into PD20 cells led to an increase of pChk1-S345, and a slight increase in pChk2-T68 but not pATM-S1981 signals, indicating the activation of ATR-Chk1 checkpoint (Fig. 4A). As the central function of ATR-Chk1 is to promote DNA damage repair (30), these data suggest that FANCD2 functions to enhance ATR-Chk1 checkpoint and suppress spontaneous DNA damage under normal growth conditions.
Figure 4. FANCD2 is Required for ATR-Chk1 and ATM-Chk2 Activation in Response to DNA Replications Stress and Damage in Lymphoblast PD20 Cells.
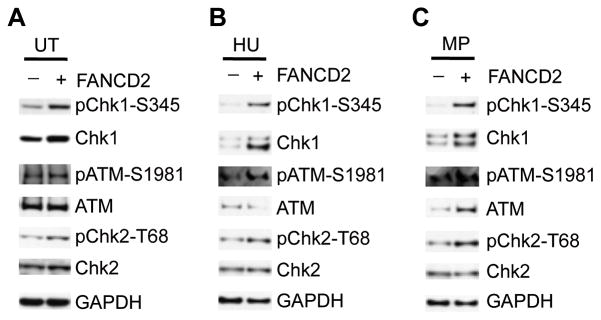
A, total protein extracts from lymphoblast PD20 cells from Fanconi anemia D2 patient stably transfected with empty vector (pMMP-Puro) or wild type FANCD2 plasmid (pMMP-wt-FANCD2) were used for immunoblotting. TU, untreated. B and C, lymphoblast PD20 cells as in A were treated with hydroxyurea (HU, 2 mM) or melphalan (MP, 2 μg/mL) for 6 hr, total protein extracts were for immunoblotting. −, pMMP-Puro; +, pMMP-wt-FANCD2.
FANCD2 is required for repair of ICL-induced DNA damage and intra-S phase checkpoint activation. To test if FANCD2 is required for intra-S phase checkpoint activation, we examined ATR-Chk1 and ATM-Chk2 activation in response to hydroxyurea (HU) in PD20 cells and the PD20 cells re-introduced with wild type FANCD2 by immunoblotting. HU treated PD20 cells displayed decreased pChk1-S345, pATM-S1981, and pChk2-T68 signals compared with those of PD20 with FANCD2 re-introduced (Fig. 4B). We additionally tested the status of ATR-Chk1 and ATM-Chk2 checkpoint in PD20 cells in response to melphalan by immunoblotting. In response to melphalan, there was apparent impairment of the activation of both ATR-Chk1 and ATM-Chk2 checkpoints in PD20 cells, demonstrated by the significant low levels of pChk1-S345, pATM-S1981 and pChk2-T68 signals compared with those of PD20 expressing wild type FANCD2 (Fig. 4C), these data demonstrate that FANCD2 is required for the full activation of both DNA replication and damage checkpoints under normal growth conditions and in response to genotoxins.
The mTOR Pathway is Essential for Proper ATM-Chk2 Checkpoint Activation in Response to ICL
The model that FANCD2 mediated nucleolytic incisions near the ICL results in DNA double strand break (DSB) (26, 27), also predicts that activation of ATR- Chk1 precedes the activation of ATM-Chk2. To explore this model, we treated Rh30 cells with melphalan for a different time points and monitored the dynamics of γH2AX and pChk1-S345 signals. Chk1 was activated in 1 hr and reached its full activation 2 hr following melphalan treatment. In contrast, phosphorylation of H2AX displayed a significant delay (Fig. 5A), which correlated with PARP1 cleavage. Similar to the delayed phosphorylation of H2AX, ATM also demonstrated delayed phosphorylation (Fig. 5B). We next knocked down FANCD2 in Rh30 cells by siRNA, transiently insulted with different concentrations of melphalan, and detected the γH2AX and pChk1-S345 signals by immunoblotting. Downregulation of FANCD2 by siRNA did not effect the activation of Chk1, but apparently attenuated the phosphorylation of H2AX by melphalan (Fig. 5C). We further examined γH2AX by immunocytochemistry of Rh30 treated with FANCD2 siRNA plus melphalan or AZD8055 plus melphalan. Both AZD8055 and FANCD2 siRNA reduced the phosphorylation of H2AX induced by melphalan (Fig. 5D). These results demonstrated that, during the early response to ICL, FANCD2 is required for the proper phosphorylation of H2AX and hence activation of ATM, but not essential for ATR-Chk1 activation, providing an evidence for the proposed model of the function of FANCD2 in response to ICL (26, 27).
Figure 5. ATR-Chk1 Precedes the Activation of ATM-Chk2 in Response to ICL.

A, Rh30 cells were treated with 2 μg/mL melphalan for the time as indicated. Total proteins were extracted for immunoblotting to detect Chk1, pChk1-S345, γH2AX and PARP1. B, Rh30 cells were treated with 2 μg/mL melphalan for the time as indicated. Total proteins were extracted for immunoblotting to detect pChk1-S345, ATM and pATM-S1981. C, Rh30 cells were transfected with control or FANCD2 siRNA. 48 hr later, melphalan with the indicated concentrations was added for 8 hr. Total proteins were extracted for immunoblotting of γH2AX, pChk1-S345 and FANCD2. D, Rh30 cells were transfected with FANCD2 siRNA for 48 hr or AZD8055 (2 μM) for 16 hr. 2 μg/mL melphalan was added alone or in the cells with AZD8055 or FANCD2 siRNA for 8 hr. FANCD2 and γH2AX was detected by immunocytochemistry. DAPI stained nuclei. Scare bar: 20 μM.
Our observations that FANCD2 is required for the timely ATM-Chk2 checkpoint activation in response to ICL and FANCD2 is controlled by mTOR signaling, suggests that the mTOR pathway is essential for the timely activation of ATM-Chk2 checkpoint in response to ICL. To test this, we treated PD20 cells with AZD8055 for 16 hr, followed by addition of melphalan for 5 hr. Whereas FANCD2 deficient PD20 cells displayed impaired Chk2 activation in response to melphalan (Fig. 4C), PD20 cells rescued by wild-type FANCD2 showed robust Chk2 activation, which was abolished by AZD8055 accompanied with reduction of FANCD2 protein (Fig. 6A). Similarly, in Rh30 cells, melphalan-induced pATM-S1981 signals were reduced by either AZD8055 or MK2206, accompanied with downregulation of FANCD2 (Fig. 6B). In addition to melphalan, AZD8055 also abolished the activation of Chk2 by mitomycin C (Fig. 6C). This reduction seems independent of the proteasome pathway because MG132 did not affect AZD8055 induced-reduction of FANCD2 and melphalan-mediated Chk2 activation (Fig. 6C). Thus, these data support the hypothesis that mTOR pathway is required for an early step(s) in ATM-Chk2 checkpoint activation in response to ICL.
Figure 6. The mTOR Pathway is Required for ATM-Chk2 Checkpoint Activation in Response to DNA Interstrand Crosslinker Induced DNA Damage.

A, lymphoblast PD20 cells stably transfected with pMMP-Puro or pMMP-wt-FANCD2 were treated with AZD8055 (2 μM) for 16 hr, then melphalan (MP, 2 μg/mL) was added alone or in combination with AZD8055 and incubated for 6 hr. Immunoblotting was done to detect FANCD2, Chk2, and pChk2-T68. −, pMMP- +, pMMP-wt-FANCD2. B, Rh30 cells were treated with rapamycin (100 ng/mL), AZD8055 (2 μM) or MK2206 (10 μM) for 16 hr. Then melphalan (MP, 2 μg/mL) was added alone or in combination as indicated and incubated for 6 hr.Immunoblotting was done to detect FANCD2 and pATM-S1981. C, Rh30 cells were treated with rapamycin (100 ng/mL), AZD8055 (2 μM) or MK2206 (10 μM) for 16 hr. Then melphalan (MP, 2 μg/mL), cisplatin (5 μM), and mitomycin C (2 μM) was added alone or in combination as indicated for 6 hr. 2 hr prior to protein extraction, MG132 (2 μM) was added as indicated. FANCD2 and pChk2-T68 were detected by Immunoblotting. GAPDH served as loading control.
Discussion
In this study, we observed that an mTOR-selective kinase inhibitor sensitized pediatric rhabdomyosarcoma xenografts to radiotherapy and rhyabdomyosarcoma cells to ICL. Control of FANCD2 transcripts and protein levels by mTOR signaling (Fig. S3A) may, at least partially, contribute to the sensitization of cancer cells. Further, FANCD2 is required for timely ATM-Chk2 checkpoint activation response to ICL in Rh30 cells, suggesting that one of the mechanisms that FA signaling promotes cell survival of ICL is through potentiation of ATM-Chk2 activation (Fig. S3B).
Importantly, chemotherapy using agents that damage DNA and radiotherapy are essential components of many clinical protocols, and form the backbone for curative treatment of several childhood malignancies. ICL based chemotherapy, such as cisplatin, or cyclophosphamide are first line drugs for treatment of many cancers. Our observation that cancer cells are sensitized to melphalan and ionizing radiation by an mTOR kinase inhibitor provides a strategy for cancer therapy by combination of targeting mTOR signaling with these DNA damaging modalities.
Our findings may provide the explanation for recent reports regarding the involvement of mTOR signaling in DDR. Several groups found that inhibition of mTOR signaling resulted in γH2AX and impairment of DNA damage response (DDR) following exposure to DNA damaging agents in cultured cells (31–33). The key function of DDR is to maximize cell survival by promoting DNA repair (30). Induction of γH2AX following inhibition of mTOR signaling suggests impairment of DNA strand break repair. Moreover, inhibition of mTOR signaling in MCF-7 cells was shown to result in impaired recruitment of BRCA1 and RAD51 to DNA repair foci (34). BRCA1 and RAD51 promote repair of DNA double-strand breaks (DSB) by homologous recombination (HR). FA signaling is essential for cells to survive DNA interstrand crosslink (ICL) by coordinating DNA damage repair through translesion DNA synthesis (TLS), nucleotide excision repair (NER) and HR (35). Therefore, downregulation of FANCD2 by inhibition of mTOR signaling may, at least partially, explain the impaired DDR in response to DNA damaging agents.
Recently it was shown that ATM is synthetically lethal with FA (28). Fifteen FANC genes have been identified in FA or FA-like patients (35, 36). FANCD2 is the key component of FA signaling. Activation of FA pathway by ICL leads to monoubiquitination of FANCI-FANCD2 by FA core E3 ubiquitin ligase complex. Activated FANCI-FANCD2 is recruited to DNA damage sites, and helps endonucleases to cut both sides of ICL to generate DNA strand breaks, and promotes TLS, NER and Rad51-medated HR together with BRCA1 and BRCA2 (FANCD1) (37). FANCD2 mediated nucleolytic incisions near the ICL result in a DNA double strand break (DSB) (26, 27), predicted to activate ATM-Chk2. In support of this model, our data demonstrate that FANCD2 is required for ATM-Chk2 activation in the early steps of FA signaling-mediated repair of ICL-induced DNA lesions.
Our observation that FANCD2 is controlled by mTOR signaling and mTOR inhibition led to attenuation of ATM-Chk2 checkpoint activation by ICL, provides the molecular mechanism by which inhibition of mTOR signaling sensitizes cancer cells to radiotherapy and chemotherapy. FANCD2 is required for repair of DSB and intra-S phase checkpoint activation (20, 35). Though FA pathway is essential for cells to survive ICL, FA pathway can be activated by most DNA damage including DSB, single strand breaks, base damage, intra- and interstrand DNA crosslinks, and DNA replication block (37). We found attenuated activation of ATR-Chk1 and ATM-Chk2 in PD20 cells under normal growth conditions, hydroxyurea induced DNA replication stress and melphalan-mediated DNA damage. ATR-Chk1 and ATM-Chk2 checkpoints are the central genome surveillance systems to maintain cell survival in response to endogenous and exogenous DNA damage. DNA damage or replication stress leads to ATR-Chk1 and ATM-Chk2 checkpoints activation to maintain cell survival by stabilizing stalled DNA replication forks, preventing the firing of late origins and promoting DNA damage repair (38, 39). Thus FANCD2 dependent activation of ATM checkpoint may be one of the mechanisms by which FA signaling promotes cell survival in response to ICL.
In addition, our data demonstrated that FANCD2 is positively regulated by CDK4 while being apparently negatively controlled by CDK6 in Rh30 cells (Fig. S2). These results could suggest a distinct role of CDK4 and CDK6 in regulating FANCD2, although this may be context specific. Finally, AZD8055 also decreased the protein levels of FANCD2 in PD20 cells (Fig. 6A), which was rescued by wild type FANCD2 via a retroviral vector. These results indicate that mTOR signaling also regulates FANCD2 at post-transcriptional levels.
In summary, we demonstrated that FANCD2 is under tight control of the mTOR pathway and FANCD2 is required for the timely activation of the ATM checkpoint in response to ICL in pediatric rhabdomyosarcoma cells. Our data provide the basis for the sensitization of cancer cells to DNA damaging agents by targeting the mTOR pathway. The observation that mTOR signaling protects cells from therapeutic modalities used in treatment of human malignancies, gives insight into potential strategies that may enhance therapeutic activity or reduce sequellae that result from high-dose therapies, particularly in children.
Supplementary Material
Acknowledgments
We thank Niall G. Howlett for plasmid pcDNA-DEST40-FANCD2, George Thomas for p70S6K1 knockout MEFs, and Alan D. D’Andrea for PD20 cells.
Grant Support
This work was supported by NIH grants CA77776 (PJH) and a Hope on Wheels grant from the Hyundai Car Company of America (CEP).
Footnotes
Conflict of Interest Statement: The authors consider that there are no actual or perceived competing financial interests.
References
- 1.Cerqueira A, Santamaria D, Martinez-Pastor B, Cuadrado M, Fernandez-Capetillo O, Barbacid M. Overall Cdk activity modulates the DNA damage response in mammalian cells. J Cell Biol. 2009;187:773–80. doi: 10.1083/jcb.200903033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. Embo J. 2004;23:4868–75. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–7. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol. 2010;5:51–75. doi: 10.1146/annurev-pathol-121808-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–30. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 6.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–9. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 7.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–48. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 10.Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol Cell. 2010;40:509–20. doi: 10.1016/j.molcel.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen C, Lancaster CS, Shi B, Guo H, Thimmaiah P, Bjornsti MA. TOR signaling is a determinant of cell survival in response to DNA damage. Mol Cell Biol. 2007;27:7007–17. doi: 10.1128/MCB.00290-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27:2278–87. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288–98. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 16.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houghton PJ, Gorlick R, Kolb EA, Lock R, Carol H, Morton CL, et al. Initial testing (stage 1) of the mTOR kinase inhibitor AZD8055 by the pediatric preclinical testing program. Pediatric Blood Cancer. 2012;58:191–9. doi: 10.1002/pbc.22935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Houghton PJ, Morton CL, Gorlick R, Lock RB, Carol H, Reynolds CP, et al. Stage 2 combination testing of rapamycin with cytotoxic agents by the Pediatric Preclinical Testing Program. Mol Cancer Ther. 2010;9:101–12. doi: 10.1158/1535-7163.MCT-09-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117:1440–9. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pichierri P, Rosselli F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004;23:1178–87. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu R, Liu D, Trink E, Bojdani E, Ning G, Xing M. The Akt-specific inhibitor MK2206 selectively inhibits thyroid cancer cells harboring mutations that can activate the PI3K/Akt pathway. J Clin Endocrinol Metab. 2011;96:E577–85. doi: 10.1210/jc.2010-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rui L, Fisher TL, Thomas J, White MF. Regulation of insulin/insulin-like growth factor-1 signaling by proteasome-mediated degradation of insulin receptor substrate-2. J Biol Chem. 2001;276:40362–7. doi: 10.1074/jbc.M105332200. [DOI] [PubMed] [Google Scholar]
- 23.Saab R, Bills JL, Miceli AP, Anderson CM, Khoury JD, Fry DW, et al. Pharmacologic inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Mol Cancer Ther. 2006;5:1299–308. doi: 10.1158/1535-7163.MCT-05-0383. [DOI] [PubMed] [Google Scholar]
- 24.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–38. [PubMed] [Google Scholar]
- 25.Hoskins EE, Gunawardena RW, Habash KB, Wise-Draper TM, Jansen M, Knudsen ES, et al. Coordinate regulation of Fanconi anemia gene expression occurs through the Rb/E2F pathway. Oncogene. 2008;27:4798–808. doi: 10.1038/onc.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joo W, Xu G, Persky NS, Smogorzewska A, Rudge DG, Buzovetsky O, et al. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. 2011;333:312–6. doi: 10.1126/science.1205805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bogliolo M, Lyakhovich A, Callen E, Castella M, Cappelli E, Ramirez MJ, et al. Histone H2AX and Fanconi anemia FANCD2 function in the same pathway to maintain chromosome stability. EMBO J. 2007;26:1340–51. doi: 10.1038/sj.emboj.7601574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sobeck A, Stone S, Costanzo V, de Graaf B, Reuter T, de Winter J, et al. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Mol Cellular Biol. 2006;26:425–37. doi: 10.1128/MCB.26.2.425-437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 31.Bandhakavi S, Kim YM, Ro SH, Xie H, Onsongo G, Jun CB, et al. Quantitative nuclear proteomics identifies mTOR regulation of DNA damage response. Mol Cell Proteomics. 2010;9:403–14. doi: 10.1074/mcp.M900326-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaur S, Chen L, Yang L, Wu X, Un F, Yen Y. Inhibitors of mTOR overcome drug resistance from topoisomerase II inhibitors in solid tumors. Cancer Lett. 2011;311:20–8. doi: 10.1016/j.canlet.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Huang S, Yang ZJ, Yu C, Sinicrope FA. Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J Biol Chem. 2011;286:40002–12. doi: 10.1074/jbc.M111.297432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Ma Z, Vanderwaal RP, Feng Z, Gonzalez-Suarez I, Wang S, et al. The mTOR inhibitor rapamycin suppresses DNA double-strand break repair. Radiat Res. 2011;175:214–24. doi: 10.1667/rr2323.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitao H, Takata M. Fanconi anemia: a disorder defective in the DNA damage response. Int J Hematol. 2011;93:417–24. doi: 10.1007/s12185-011-0777-z. [DOI] [PubMed] [Google Scholar]
- 37.Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–94. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 39.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.