

Abstract
LD22-4, an 86 amino acid fragment of basic fibroblast growth factor, is an inhibitor of cell migration. LD22-4 inhibits the migration of various tumor cells, endothelial cells, and fibroblasts in vitro and suppresses tumor growth and angiogenesis in vivo. LD22-4 is effective in the presence of multiple growth factors, either alone or in combination, as well as haptotactic factors. LD22-4 inhibits the rate of malignant gliomas prepared from U87MG cells in an orthotopic mouse model by 90% compared to untreated mice. Employing U87MG cells, we identified the LD22-4 membrane receptor as neuropilin 1 (NRP1). The identification of NRP1 as the LD22-4 receptor was based upon mass spectrometric analysis of proteins that bind to LD22-4, immunoprecipitation of an NRP1-LD22-4 complex formed during incubation of LD22-4 with U87MG cells, LD22-4-NRP1 co-immunoprecipitation studies, and binding of LD22-4 to HEK293 cells expressing NRP1. In contrast, NRP1 binding of an inactive mutant of LD22-4 was substantially reduced. As is typical of NRP1 binding proteins, LD22-4 itself binds to heparin and requires heparan sulfate for binding to cells. Heparin addition to migration assays increased the inhibitory activity of LD22-4. In addition to a heparin binding region, LD22-4 contains a five amino acid C-terminus that matches an NRP1 consensus binding sequence. Thus, direct binding experiments, dependence on heparan sulfate, and the presence of a NRP1 consensus binding sequence indicate that NRP1 is the binding site of LD22-4 and mediates inhibition of cell migration.
Keywords: Neuropilin 1, glioma cells, receptor, LD22-4, migration
Introduction
LD22-4 (previously referred to as ATE+31), an 86 amino acid N-terminal fragment of basic fibroblast growth factor (FGF2), is an inhibitor of cell migration (1;2). LD22-4 inhibits the migration of tumor cells, endothelial cells, and fibroblasts in vitro and is effective in the presence of various growth factors that promote enhanced migration [vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF), epidermal growth factor (EGF), platelet derived growth factor (PDGF)], complex mixtures of growth factors and cytokines (serum), as well as haptotactic factors (fibronectin). LD22-4 suppresses tumor growth and angiogenesis by >90% in animal models employing breast, prostate, and lung carcinoma-derived tumors (1;2). LD22-4 is not cytotoxic, does not induce apoptosis, nor does it have a significant effect on proliferation rates of endothelial cells and tumor cells. LD22-4 has a highly basic N-terminal domain containing three clusters of three arginines each, and arginine to alanine substitution in the amino terminal two clusters diminishes LD22-4 activity. Mechanistically, LD22-4 blocks focal adhesion kinase (FAK) phosphorylation in response to growth factors, specifically the phosphorylation of Tyr407-FAK and Ser732-FAK (3). FAK-tyr407 is required to recruit paxillin and vinculin to FAK and to ensure formation of focal adhesions (4). LD22-4 does not affect the phosphorylation of FAK-tyr861, tyr 925, tyr 577, or tyr397 nor the phosphorylation of PYK2, src kinases, Erk1/2, or AKT following growth factor treatment (3–5). Thus, LD22-4 appears to target a highly specific phosphorylation event necessary for cell migration. The failure of growth factors to phosphorylate FAK-tyr407 in the presence of LD22-4 occurs simultaneously with the failure of FAK, focal adhesion plaques, and actin stress fibers to redistribute within the cell cytoplasm and periphery suggesting that LD22-4 causes a systemic failure in the mechanisms promoting cell migration (3).
Neuropilin 1 (NRP1) is a single-pass transmembrane glycoprotein with multiple ligands (6–8). The primary role of NRP1 is the regulation of cell motility, particularly with respect to neural and vascular development (7–12). NRP1 forms a co-receptor complex with VEGFR2 through the bridging of the two receptors by VEGF (6;9;13–16). NRP1-VEGF-VEGFR2 complex formation leads to enhanced VEGFR2 activation, actin reorganization, and the stimulation of cell migration, and blocking VEGF binding to NRP1 diminishes the rate of cell migration while having no effect on cell growth (9;17). Thus, NRP1 has been associated with the VEGF-dependent stimulation of cell migration. Blocking VEGF binding to NRP1 does not affect VEGF-induced phosphorylation of Erk1/2 or Akt indicating that NRP1 is not required for the activation of all of the VEGF signaling pathways and that some occur exclusively through VEGFR-VEGF interaction (17). In addition to VEGF, NRP1 also interacts with other growth factors including FGF2, hepatocyte growth factor (HGF/SF), PDGF, and placental growth factor (PlGF) (10;18–22). The NRP1 binding sites for FGF2 and HGF are distinct from that of VEGF; an antibody that blocks VEGF binding to NRP1 does not interfere with cell migration promoted by FGF2 or HGF (22). NRP1 is also required for p130cas phosphorylation in response to HGF and PDGF in malignant glioma cells (10).
NRP1 is expressed by human tumor cell lines and tumor cells derived from lung, breast, prostate, pancreatic, and colon carcinomas, but is not found in the corresponding normal tissues (7;17;23–26). Clinical studies suggest that NRP1 plays a role in tumor growth and disease progression. It is preferentially expressed in metastatic cells, and is associated with invasive behavior and metastatic potential. Overexpression of NRP1 in prostate and colon cancer cells enhances angiogenesis and tumor growth in animals. We present here evidence that NRP1 acts as the receptor for LD22-4, and that the binding characteristics and subsequent effects are consistent with those defined for protein-NRP1 interaction.
Materials and Methods
Cell culture
U87MG and HEK293 cell lines were purchased from ATCC and cultured as instructed. U87MG cells expressing luciferase (U87MG-luc) were provided by Dr. Patrick McConville (Molecular Imaging, Inc. Ann Arbor, Michigan). The cell lines used in the manuscript have not been tested or authenticated.
Purification and Biotinylation of LD22-4
LD22-4 was prepared as described previously (2). Biotinylation of LD22-4 was performed with 0.8 mg/ml LD22-4 in the presence of 5mM Sulfo-NHS-LC-Biotin (EZ-Link Sulfo-NHS-LC-Biotin (Pierce)) in PBS for 1 hr at room temperature. The reaction was quenched by the addition of 100mM Tris-HCl, pH 7.4, and the reaction mixture was dialyzed against PBS. LD63-6, a functionally deficient mutant of LD22-4 in which the arginines within the first 22 amino acids were converted to alanines, was prepared and biotinylated in the same way. Both LD22-4 and LD63-6 were generated with a His-tag inserted at the C-terminal end to allow for immunodetection with equal avidity.
Migration Assay
Cell migration assays were performed using modified Boyden chambers with a 6.5-mm diameter and a 8.0 μm pore size polycarbonate membrane separating the two chambers (Nalge Nunc International) as described (1). Both sides of the membrane were coated with vitronectin at 1μg/ml for 2hr at room temperature. The results were expressed as a percentage of the number of cells migrating in the presence of PDGF alone ±S.D. All experiments were performed in triplicate.
Binding Experiments
U87MG cells (5× 104 cells) were plated on glass coverslips (18mm) coated with Poly-L-lysine (Sigma) incubated with 2.5 μM biotinylated-LD22-4 or biotinylated-LD63-6 in PBS, fixed in 4% (w/v) paraformaldehyde, and incubated with FITC-Streptavidin.
Fractionation of U87MG plasma membrane and isolation of LD22-4 binding proteins
U87MG glioma cells were disrupted by Dounce homogenization, and centrifuged at 800 × g for 20 min to remove the nuclear fraction. The remaining cell lysates underwent centrifugation at 45,000 × g for 20 min at 4°C, and the pellet was resuspended in 1 ml of lysis buffer containing 1% octyl glucoside (OG) for 2 hr at 4°C. The remaining insoluble material was removed by centrifugation at 45,000 × g for 20 min at 4°C. To isolate LD22-4 binding proteins, LD22-4 or LD63-6-Sepharose was incubated with 100μl U87MG membrane extract 2 hrs at 4°C. The Sepharose-protein complex was removed from the reaction mixture by centrifugation, and treated with 5 mM β-Mercaptoethanol and MS grade trypsin (Invitrogen) overnight at 37°C. Sepharose beads were removed by centrifugation and the digest analyzed by nanoLC-MS/MS on an LTQ Mass Spectrometer at The Scripps Research Institute Center for Mass Spectrometry. The raw data were screened using the MASCOT NCBInr Homo Sapiens data base.
Generation of HEK293 cells expressing NRP1
HEK293 cells were transfected with a plasmid consisting of the full length human NRP1 cDNA in a pENTR221 mammalian expression vector (GeneCopoeia, Inc).
LD22-4-NRP1 Co-Immunoprecipitation Analysis
Binding of LD22-4 to NRP1 was analyzed by three methods: 1) U87MG cells were treated with 2.5 μM LD22-4 or LD63-6 for 10 min at 37°C, washed with cold PBS twice, and lysed with immunoprecipitation buffer containing 1% OG plus protease inhibitors. Equal amounts of protein were treated with anti-neuropilin-1 antibody overnight at 4°C, the immune complexes precipitated with protein A/G agarose (Calbiochem) and samples were analyzed by SDS-PAGE and immunoblotting with biotinylated anti-His antibody; 2) Cell membrane extracts were prepared and 2.5 μM LD22-4-Sepharose or LD63-6-Sepharose was incubated with 100μl U87MG membrane extract for 4 hrs at 4°C. The complexes were removed by centrifugation and analyzed by SDS-PAGE and immunoblotting with α-NRP1 antibodies; 3) U87MG cell membrane extracts were prepared and 100μl incubated with α-NRP1-agarose conjugates overnight at 4°C. The NRP1-α-NRP1-agarose was incubated with 2.5μM LD22-4 or LD63-6 for 4 hrs at 4°C. Complexes were precipitated by centrifugation and analyzed by SDS-PAGE and immunoblotting with α-His and α-NRP1 antibodies.
In vivo analysis of LD22-4 activity
All experiments were performed with CD-1 nu/nu female mice, 6–7 weeks old. All procedures carried out in this experiment were conducted in compliance with all the laws, regulations and guidelines of the National Institutes of Health (NIH) and with the approval of the Institutional Animal Care and Use Committee. Each animal underwent surgery for implantation of a stainless steel cannula (13 mm long and 0.635 mm in diameter) 3 mm into the right frontal brain. Animals were allowed to recover from surgery for one week before tumor cell inoculation. Mice were treated with 0, 0.44 or 4.4 μg LD22-4 every four days and BLI performed every 7 days.
RESULTS
Inhibition of glioma migration and tumor growth by LD22-4
LD22-4 inhibited U87MG cell migration in a dose dependent manner with maximum inhibition (26.2±8.5% residual activity) occurring at 2.5 μM (Fig. 1A). No further inhibition occurred at higher concentrations of LD22-4. This maximum inhibition is consistent with that observed with all cell types and chemoattractants in previous studies (1–3). LD 63-6, the LD22-4 mutant containing arginine to alanine substitutions in the N-terminus (1), was less effective in inhibiting cell migration than LD22-4, reducing migration by only 20% (to 80.3 ± 7.8% of control cultures) at the same concentration (Fig. 1B). Little difference in the inhibitory activity between biotinylated-LD22-4 and unlabeled LD22-4 was observed (Fig. 1C).
Figure 1.
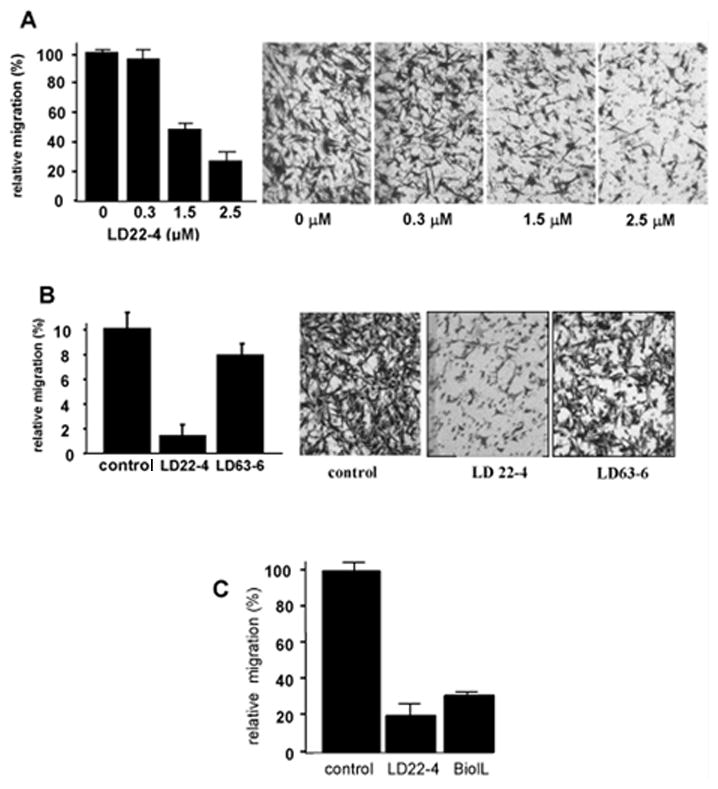
LD22-4 inhibits the migration of U87MG cells. A. U87MG cells were treated with increasing concentrations of LD22-4 and the effect on migration in response to 10 ng/ml PDGF tested. B. The effect of 2.5 μM LD63-6 on U87MG cell migration compared to cells treated with vehicle or 2.5 μM LD22-4. C. The effect of biotinylated-LD22-4 (B-LD22-4) on U87MG cell migration. Both biotinylated-LD22-4 and native LD22-4 were added at a concentration of 2.5μM. All results represent an average (n=3, each performed in triplicate) of the number of cells migrating to the bottom of the filter and expressed as a percentage ±S.D of the number of cells migrating in the presence of PDGF alone.
The effect of LD22-4 on the growth of malignant gliomas was tested in an orthotopic mouse model. Animals were treated with 0, 0.44 μg (17.6 μg/kg), or 4.4 μg (176 μg/kg) LD22-4 (Fig. 2A) and tumor growth measured by BLI every 7 days (arrows). In all three cohorts, tumor growth remained static for 20 days followed by a comparable increase from 20 to 27 days (Fig. 2A). Tumor size continued to increase in the untreated and 0.44 μg LD22-4-treated animals, but animals treated with 4.4 μg LD22-4 showed no further increase in mean BLI intensity; at day 35, the average tumor size in these animals was only 21±4% that of the control group (p=0.016). Tumors in the group treated with 0.44 μg were 65±16% that of the untreated group although this decrease was not statistically significant (p=0.29). The group medians were 13% and 84% of the control group in the animals treated with 4.4 μg and 0.44 μg LD22-4, respectively (Fig. 2B). The experiment was repeated with 4.4 μg LD22-4 and similar results were observed (Fig. 2C); at day 35 the tumors were 10±2% (group mean; p=0.017) or 9% (group median) of those in the untreated group. No treatment-related deaths occurred during the study, and changes in the weights of control and LD22-4-treated animals were similar (data not shown). Histological analysis of the brains at day 35 confirmed the BLI results (Fig. 2D); 4.4 μg LD22-4-treated animals showed only traces of residual tumor (circled area) compared to the large tumors present in the brains of untreated mice.
Figure 2.
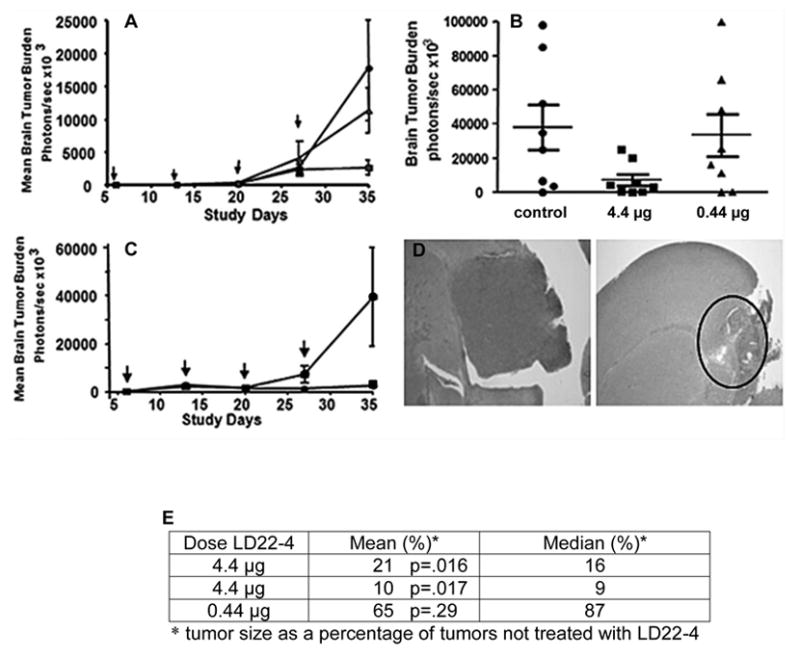
Effect of LD22-4 on malignant glioma tumors in a mouse orthotopic model. CD-1 nu/nu female mice were implanted with 1 × 105 U87MG tumor cells expressing luciferase. One week later, 20μl of saline containing 0 (●), 0.44 μg (▲), or 4.4 μg (■) LD22-4 was infused at the site of the tumor. Treatment was repeated every four days and tumor size was measured every 7 days by BLI (arrows). Treatment with LD22-4 was terminated at day 23 and the animals sacrificed at day 35. A. Tumor response to LD22-4. Mean BLI values ± S.D. at each time point. B. Scatter plot of BLI values at day 35. C. Repeat of the experiment presented in A with 4.4 μg LD22-4. D. Histological evaluation of tumors after treatment with 4.4 μg LD22-4. Control animals, left panel; treated animals, right panel. The location of tumor cell implantation is circled. Magnification, 4X.
LD22-4 Binding to U87MG cells
U87MG cells were incubated with 2.5 μM biotinylated-LD22-4 in the presence or absence of unlabeled LD22-4 (Fig. 3). LD22-4 was uniformly distributed throughout the cell surface (Fig. 3A). To verify that binding was specific, unlabeled LD22-4 at a 20-fold (Fig. 3B) and 60-fold (Fig. 3C) molar excess was included in the binding solution. A 20-fold excess of unlabeled LD22-4 significantly reduced fluorescence intensity while little detectable fluorescence remained at 60-fold excess. In contrast, binding of LD63-6 was significantly reduced, but not completely eliminated (Fig. 3D, control; 3E, LD63-6). No binding was observed with trypsin-treated biotinylated-LD 22-4 indicating that the positive signal were not due to the biotinylation of cell surface molecules by residual biotin reagents (data not shown).
Figure 3.
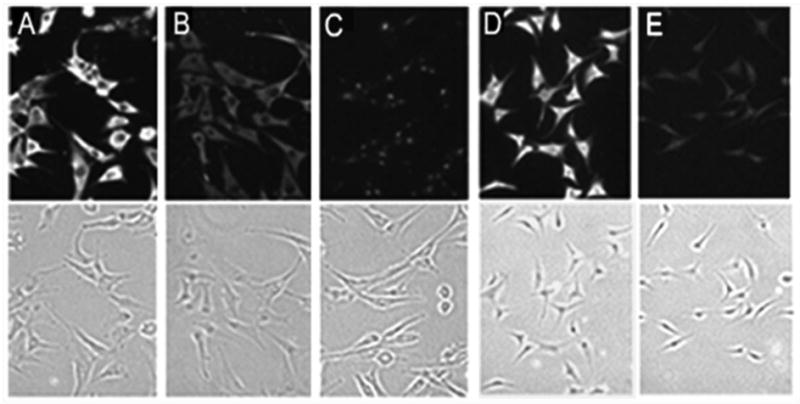
Binding of LD22-4 to U87MG cells. Competition binding studies were performed with 2.5 μM biotinylated-LD22-4 in the absence (A) or presence of 20-fold (B) or 60-fold (C) unlabeled LD22-4. Fluorescence images are shown in the upper panels and transmission images are shown in the bottom panels. In separate experiments, binding studies were performed with 2.5 μM biotinylated-LD22-4 (D) or biotinylated-LD63-6 (E). Magnification: 100X.
Identification of the LD22-4 binding protein
Isolation of an LD22-4 cell surface binding protein was performed by subjecting U87MG cell membrane extracts to affinity chromatography on LD22-4-Sepharose and LD63-6-Sepharose, digestion of bound proteins trypsinization, and the digest analyzed by mass spectrometry-based proteomics. Of the proteins binding specifically to LD22-4, only one was identified as a known surface receptor with a transmembrane sequence and a biological role in the regulation of cell migration, NRP1. Table 1 lists the peptides corresponding to NRP1 that were obtained when the LD22-4 binding proteins were subjected to mass spectrometry.
Table 1.
| Mr (exp) | Mr (calc) | Expect | Score | Peptides | Neuropilin 1 |
|---|---|---|---|---|---|
| 1897.4258 | 1896.8278 | 2.8×10−4 | 65 | R.LNYPENGWTPGEDSYR.E | 307 RLNYPENGWTPGEDSYRE 324 |
| 2090.3026 | 2088.8813 | 0.24 | 35 | K.YDYVEVFDGENEGHFR.G | 83 KYDYVEVFDGENENGHFRG 101 |
| 2535.9037 | 2534.1310 | 1.4×10−4 | 67 | K.TGPIQDHGTGDGNFIYSQADENQKG | 679 KTGPIQDHTGDGNFIYSQADENQKG 703 |
Mr (exp), observed peptide mass; Mr (calc), calculated peptide mass; Expect value, frequency this match occurs by chance, <0.05= statistical confidence at the 5% threshold; Score, ion score.
Confirmation that LD22-4 specifically bound to NRP1 was obtained with HEK293 cells expressing human NRP1 (HEK293-NRP1) (Fig. 4). Expression of NRP1 by the HEK293 transfected cells was validated by FACS analysis of cells transfected with the NRP1 gene (Fig. 4B, α-NRP1). For LD22-4 binding studies, 0, 100 and 1000 nM biotinylated-LD22-4 were incubated with HEK293 or HEK293-NRP1; no binding of LD22-4 was observed at 100nM while 1000 nM LD22-4 caused a shift in fluorescence demonstrating that the presence of NRP1 on the cell surface promotes LD22-4 binding. Binding of LD22-4 to HEK293-NRP1 but not HEK293 parental cells was also demonstrated by immunohistochemical analysis and fluorescent microscopy (Fig. 4A).
Figure 4.

Binding of LD22-4 to NRP1 expressing HEK293 cells. A. The expression of NRP1 by HEK293-NRP1 cells was confirmed by FACS analysis (α-NRP1). Biotinylated-LD22-4 at 0, 100, 1000 nM was incubated with the cells and the presence of biotinylated-LD22-4 was assessed by incubating cells with fluorescein-conjugated streptavidin. The cells were analyzed by FACS analysis for specific LD22-4 binding which fell within the gated region (M1). A. native HEK293 cells; B. HEK293-NRP1 cells. B. Biotinylated-LD22-4 was incubated with HEK293-NRP1 and native HEK293 cells and bound LD22-4 visualized by fluorescence microscopy.
Further evidence that LD22-4 binds to NRP1 was sought by co-immunoprecipitation experiments using U87MG membrane NRP1. Three approaches were taken: 1) LD22-4-Sepharose or LD63-6-Sepharose was incubated with U87MG cell membrane extracts, and the presence of NRP1 evaluated by immunoblotting (Fig. 5A). NRP1 in crude membrane extracts appeared as a doublet with an upper band representing a molecular weight of 130 kDa, the defined molecular weight of NRP1, and a lower band of approximately 110 kDa. NRP1 has also been observed as a doublet in extracts of U87MG cells, smooth muscle cells, endothelial cells, and tissue removed from glioma tumors in previous studies (10;19;27). No explanation of the source of the lower band has been presented, and it is unclear whether it is the result of proteolytic cleavage or regulated transcriptional or translational modification. Immunoblot analysis of LD22-4- and LD63-6-Sepharose binding proteins showed NRP1 had associated with both although a much smaller amount was observed in samples containing LD63-6-Sepharose (Fig. 5A). However, only the upper NRP1 band of the doublet was observed in both samples. Since it is not clear whether the difference in molecular weight is due to protein size or post-translational modification, the apparent difference in affinity cannot be explained. 2) Formation of an NRP1- LD22-4 complex was evaluated by isolating NRP1 from U87MG membrane extracts with α-NRP1-agarose and then incubating the immune complex with LD22-4 or LD63-6 (Fig. 5B). Immunoblot analysis of the resulting agarose-αNRP1 associated proteins revealed the presence of LD22-4. LD63-6 was also detected but at lower levels than LD22-4. To ensure that the bound NRP1 was presented to LD22-4 and LD63-6 in equal amounts, aliquots of agarose-αNRP1 were removed after incubation with the membrane extracts and the relative amount of NRP1 determined. The results showed little difference in the amount of NRP1 available to LD22-4 and LD63-6 (Fig. 5B, right panel). 3) Direct cell surface binding experiments were performed by incubating LD22-4 or LD63-6 with intact U87MG cells and isolating the complexes after cell extraction by immunoprecipitation with α-NRP1-agarose (Fig. 5C). LD22-4 was present while LD63-6 was barely detectable (left panel). Comparison of the amount of NRP1 captured by αNRP1-agarose in extracts containing LD22-4 or LD63-6 showed that they were equal.
Figure 5.

Co-Immunoprecipitation of LD22-4 and NRP1. A. LD22-4 or LD63-6-Sepharose was incubated with U87MG membrane extracts and the bound protein analyzed for NRP1 by Western blot (left panel). NRP1 in U87MG plasma membrane extracts (PM) or whole cell extracts (CE) were visualized by Western blot (right panel). B. NRP1 was isolated from U87MG membrane extracts by αNRP1-agarose and the agarose-αNRP1-NRP1 complex incubated with LD22-4 or LD63-6. Western blot analysis was performed with α-His antibodies (left panel) to detect LD22-4 and LD63-6. The relative amount of NRP1 presented to LD22-4 and LD63-6 is shown in the right panel. C. LD22-4 or LD63-6 was incubated with intact U87MG cells, the cells extracted, and the extracts incubated with αNRP1-agarose. Agarose complexes were analyzed for LD22-4 and LD63-6 by Western blot. (left panel). The relative amounts of NRP1 captured by the αNRP1-agarose was measured by removing an equal aliquot of agarose after incubation with cell extracts and analyzing them for NRP1 (right panel).
LD22-4 binds heparin
Heparin plays a crucial role in the interaction between NRP1 and its binding proteins. VEGF, FGF2, PDGF, AND HTLV all bind to heparin and removal of heparan sulfates from the cell surface limit the binding of the proteins to NRP1(21;22;28;29). To determine whether LD22-4 has an affinity for heparin, binding experiments were performed with LD22-4 and heparin-agarose (Fig. 6). Under physiologic salt concentrations, LD22-4 bound to heparin-agarose and was only partially eluted with 350 mM NaCl. Complete elution of the protein from heparin required 500 mM NaCl (Fig. 6A). Elution of LD22-4 from the heparin-agarose could also be achieved with free heparin with 50% removal at 125 μg/ml heparin and complete removal at 625 μg/ml. (Fig. 6B, left panel). Little binding of LD63-6 to heparin was observed (Fig. 6B, right panel) suggesting that the arginine-rich N-terminus of LD22-4 is required for heparin binding. Heparinase treatment prior to adding the protein resulted in a decline in LD22-4 binding which was not further reduced with additional lyase. To determine whether cell surface heparan sulfate was required for LD22-4 binding to U87MG cells, cells were treated with heparitinase I (Fig. 6C,D). LD22-4 binding was reduced but not completely eliminated. Immunological detection showed that heparan sulfate could not be detected on the cell surface following digestion (Fig. 6E). Lastly, to metabolically block the sulfation of GAGs, cells were treated with sodium chlorate for 20 hrs and binding experiments repeated. Incubation with 20mM sodium chlorate reduced LD22-4 binding although some residual binding could be observed (Fig. 6F). Cell viability following sodium chlorate treatment was 85±6%.
Figure 6.

LD22-4 binds heparin sulfate. A. LD22-4 was mixed with heparin-agarose and then treated with 150 mM, 350 mM, or 500 mM NaCl. LD22-4 remaining on the heparin-agarose and present in the eluates was detected by Western blot analysis. 1, eluate; 2, heparin-agarose. B. LD22-4 (left panel) was mixed with heparin-agarose and heparin-bound LD22-4 treated with vehicle, 125μg/ml, or 625μg/ml heparin. LD22-4 remaining bound to the heparin-agarose or present in the eluate was detected by Western blot. 1, heparin-agarose; 2, supernatant. The experiment was repeated with LD63-6 (right panel). C, D. U87MG cells were treated with 2.5 and 10 U/ml heparinase I (C) or 6mU/ml heparitinase I (D) and LD22-4 binding studies performed. Magnification: 100X. E. U87 cultures treated heparinase or heparitinase were stained with anti-heparan sulfate antibodies to assess the effect of the digestion. F. U87MG cultures were treated with 10 or 20mM sodium chlorate and LD22-4 binding studies performed. G. The effect of heparin on the inhibition of migration by LD22-4. Cell were treated with 2.5 μM LD22-4 in the presence or absence of 10 μg/ml heparin and the effects on migration determined. The results are presented as the average (n=3, each performed in triplicate) of the number of cells migrating to the bottom of the filter for each condition and the results expressed as a percentage ±S.D of the number of cells migrating in the presence of PDGF alone.
The functional effect of heparin on LD22-4 activity was assessed by treating glioma cells with LD22-4 in the presence or absence of 10 μg/ml heparin (Fig. 6G). Heparin itself at a concentration of up to 625 μg/ml had no effect on U87MG cell migration (data not shown). However, when cells were treated with 2.5μM LD22-4 and 10μg/ml heparin, the rate of cell migration declined from 17.5+6.6% to 6.6+4.3% of untreated cultures.
DISCUSSION
In this report, we identify NRP1, a growth factor co-receptor linked to the control of cell migration, as a cell surface binding site for LD22-4. This conclusion is supported by mass spectrometric analysis of proteins that bind to LD22-4, immunoprecipitation of an NRP1-LD22-4 complex formed during incubation of U87MG cells with LD22-4, co-immunoprecipitation of the two proteins, and binding of LD22-4 to HEK293 cells expressing NRP1.
There are several characteristics of LD22-4 that are consistent with a protein that binds to NRP1. One of the most striking is the C-terminal sequence of LD22-4. Studies employing phage display screens to identify a class of peptides that induce cell binding and internalization showed that those with the greatest binding affinity had a terminal consensus R/KXXR/K sequence (30). Most efficient were peptides with a C-terminal arginine residue preceded by an arginine or lysine. Optimum binding affinity occurred when two non-basic amino acids separated the R/K at either end of the consensus sequence. For example, a peptide containing the sequence TKPPR (enhanced tuftsin) binds to endothelial cells around 20-fold more avidly than the peptide TKPR (tuftsin) (31). NRP1 was identified as the receptor for the peptides containing this motif. VEGF165 interacts with NRP-1 through a C-terminal consensus-like sequence (CRCDKPRR) and tuftsin, enhanced tuftsin, and A7R (ATWLPPR), compete with VEGF165 for NRP-1 binding (31–33). This occurred in tumor cell lines, vascular endothelial cells, and in cells isolated from normal mouse organ. In addition, this consensus sequence is found within the HTLV surface unit, and is required for virus binding to NRP1. The C-terminus of LD22-4, KDPKR, is identical to the consensus binding sequence defined by this class of NRP1 binding proteins. Thus, it is likely that LD22-4 is one of a class of proteins that binds to NRP1 through this specific sequence. In fact, deletion mutagenesis studies showed that the absence of the C-terminal 11 amino acids of LD22-4 (which includes the KDPKR sequence) reduced the inhibition of cell migration by half. Therefore, we propose that maximum binding of LD22-4 to NRP1 is dependent on both the C-terminal sequence and the heparin binding region.
Heparin binding to growth factors and NRP1 itself is an essential part of NRP1 interaction, dimerization, and signal transduction (6;21;22;32;34). Both VEGF165 and NRP1 bind heparin and heparan sulfate which enhance the interaction between the two proteins by 100-fold. LD22-4-heparin binding studies show that the protein has an affinity for heparin that withstands salt concentrations greater than physiologic levels, and requires an excess of heparin for dissociation. The reduction of LD22-4 binding following heparinase digestion of the cell surface and the increased inhibition of migration in the presence of added heparin indicates that LD22-4-heparin binding has a functional consequence leading to increased LD22-4 activity. Heparan sulfates on the cell surface also appear to play some role in LD22-4 binding since enzymatic digestion by heparitinase reduces LD22-4 binding. The apparent difference in binding of LD22-4 following digestion with each lyase is probably due to differing requirements for the specific GAGs affected by the enzymes. In both cases of GAG degradation, LD22-4 binding is not completely eliminated, suggesting that there are multiple factors required for optimum binding. The NRP1 C-terminal consensus binding sequence is a likely candidate for a secondary binding site that acts in concert with the GAGs.
The arginine rich N-terminal sequence on LD22-4 is consistent with a heparin binding site. The consensus sites in many glycosaminoglycan binding proteins have been defined as arrays of basic amino acid clusters (arginine, lysine and/or histidine) with arginine containing peptides having the greatest affinity for heparin. The most commonly observed spacing pattern is a single non-basic amino acid between basic amino acid clusters (35). The LD22-4 N-terminus contains three regions of three arginines apiece with each arginine separated by a glycine. The failure of the mutant (LD63-6) to bind heparin and its reduced inhibitory activity suggests that the arginine-rich region of LD22-4 is responsible for heparin binding and is associated with maximum inhibition of migration.
The mechanism by which LD22-4 blocks cell migration through NRP1 binding remains to be elucidated. Simple competition for growth factor binding to NRP1 would involve LD22-4 blocking the interaction of multiple growth factors which do not bind to the same locus on NRP1. In addition, neither IGF1 nor fibronectin have been found to bind to NRP1. An alternative to directly interfering with growth factor binding is blocking the transmission of signals that promote migration. Proteins involved in the formation of signaling receptor complexes are often single transmembrane domain proteins which form homo- or hetero-dimers and oligomeric complexes that are crucial for signal transduction (36–38). NRP1 fits this description and has a putative dimerization motif found in the transmembrane domain (39;40). If NRP1 signaling is dependent on the formation of NRP1 dimers or oligomers as this model predicts, then LD22-4 could disrupt NRP1 function by interfering with NRP1 dimerization or its interaction with other membrane receptors. Lastly, the interaction between LD22-4 and NRP1 may directly interfere with signal transduction by suppressing the association of NRP1 with cytoplasmic proteins that initiate intracellular signaling pathways. There are also studies that suggest that NRP1does not necessarily function solely as a co-receptor, but can signal independently of growth factor receptor interaction through NRP1 cytoplasmic domain interaction with non-receptor intracellular proteins (8;10;16;41;42). Deletion mutagenesis studies show that the C-terminal three amino acids, serine-glutamic acid-alanine, are essential for NRP-1-mediated HUVEC migration and that this involves the RGS-GAIP-interacting protein which binds to NRP1 through the SEA-COOH motif (16;43). Thus, another mechanism by which LD22-4 may inhibit migration is through interference or disruption of interaction between the NRP1-cytoplasmic domain and intracellular proteins.
Taken together, the data presented in this study are consistent with NRP1 acting as the cell surface receptor for LD22-4. The fact that NRP1 regulates cell migration is also consistent with it being the target of a protein such as LD22-4 that acts as an inhibitor of cell motility. The efficacy of LD22-4 in the presence of various growth factors and the identification of NRP1 as the target for LD22-4-cell interaction suggest that there is a binding site on NRP1 for LD22-4 that can be exploited to inhibit cell migration regardless of its stimulus. Identification of the precise binding site on NRP1 and the mechanism by which it effects migration will allow for development of more efficacious compounds to control tumor growth and angiogenesis.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by NIH grant RO1 CA081209 to EGL.
Footnotes
The authors disclose no potential conflicts of interest
Reference List
- 1.Ding L, Donate F, Parry GC, Guan X, Maher P, Levin EG. Inhibition of cell migration and angiogenesis by the amino-terminal fragment of 24kD basic fibroblast growth factor. J Biol Chem. 2002;277:31056–61. doi: 10.1074/jbc.M203658200. [DOI] [PubMed] [Google Scholar]
- 2.Levin EG, Sikora L, Ding L, Rao SP, Sriramarao P. Suppression of tumor growth and angiogenesis in vivo by a truncated form of 24-kd fibroblast growth factor (FGF)-2. Am J Pathol. 2004;164:1183–90. doi: 10.1016/S0002-9440(10)63206-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin AH, Eliceiri BP, Levin EG. FAK mediates the inhibition of glioma cell migration by truncated 24 kDa FGF-2. Biochem Biophys Res Commun. 2009;382:503–7. doi: 10.1016/j.bbrc.2009.03.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin EG. Cancer therapy through control of cell migration. Curr Cancer Drug Targets. 2005;5:505–18. doi: 10.2174/156800905774574048. [DOI] [PubMed] [Google Scholar]
- 5.Piotrowicz RS, Maher PA, Levin EG. Dual activities of 22–24 kDA basic fibroblast growth factor: inhibition of migration and stimulation of proliferation. J Cell Physiol. 1999;178:144–53. doi: 10.1002/(SICI)1097-4652(199902)178:2<144::AID-JCP3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 7.Pellet-many C, Frankel P, Jia H, Zachary I. Neuropilins: structure, function and role in disease. Biochem J. 2008;411:211–26. doi: 10.1042/BJ20071639. [DOI] [PubMed] [Google Scholar]
- 8.Zachary IC, Frankel P, Evans IM, Pellet-many C. The role of neuropilins in cell signaling. Biochem Soc Trans. 2009;037:1171–8. doi: 10.1042/BST0371171. [DOI] [PubMed] [Google Scholar]
- 9.Pan Q, Chathery Y, Wu Y, Rathore N, Tong RK, Peale F, et al. Neuropilin-1 Binds to VEGF121 and Regulates Endothelial Cell Migration and Sprouting. J Biol Chem. 2007;282:24049–56. doi: 10.1074/jbc.M703554200. [DOI] [PubMed] [Google Scholar]
- 10.Pellet-many C, Frankel P, Evans IM, Jünemann-Ramírez M, Zachary I. Neuropilin-1 mediates PDGF stimulation of vascular smooth muscle cell migration and signalling via p130C as. Biochemical Biochem J. 2011;435:609–618. doi: 10.1042/BJ20100580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bagnard D, Vaillant C, Khuth ST, Dufay N, Lohrum M, Puschel AW, et al. Semaphorin 3A-vascular endothelial growth factor-165 balance mediates migration and apoptosis of neural progenitor cells by the recruitment of shared receptor. J Neurosci. 2001;21:3332–41. doi: 10.1523/JNEUROSCI.21-10-03332.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia H, Cheng L, Tickner M, Bagherzadeh A, Selwood D, Zachary I. Neuropilin-1 antagonism in human carcinoma cells inhibits migration and enhances chemosensitivity. Br J Cancer. 2010;102:541–52. doi: 10.1038/sj.bjc.6605539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Favier B, Alam A, Barron P, Bonnin J, Laboudie P, Fons P, et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood. 2006;108:1243–50. doi: 10.1182/blood-2005-11-4447. [DOI] [PubMed] [Google Scholar]
- 14.Fuh G, Garcia KC, de Vos AM. The Interaction of Neuropilin-1 with Vascular Endothelial Growth Factor and Its Receptor Flt-1. J Biol Chem. 2000;275:26690–5. doi: 10.1074/jbc.M003955200. [DOI] [PubMed] [Google Scholar]
- 15.Gu C, Limberg BJ, Whitaker GB, Perman B, Leahy DJ, Rosenbaum JS, et al. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J Biol Chem. 2002;277:18069–76. doi: 10.1074/jbc.M201681200. [DOI] [PubMed] [Google Scholar]
- 16.Prahst C, Héroult M, Lanahan AA, Uziel N, Kessler O, Shraga-Heled N, et al. Neuropilin-1-VEGFR-2 Complexing Requires the PDZ-binding Domain of Neuropilin-1. J Biol Chem. 2008;283:25110–4. doi: 10.1074/jbc.C800137200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, et al. Blocking Neuropilin-1 Function Has an Additive Effect with Anti-VEGF to Inhibit Tumor Growth. Cancer Cell. 2007;11:53–67. doi: 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 18.Ball SG, Bayley C, Shuttleworth CA, Kielty CM. Neuropilin-1 regulates platelet-derived growth factor receptor signalling in mesenchymal stem cells. Biochem J. 2010;427:29–40. doi: 10.1042/BJ20091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans IM, Yamaji M, Britton G, Pellet-many C, Lockie C, Zachary IC, et al. Neuropilin-1 Signaling through p130Cas Tyrosine Phosphorylation Is Essential for Growth Factor-Dependent Migration of Glioma and Endothelial Cells. Mol Cell Biol. 2011;31:1174–85. doi: 10.1128/MCB.00903-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu B, Guo P, Bar-Joseph I, Imanishi Y, Jarzynka MJ, Bogler O, et al. Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF//SF autocrine pathway. Oncogene. 2007;26:5577–86. doi: 10.1038/sj.onc.1210348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mamluk R, Gechtman Z, Kutcher ME, Gasiunas N, Gallagher J, Klagsbrun M. Neuropilin-1 Binds Vascular Endothelial Growth Factor 165, Placenta Growth Factor-2, and Heparin via Its b1b2 Domain. J Biol Chem. 2002;277:24818–25. doi: 10.1074/jbc.M200730200. [DOI] [PubMed] [Google Scholar]
- 22.West DC, Rees CG, Duchesne L, Patey SJ, Terry CJ, Turnbull JE, et al. Interactions of Multiple Heparin Binding Growth Factors with Neuropilin-1 and Potentiation of the Activity of Fibroblast Growth Factor-2. J Biol Chem. 2005;280:13457–64. doi: 10.1074/jbc.M410924200. [DOI] [PubMed] [Google Scholar]
- 23.Bagri A, Tessier-Lavigne M, Watts RJ. Neuropilins in Tumor Biology. Clin Cancer Res. 2009;156:1860–4. doi: 10.1158/1078-0432.CCR-08-0563. [DOI] [PubMed] [Google Scholar]
- 24.Ellis LM. The role of neuropilins in cancer. Molecular Cancer Therapeutics. 2006;5:1099–107. doi: 10.1158/1535-7163.MCT-05-0538. [DOI] [PubMed] [Google Scholar]
- 25.Hersey P, Bastholt L, Chiarion-Sileni V, Cinat G, Dummer R, Eggermont AM, et al. Small molecules and targeted therapies in distant metastatic disease. Ann Oncol. 2009 Aug;20(Suppl 6):vi35–vi40. doi: 10.1093/annonc/mdp254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong TM, Chen YL, Wu YY, Yuan A, Chao YC, Chung YC, et al. Targeting Neuropilin 1 as an Antitumor Strategy in Lung Cancer. Clin Cancer Res. 2007;13:4759–68. doi: 10.1158/1078-0432.CCR-07-0001. [DOI] [PubMed] [Google Scholar]
- 27.Shintani Y, Takashima S, Asano Y, Kato H, Liao Y, Yamazaki S, et al. Glycosaminoglycan modification of neuropilin-1 modulates VEGFR2 signaling. EMBO J. 2006;25:3045–55. doi: 10.1038/sj.emboj.7601188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones KS, Lambert S, Bouttier M, Bénit L, Ruscetti FW, Hermine O, et al. Molecular Aspects of HTLV-1 Entry: Functional Domains of the HTLV-1 Surface Subunit (SU) and Their Relationships to the Entry Receptors. Viruses. 2011;3:794–810. doi: 10.3390/v3060794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiodelli P, Mitola S, Ravelli C, Oreste P, Rusnati M, Presta M. Heparan Sulfate Proteoglycans Mediate the Angiogenic Activity of the Vascular Endothelial Growth Factor Receptor-2 Agonist Gremlin. Arterio Thromb Vasc Biol. 2011;31:e116–e127. doi: 10.1161/ATVBAHA.111.235184. [DOI] [PubMed] [Google Scholar]
- 30.Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci U S A. 2009;106:16157–62. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Wronski MA, Raju N, Pillai R, Bogdan NJ, Marinelli ER, Nanjappan P, et al. Tuftsin binds neuropilin-1 through a sequence similar to that encoded by exon 8 of vascular endothelial growth factor. J Biol Chem. 2006;281:5702–10. doi: 10.1074/jbc.M511941200. [DOI] [PubMed] [Google Scholar]
- 32.Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD, Leahy DJ. Structural basis for ligand and heparin binding to neuropilin B domains. Proc Natl Acad Sci U S A. 2007;104:6152–7. doi: 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starzec A, Ladam P, Vassy R, Badache S, Bouchemal N, Navaza A, et al. Structure function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF165 binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides. 2007;28:2397–402. doi: 10.1016/j.peptides.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 34.Cébe Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, et al. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell Mol Life Sci. 2006;63:2067–77. doi: 10.1007/s00018-006-6254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arterio Thromb Vasc Biol. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- 36.Breitwieser GE. G Protein Coupled Receptor Oligomerization. Circ Res. 2004;94:17–27. doi: 10.1161/01.RES.0000110420.68526.19. [DOI] [PubMed] [Google Scholar]
- 37.Davis MM, Krogsgaard M, Huppa JB, Sumen C, Purbhoo MA, Irvine DJ, et al. Dynamics of Cell Surface Molecules During T Cell Recognition. Annu Rev Biochem. 2003;72:717–42. doi: 10.1146/annurev.biochem.72.121801.161625. [DOI] [PubMed] [Google Scholar]
- 38.Schlessinger J. Ligand-Induced, Receptor-Mediated Dimerization and Activation of EGF Receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 39.Roth L, Nasarre C, rrig-Grosch S, Aunis D, Cremel G, Hubert P, et al. Transmembrane domain interactions control biological functions of neuropilin-1. Mol Biol Cell. 2008;19:646–54. doi: 10.1091/mbc.E07-06-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nasarre C, Roth M, Jacob L, Roth L, Köncina E, Thien A, et al. Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo. Oncogene. 2010;29:2381–92. doi: 10.1038/onc.2010.9. [DOI] [PubMed] [Google Scholar]
- 41.Valdembri D, Caswell P, Anderson K, Schwarz J, Konig I, et al. Neuropilin-1/GIPC1 Signaling Regulates α5β1 Integrin Traffic and Function in Endothelial Cells. Plos Biology. 2009;7:25. doi: 10.1371/journal.pbio.1000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murga M, Fernandez-Capetillo O, Tosato G. Neuropilin-1 regulates attachment in human endothelial cells independently of vascular endothelial growth factor receptor-2. Blood. 2005;105:1992–9. doi: 10.1182/blood-2004-07-2598. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Mukhopadhyay D, Xu X. C terminus of RGS-GAIP-interacting protein conveys neuropilin-1-mediated signaling during angiogenesis. FASEB Journal. 2006;20:1513–5. doi: 10.1096/fj.05-5504fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.