

Abstract
Context:
Inactivating mutations in HNF1A and HNF4A cause the maturity-onset diabetes of youth (MODY)-3 and MODY1 forms of monogenic diabetes, respectively. Children carrying HNF4A (MODY1) mutations can present in early infancy with macrosomia and diazoxide-responsive hyperinsulinism.
Objective:
Our objective was to describe three novel cases of hyperinsulinism associated with MODY1 and MODY3 mutations.
Research Design and Methods:
Clinical data were obtained from chart review. Gene sequencing was performed on genomic DNA.
Results:
Case 1 was diagnosed at 20 months with persistent hyperinsulinemic hypoglycemia and was found to have a novel MODY3 HNF1A mutation, carried by her father who had diabetes. Case 2 was diagnosed with diazoxide-responsive hyperinsulinism at 3 months of age and had complete resolution of hyperinsulinism by 4 yr. She was found to have a novel MODY3 HNF1A missense mutation, also carried by her father. Case 3 presented as a newborn with diazoxide-responsive hyperinsulinism and later developed renal Fanconi syndrome, hypophosphatemic rickets, and hepatic glycogenosis. Although the latter's features suggested Fanconi-Bickel syndrome, sequencing of the SLC2A2 gene was normal. The patient was found to have a known MODY1 mutation in HNF4A. In all cases, the hyperinsulinism improved with age.
Conclusions:
The first two cases demonstrate that mutations in HNF1A (MODY3) can cause hyperinsulinism early in life and diabetes later, similar to the phenotype recently reported for HNF4A (MODY1) mutations. Case 3 indicates that the effects of HNF4A mutations in infancy may extend beyond pancreatic β-cells to produce a disorder similar to glucose transporter 2 deficiency involving both liver glycogen metabolism and renal tubular transport.
Familial monogenic diabetes [formerly known as maturity-onset diabetes of youth (MODY)] is an autosomal dominantly inherited disorder that usually becomes manifest in late teenage to early adult years. The MODY1 and MODY3 forms involve inactivating mutations of two related transcription factors, hepatocyte nuclear factor (HNF)-1α and HNF4α (1). Recently, HNF4A mutations associated with MODY1 were reported to cause macrosomia and hyperinsulinism that may persist for several months after birth before evolving to diabetes later in life (2–4). In contrast, HNF1A mutations associated with MODY3 have not been shown to lead to increased birth weight or neonatal hypoglycemia (2).
In this report, we describe three unusual cases of congenital hyperinsulinism associated with mutations in HNF4A and HNF1A. These cases expand the links between hyperinsulinism and these two genes and highlight their complex transcriptional interactions regulating glucose metabolism in humans.
Materials and Methods
Clinical data
Clinical information was obtained by chart review with approval from The Children's Hospital of Philadelphia Institutional Review Board.
DNA mutation analysis
Mutation analysis was completed by amplification of genomic DNA by PCR and direct sequencing (Applied Biosystems, Foster City, CA). Coding exons of HNF4A and HNF1A along with intron/exon boundaries were included. Sequences were analyzed (Gene Codes Corp., Ann Arbor, MI) and compared with the published sequences for HNF1A (NM_000545) and HNF4A (NM_000457: exons 1a, 2–10; AY680697: exon 1d only; and U72959: exons 1b and 1c) (http://www.ncbi.nlm.nih.gov/).
Results
Case 1 was a full-term female, large for gestational age (93rd percentile). She had neonatal hypoglycemia that appeared to resolve after 72 h of life. However, at 20 months of age, after stopping frequent breastfeeding, she had symptoms suggestive of hypoglycemia. A diagnosis of hyperinsulinism was established based on characteristic findings at the time of hypoglycemia [blood glucose 2.6 mmol/liter (47 mg/dl])]: inappropriately suppressed β-hydroxybutyrate (1.1 mmol/liter) and inappropriate glycemic response to glucagon [increase of 3.9 mmol/liter (71 mg/dl)], all indicating increased insulin action (5). The plasma insulin level was undetectable, but this does not preclude the diagnosis of hyperinsulinism, because increased insulin levels are not always present at the time of hypoglycemia (5). The child was treated with diazoxide, a KATP channel agonist, for 2 months with partial improvement of fasting tolerance.
By age 6, the follow-up fasting test showed complete resolution of the hyperinsulinism: appropriate activation of ketogenesis and loss of the inappropriate glycemic response to glucagon. Over the following years, she did not have further symptoms of hypoglycemia.
Sequencing of ABCC8, KCNJ11, GCK, and GLUD1 revealed no mutations. However, a family history of adult-onset diabetes in the father, paternal uncle, and paternal grandmother prompted analysis of MODY genes. The proband and her father were found to be heterozygous for a novel, nonsense mutation in HNF1A (p.Glu32X, c.94G→T) affecting the dimerization domain of the gene. The other family members were not tested.
Case 2 was a full-term female, with normal birth weight (7th percentile). At 3 months of age, she had a hypoglycemic seizure with a blood glucose concentration of 1.1 mmol/liter (20 mg/dl). Evaluation at the time of hypoglycemia revealed suppressed ketone production and a positive response to glucagon [blood glucose increase of 3.3 mmol/liter (60 mg/dl)]. Based on these findings, a diagnosis of hyperinsulinism was made, and she was treated with diazoxide and octreotide. The latter was stopped, and the hyperinsulinism was controlled on diazoxide. Repeat evaluation at 10 months of age, on diazoxide, showed hypoglycemia after 18 h of fasting, with ketone production but a positive glucagon stimulation test [blood glucose increased by 1.6 mmol/liter (30 mg/dl)]. After 3 yr, diazoxide was discontinued. Off therapy, an evaluation at 42 months of age showed a normal adaptation to fasting, with no additional evidence of hyperinsulinism.
Genetic mutation testing was negative for the most common hyperinsulinism genes but showed a missense mutation in HNF1A (Pro291Ser, c.871 C→T), inherited from the father, who had not developed diabetes by age 30. This mutation has been reported in a family with MODY3 (6).
Case 3 was a full-term female, slightly large for gestational age (87th percentile) who developed severe hypoglycemia [0.7 mmol/liter (13 mg/dl)] in the first hours of life. She required iv dextrose for 1 wk. Hypoglycemia [2–3.7 mmol/liter (36–66 mg/dl)] persisted after discharge from the nursery but was not treated.
During a hypoglycemic event at 4 wk of age, plasma samples showed inappropriately suppressed β-hydroxybutyrate and free fatty acids (0.1 mmol/liter and 1.6 mmol/liter, respectively), plasma insulin level of 52 pmol/liter (7.5 μU/ml), and an inappropriate glycemic response to glucagon [an increase in glucose of 1.7 mmol/liter (31 mg/dl)], consistent with a diagnosis of hyperinsulinism. The hyperinsulinism was well controlled on diazoxide therapy. Over the following years, she required lower doses to maintain normoglycemia, and treatment was discontinued at age 4.
During the first 6 months, additional abnormalities were noted. Hepatomegaly with increased serum levels of transaminases became apparent at 3 months of age. A liver biopsy showed abundant cytoplasmic glycogen with mild portal inflammation and fibrosis (Fig. 1). By age 6, although clinically improved, mildly elevated liver transaminases and mild hepatomegaly were still present.
Fig. 1.

Liver biopsy specimen in case 3 with HNF4A mutation. Periodic acid-Schiff stain (A) showing intense cytoplasmic positivity within hepatocytes was abolished with diastase (B) and consistent with abundant cytoplasmic glycogen (×10 magnification).
By age 1, she developed radiological evidence of rickets, elevated plasma alkaline phosphatase, and low plasma concentrations of phosphorus. Supplementation with oral sodium citrate, potassium, sodium phosphate, and calcitriol led to rickets improvement, but the elevation of alkaline phosphatase persisted. In addition, the child had persistent glycosuria, in the absence of hyperglycemia, as well as stable proteinuria and metabolic acidosis.
The clinical phenotype suggested the possibility of Fanconi-Bickel syndrome. However, sequencing of genomic DNA revealed no mutation of the gene encoding glucose transporter 2 (GLUT2), SLC2A2. Mutation analyses for the known hyperinsulinism genes GLUD1, GCK, ABCC8, KCNJ11, UCP2, and HADH were also negative. However, sequencing of HNF4A revealed a missense, de novo mutation (p.Arg76Trp, c.226C→T) that has previously been reported in one other case of congenital hyperinsulinism (7). There was no family history of diabetes or Fanconi syndrome.
Discussion
This report describes three unusual cases of congenital hyperinsulinism associated with mutations in HNF1A and HNF4A. Cases 1 and 2 had hypoglycemia within the first 3 months of life. Case 1 responded only partially to diazoxide, whereas case 2 was successfully treated with diazoxide. Both of these children inherited a mutation in HNF1A from their fathers, but only in one case has the father been diagnosed with diabetes. Case 3 presented as a newborn with diazoxide-responsive hyperinsulinism and subsequently developed unusual features of renal Fanconi syndrome and hepatic glycogenosis. She was found to have a known disease-causing MODY1 mutation in HNF4A. In all three cases, the hyperinsulinism appeared to resolve with age. Although mutations in HNF4A have previously been reported to cause congenital hyperinsulinism, hyperinsulinism has not been described in HNF1A mutations.
The phenotype of congenital hyperinsulinism in cases 1 and 2 is similar to that described in HNF4A mutation carriers: evidence of insulin excess in utero (large for gestational age) in at least one of the cases and early-onset hyperinsulinemic hypoglycemia that resolved with age (2). Mutations in the dimerization region and in the DNA-binding domain of the HNF1A gene, as seen in case 1, have been associated with an earlier age of diagnosis of diabetes when compared with mutations in the transactivation domain, as seen in case 2 (8). We speculate that the location of the nonsense mutations in the dimerization domain (case 1) or transactivation domain (case 2) of HNF1A leads to the formation of a functionally impaired, possibly dominant-negative HNF1α protein. This might cause dysregulated expression of HNF4α (9) and other HNF1α or HNF4α transcriptional targets, such as Kir6.2 or upcoupling protein-2 (10, 11). A diabetes phenotype was seen only in the carrier father of case 1. This is similar to other families in which only a few of the parents of children with hyperinsulinism, carrying mutations in HNF4A, had diabetes (7). In parallel to MODY (12), it is possible that the hyperinsulinism phenotype has incomplete penetrance and variable clinical severity, implicating environmental or additional genetic factors.
The phenotype of case 3 with the HNF4A mutation illustrates the wide regulatory network of HNF4α, not only in pancreatic β-cells and the liver but also in the kidney. The HNF4A mutation in this child has been previously described in cases of MODY1 and hyperinsulinism (7). The diazoxide-responsive hyperinsulinism in this case was similar to that associated with other HNF4A mutations (2, 3). However, the association of this mutation with renal Fanconi tubulopathy and hepatomegaly with increased glycogen stores has not been previously described.
Inactivating mutations of SLC2A2 (Fanconi-Bickel syndrome) lead to hepatomegaly due to glycogen accumulation, hypoglycemia, renal Fanconi syndrome, and galactose intolerance. In these cases, hypoglycemia is due to impaired hepatic release of glucose as a result of nonfunctional GLUT2 in hepatocytes (13). The association with congenital hyperinsulinism was reported in a child with mutations of both SLC2A2 and ABCC8 (14). Our case 3 did not have the pattern of postprandial hyperglycemia and fasting ketotic hypoglycemia as in Fanconi-Bickel syndrome. Moreover, decreased hepatic glucose release due to GLUT2 deficiency did not play a role in the etiology of hypoglycemia, because the response to glucagon was consistent with excessive insulin action and not with a complete deficiency of GLUT2. The hepatomegaly improved in the first 2–3 yr of life, much earlier than expected of inactivating mutations of SLC2A2 (13). The tubulopathy, with glucosuria and proteinuria, but normal glomerular function, was stable and similar to other described cases of Fanconi-Bickel syndrome (13), suggesting that GLUT2 expression was more severely affected in the kidney. It is possible that this liver and kidney phenotype is present in other children with mutations in HNF4A but is transient or has a variable penetrance and severity of presentation and might be harder to recognize.
Both HNF4α and HNF1α could regulate the expression of SLC2A2 (15, 16). Hence, the clinical phenotype in case 3 can be explained by the impact of her HNF4A mutation on decreasing the expression of SLC2A2 in both liver and kidney, either by impaired binding of mutated HNF4α to the SLC2A2 promoter or by impaired interaction of the mutant HNF4α with the HNF1A promoter, thus further decreasing the activation of the SLC2A2 promoter (Fig. 2).
Fig. 2.
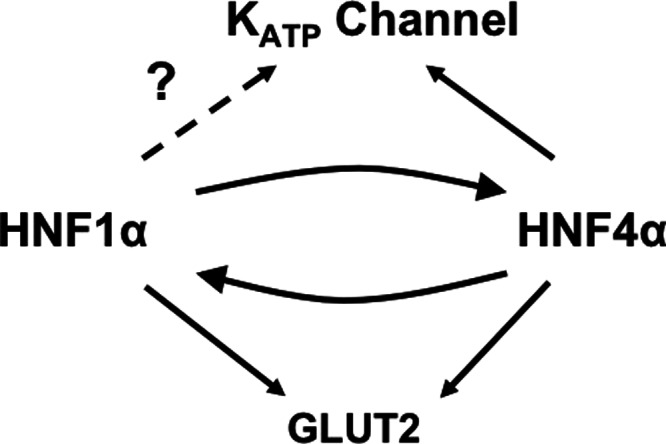
Proposed relationship between HNF4α, HNF1α, and their targets. HNF4α and HNF1α can bind and activate each other's promoter; both can also regulate the expression of GLUT2. Although there is evidence for only HNF4α regulating the expression of KATP subunit Kir6.2, it is possible HNF1α might also have the same function.
Flanagan et al. (7) have reported that HNF4A mutations were the third most common genetic cause of diazoxide-responsive congenital hyperinsulinism. We suggest that mutations in HNF1A should also be considered in infants with diazoxide-responsive or other forms of congenital hyperinsulinism. Furthermore, it is important to recognize that the manifestations of mutations affecting the HNF4A and HNF1A network could extend beyond pancreatic β-cells, including impairment of GLUT2 activity. It is not known how the initial hyperinsulinism phenotype impacts the long-term risk of diabetes. However, the early identification of these children may result in an earlier diagnosis of diabetes, earlier intervention, and possibly better long-term outcomes.
Acknowledgments
The project described was supported by National Institutes of Health Grants R37 DK056268 and RR024134. D.D.D.L received support from R56DK083670.
D.E.S. researched data, contributed to discussion, wrote manuscript, and reviewed/edited manuscript. N.H. researched data. B.K., C.A.S., and D.D.D.L. contributed to discussion and reviewed/edited manuscript.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- GLUT2
- Glucose transporter 2
- HNF
- hepatocyte nuclear factor
- MODY
- maturity-onset diabetes of youth.
References
- 1. Stride A, Hattersley AT. 2002. Different genes, different diabetes: lessons from maturity-onset diabetes of the young. Ann Med 34:207–216 [PubMed] [Google Scholar]
- 2. Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, Ellard S, Ferrer J, Hattersley AT. 2007. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med 4:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, Ellard S, Hussain K. 2008. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes 57:1659–1663 [DOI] [PubMed] [Google Scholar]
- 4. Pingul MM, Hughes N, Wu A, Stanley CA, Gruppuso PA. 2011. Hepatocyte nuclear factor 4α gene mutation associated with familial neonatal hyperinsulinism and maturity-onset diabetes of the young. J Pediatr 158:852–854 [DOI] [PubMed] [Google Scholar]
- 5. Palladino AA, Bennett MJ, Stanley CA. 2008. Hyperinsulinism in infancy and childhood: when an insulin level is not always enough. Clin Chem 54:256–263 [DOI] [PubMed] [Google Scholar]
- 6. Bazalová Z, Rypácková B, Broz J, Brunerová L, Polák J, Rusavý Z, Treslová L, Andel M. 2010. Three novel mutations in MODY and its phenotype in three different Czech families. Diabetes Res Clin Pract 88:132–138 [DOI] [PubMed] [Google Scholar]
- 7. Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, Siahanidou T, Banerjee I, Akcay T, Rubio-Cabezas O, Shield JP, Hussain K, Ellard S. 2010. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol 162:987–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bellanné-Chantelot C, Carette C, Riveline JP, Valéro R, Gautier JF, Larger E, Reznik Y, Ducluzeau PH, Sola A, Hartemann-Heurtier A, Lecomte P, Chaillous L, Laloi-Michelin M, Wilhem JM, Cuny P, Duron F, Guerci B, Jeandidier N, Mosnier-Pudar H, Assayag M, Dubois-Laforgue D, Velho G, Timsit J. 2008. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes 57:503–508 [DOI] [PubMed] [Google Scholar]
- 9. Gragnoli C, Lindner T, Cockburn BN, Kaisaki PJ, Gragnoli F, Marozzi G, Bell GI. 1997. Maturity-onset diabetes of the young due to a mutation in the hepatocyte nuclear factor-4α binding site in the promoter of the hepatocyte nuclear factor-1α gene. Diabetes 46:1648–1651 [DOI] [PubMed] [Google Scholar]
- 10. Thomas P, Ye Y, Lightner E. 1996. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet 5:1809–1812 [DOI] [PubMed] [Google Scholar]
- 11. González-Barroso MM, Giurgea I, Bouillaud F, Anedda A, Bellanné-Chantelot C, Hubert L, de Keyzer Y, de Lonlay P, Ricquier D. 2008. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One 3:e3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fajans SS, Bell GI. 2006. Phenotypic heterogeneity between different mutations of MODY subtypes and within MODY pedigrees. Diabetologia 49:1106–1108 [DOI] [PubMed] [Google Scholar]
- 13. Santer R, Steinmann B, Schaub J. 2002. Fanconi-Bickel syndrome: a congenital defect of facilitative glucose transport. Curr Mol Med 2:213–227 [DOI] [PubMed] [Google Scholar]
- 14. Hoffman TL, Blanco E, Lane A, Galvin-Parton P, Gadi I, Santer R, DeLeón D, Stanley C, Wilson TA. 2007. Glucose metabolism and insulin secretion in a patient with ABCC8 mutation and Fanconi-Bickel syndrome caused by maternal isodisomy of chromosome 3. Clin Genet 71:551–557 [DOI] [PubMed] [Google Scholar]
- 15. Ban N, Yamada Y, Someya Y, Miyawaki K, Ihara Y, Hosokawa M, Toyokuni S, Tsuda K, Seino Y. 2002. Hepatocyte nuclear factor-1α recruits the transcriptional co-activator p300 on the GLUT2 gene promoter. Diabetes 51:1409–1418 [DOI] [PubMed] [Google Scholar]
- 16. Stoffel M, Duncan SA. 1997. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4α regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci USA 94:13209–13214 [DOI] [PMC free article] [PubMed] [Google Scholar]