

Abstract
Multiple lines of evidence suggest a functional link between the androgen receptor (AR) and the serine/threonine kinase Akt in the development and progression of prostate cancer. To investigate the impact of Akt activity on AR homeostasis, we treated androgen-dependent LNCaP and LAPC-4 prostate cancer cells with Akt inhibitor. Akt inhibition decreased AR expression, suggesting that Akt activity was required for regulation of AR protein levels. However, while androgen-independent LNCaP-abl cells also showed diminished AR protein levels in response to Akt inhibition, treatment of androgen-independent LNCaP-AI cells failed to alter AR protein levels upon similar treatment, suggesting that AR protein levels in these androgen-independent prostate cells were regulated by mechanisms independent of Akt activation. Regulation of AR, downstream of activated Akt, also was observed in vivo when examining transgenic mice that overexpress constitutively active mutant myristoylated (myr)-Akt1 in the prostate. Transgenic mice animals expressing activated myr-Akt1 exhibited higher levels of AR mRNA and protein. Expression of activated myr-Akt1 did not alter prostate cell growth and no significant size differences between prostate tissues derived from transgenic animals were observed when comparing transgenic to wild-type mice. Still, transgenic mice overexpressing Akt exhibited higher levels of γH2AX and phosphorylated Chk2 in prostate tissue. These changes in markers associated with oncogene-induced senescence confirmed significant altered signaling in the transgenic mouse model. Overall, results presented here suggest that AR levels are regulated by the Akt pathway.
Keywords: Akt, AR, prostate cancer, hormone-refractory, androgen-independent
Introduction
The androgen receptor (AR) directs prostate growth and differentiation and, for this reason, anti-androgens are commonly used to treat prostate cancer. The importance of understanding the mechanism of AR gene and protein regulation is underscored by the finding that prostate cancer is reliant on the expression of AR even after progressing to anti-androgen resistant disease and increased expression of the androgen receptor is the major factor driving prostate cancer recurrence (Chen et al., 2004). Other factors contribute to disease progression, notably, loss of function of PTEN and activation of Akt which are strongly correlated with prostate cancer (Di Cristofano et al., 1998; Wang et al., 2003). Synergistic interactions between AR and Akt in an in vivo prostate regeneration model (Xin et al., 2006) provide evidence that the phosphoinositide 3-kinase (PI 3-kinase)/phosphatase and tensin homolog (PTEN)/Akt and AR pathways may be linked mechanistically.
It has been previously reported that overexpression of myristoylated Akt in prostate results in Prostate Intraepithelial Neoplasia (PIN) (Majumder et al., 2003). In addition, the PIN lesions occurring in Akt overexpressing transgenic animals invoked an increase in cellular levels of p27/kip1 resulting in cellular senescence (Majumder et al., 2008); consistent with reports that cellular senescence is often seen in early or pre-invasive stages of cancer (Chen et al., 2005; Collado et al., 2005; Michaloglou et al., 2005). To explore the link between PI 3-Kinase/PTEN/Akt and AR pathways, we examined the impact of Akt activity on AR protein levels in cultured prostate cells and a transgenic mouse model. Our findings indicate that AR expression is regulated by Akt in both models, but can be Akt-dependent or Akt-independent in androgen-independent cell lines depending on their individual characteristics.
Materials and Methods
Generation of transgenic lines expressing active Akt
The ARR2PB rat probasin promoter (Zhang et al., 2000), a SV40 t-intron and poly A sequence were separately subcloned into pBluescript II SK. The minimal ARR2 probasin promoter is a composite of two androgen response regions (ARR) of the probasin promoter (ARR 1 (−244 to −96) placed 5′ to ARR 2 (−286 to +24)). A cDNA insert encoding a myristoylated mutant of mouse Akt1 (myr-Akt1-HA) was liberated from pCMV6 by double digestion with HindIII and BamHI, blunted, and ligated into the EcoRI site of pBluescript vector between the probasin promoter and the SV40 poly-A sequence. Southern blot analysis of tail DNA derived from transgenic mice identified three founder animals from the probasin-driven Akt construct (ARR2-myr-Akt). Mice were maintained in accordance with the guidelines for the Care and Use of Laboratory Animals.
Cell Culture, Transfection, and Reagents
LNCaP and VCaP cell lines were obtained from the ATCC (Manassas, VA) and cultured in RPMI-1640 and DMEM, respectively. Media was supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin. LNCaP cells used in this study were from passages 23–30. LAPC-4 cells (gift from R. Reiter, UCLA) and androgen-independent LNCaP-AI cells (gift from A. Ferrari, NYU) were cultured in IMDM and RPMI-1640, respectively. LNCaP-AI culture media was supplemented with 10% (v/v) charcoal stripped FBS (cFBS) and 5μg/ml insulin (Sigma; St. Louis, MO). Androgen independent LNCaP-abl cells (gift from Z. Culig) were maintained in RPMI-1640 supplemented with 10% charcoal stripped serum (cFBS). Androgen independent LNCaP-AI and LNCaP-abl cell lines were both derived from long-term culture in 10% cFBS. (Culig et al., 1999; Gao et al., 1999). R1881 (methyltrienolone) was purchased from Perkin Elmer (Waltham, MA). Akt inhibitor VIII and PI3-K inhibitor LY294002 were purchased from EMD Chemicals (Gibbstown, NJ) and reconstituted in DMSO.
VCaP cells were transiently transfected using Lipofectamine 2000 (Invitrogen; Carlsbad, CA) with vector or pCMV6-myr-Akt1-HA according to the manufacturer’s recommendations. 24 hours post-transfection, cells were washed with Dulbecco’s phosphate buffered saline (PBS) and treated overnight with low serum and androgen-free media (0.05% (v/v) cFBS), followed by treatment with R1881 for 2 hours. Cells were lysed as described below 48 hours post-transfection.
Hematoxylin and Eosin Staining
Prostate tissues were dissected and fixed in formalin overnight at 4° C, dehydrated and embedded in paraffin. 5 micron sections were cut, placed on charged slides, and baked for 1.5 hours at 60° C. Samples were deparaffinized in xylene and rehydrated in ethanol and water. Sections were stained in non-aqueous hematoxylin and eosin. Samples were dehydrated in ethanol and xylene, allowed to air dry, mounted (Permount) and coverslipped.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections were dewaxed in xylene, rehydrated and washed in phosphate-buffered saline (PBS), pH7.4. For antigen retrieval, paraffin sections were heated in a microwave oven (900 W) in 1X citrate buffer, followed by treatment with 3% (v/v) H2O2 and blocked with 20% (v/v) normal goat serum. Sections were incubated with phospho-Chk2 Thr68 (1: 50) and γ-H2AX Ser139 (1:50) (Cell Signaling; Danvers, MA), followed by incubation with biotinylated rabbit secondary antibody (1:1000 in 2% (w/v) BSA; Vector Labs, Burlingame, CA) and Strepdavidin Peroxidase. Staining was visualized with DAB (Vector Labs) and sections were counterstained with Hematoxylin (Vector Labs) prior to mounting with Crystal/Mount (Electron Microscopy Sciences; Hatfield, PA). Microscopy was performed using a Zeiss Axio Imager A2 microscope. Images were acquired with QCapture Prosoftware.
Protein Extraction and Western blot Analysis
Whole prostates were dissected from age-matched C57BL/6 wildtype (Taconic; Hudson, NY) and transgenic ARR2-myr-Akt mice. A Tissue Tearor (BioSpec Products) was used to homogenize tissues. Proteins were extracted using TNN lysis buffer (50mmol l−1 Tris, pH 7.4, 150mmol l−1 NaCl, 0.5% (v/v) NP-40, 5mmol l−1 EDTA, 5mmol l−1 NaF, 1mmol l−1 DTT, 5μg/ml aprotinin, 5μg/ml leupeptin, 1mmol l−1 PMSF, 1mmol l−1 sodium orthovanadate). Lysates were incubated on ice for 30 min. then centrifuged and extracted protein collected.
To extract protein from cell lines, cells were lysed in Triton X-100 lysis buffer (50mmol l−1 HEPES, pH 7.5, 150mmol l−1 NaCl, 1mmol l−1 EDTA, 1mmol l−1 EGTA, 10% (v/v) glycerol, 1% (v/v) Triton X-100, 25mmol l−1 NaF, 10μmol l−1 ZnCl2, 10 μg/ml aprotinin, 10μg/ml leupeptin, 1mmol l−−1 PMSF, 1mmol l−1 sodium orthovanadate).
Protein lysates were quantified using BioRad Protein Assay Dye and normalized for total protein concentrations. Total protein was subjected to SDS-PAGE and immunoblotted with the following antibodies: phospho-AR S213 (Taneja et al., 2005), AR (441) and ERK-1 (K-23) (Santa Cruz Biotechnology; Santa Cruz, CA); phospho-Akt S473 and Akt (Total) (Cell Signaling; Danvers, MA); Tubulin, HA-epitope tag, and Keratin 14 (Covance; Denver, PA); and HA conjugated to HRP (Roche; Indianapolis, IN). Protein expression levels were quantified using ImageJ 1.42q software (National Institutes of Health).
Quantitative RT-PCR
Total RNA was extracted from mouse prostates using RNeasy (Qiagen; Valencia, CA) according to the manufacturer’s recommendations. RNA (1ug) was reverse transcribed using Superscript III Reverse Transcriptase (Invitrogen) and then 2.5% (v/v) of the resulting cDNA was used for quantitative PCR (qPCR). qPCR was performed using SYBR green Taq ready mix (Sigma) and a LightCyler (Roche). Data was analyzed by the ΔΔCT method (Bookout and Mangelsdorf, 2003) using RPL19 or mouse keratin14 as control genes, then normalized to naive samples arbitrarily set to 1. The primers used for the qPCR are: Mouse AR F 5′ TACCAGCTCACCAAGCTCCT; Mouse AR R 5′ GAACTGATGCAGCTCTCTTGC; RPL19 F 5′CACAAGCTGAAGGCAGACAA; RPL19 R 5′ GCGTGCTTCCTTGGTCTTAG; Mouse Keratin 14 F 5′ TCCCAATTCTCCTCATCCTC; Mouse Keratin 14 R 5′ GGTTGGTGGAGGTCACATCT; Mouse Keratin 18 F 5′ CTTGCTGGAGGATGGAGAAG; and Mouse Keratin 18 R 5′ CTGCACAGTTTGCATGGAGT.
Results
Akt regulation of AR protein levels in prostate cancer cells
To determine the impact of Akt activity on AR protein levels, we treated LNCaP (PTEN-deficient), LAPC-4 (PTEN wild-type), and VCaP (PTEN wild-type) prostate cancer cells with an inhibitor of Akt isoforms 1 and 2 (Akti). Figure 1A shows Western blot analysis of lysates from LNCaP cells treated with or without the synthetic androgen, R1881, in the presence or absence of Akt inhibitor. The results indicate that Akti treatment completely abolished phosphorylation of Akt at S473 (P-Akt S473), but did not affect total protein levels of Akt. Interestingly, inhibition of Akt activity by Akti resulted in decreased AR protein levels compared to cells treated with vehicle alone. While this decrease may be more apparent in the absence of R1881, both R1881 treated and untreated cells showed diminished AR in the presence of the Akt inhibitor (Figure 1A, AR panel, 2nd and 4th lane). This result was not specific to one cell type or due to the AR T877A mutation in LNCaP cells. LAPC-4 prostate cancer cells, which express wildtype AR, also showed diminished AR protein levels following treatment with the PI 3-kinase inhibitor LY 294002 (LY) or Akti. Furthermore, the decrease in AR protein levels in the presence of the Akt inhibitor exceeded the effect that was observed after treatment with LY 294002 which correlates a greater suppression of phosphorylation of Akt S473 by Akti (Figure 1B).
Figure 1. Effect of Akt inhibition on prostate cancer cell lines.

LNCaP androgen-dependent (A) and androgen-independent LNCaP-AI (C) and LNCaP-abl (D) cells were serum and androgen starved overnight in 0.05% cFBS before pretreatment with 20μM Akti or vehicle for 30 minutes. After pretreatment, 10nM R1881 or vehicle was added for 2 hours. B) LAPC-4 cells were serum and androgen starved overnight in 0.05% cFBS then pretreated with 50μM LY 294002 (LY), 20μM Akti, or vehicle for 30 minutes. After pretreatment, 10nM R1881 or vehicle was added for 2 hours. The vehicle for Akti and LY 294002 was 0.1% and 0.05% DMSO, respectively and for R1881, 0.1% ethanol. Total protein was extracted and subjected to Western blot with antibodies against AR, phospho-Akt S473, Akt (total), and ERK-1 (loading control). Quantitation of AR protein levels are shown in the boxed area. AR levels are normalized to the untreated lane arbitrarily set to 1.
In contrast, in the androgen-independent LNCaP subline (LNCaP-AI), Akti inhibited P-Akt S473 to the same extent as in the androgen-dependent LNCaP cells but did not decrease AR protein expression (Figure 1C). This suggests that in androgen-dependent LNCaP and LAPC-4 cells, AR protein levels are regulated through Akt and that this homeostasis is altered in the LNCaP-AI prostate cancer model. In another model of androgen-independent prostate cancer, LNCaP-abl, which was derived in a comparable manner as LNCaP-AI cells (Culig et al., 1999; Gao et al., 1999), treatment with Akti decreased expression of AR (Figure 1D), similar to the parental androgen-dependent LNCaP cells. The different responses to Akt inhibition in the androgen-independent models suggest that AR is regulated by different mechanisms even though both LNCaP-AI and LNCaP-abl are capable of growing in the absence of androgen.
The relationship between Akt activity and AR expression was also examined in the androgen-dependent VCaP prostate cancer cell line that expresses wild-type AR. These cells differ from LNCaP and LAPC-4 cells in that basal levels of P-Akt S473 are very low. In fact, when the cells were grown in 0.05% charcoal stripped FBS (cFBS), similar to the experiments shown in Figure 1, no phosphorylated Akt was observed (Figure 2A). In 10% FBS, a small amount of P-Akt S473 was observed, however, Akti did not decrease AR levels despite complete inhibition of phosphorylated Akt. Thus, regulation of phospho-Akt appears tightly regulated in VCaP cells where serum withdrawal is sufficient to suppress Akt activity. However, while inhibition of the low level of endogenous Akt kinase activity did not affect AR protein levels in VCaP cells, overexpression of Akt resulted in increased levels of AR protein. Figure 2B shows that transient transfection of VCaP cells with myr-Akt1-HA resulted in a small, reproducible increase in AR protein in response to increasing amounts of overexpressed myr-Akt in both the presence and absence of R1881 (compare AR panel, 0 μg versus 0.5 and 1 μg myr-Akt1-HA). There was at least a two-fold increase in AR protein expression levels in the presence of overexpressed myr-Akt1-HA (Figure 2C).
Figure 2. VCaP response to Akt inhibition and expression of activated myr-Akt1.

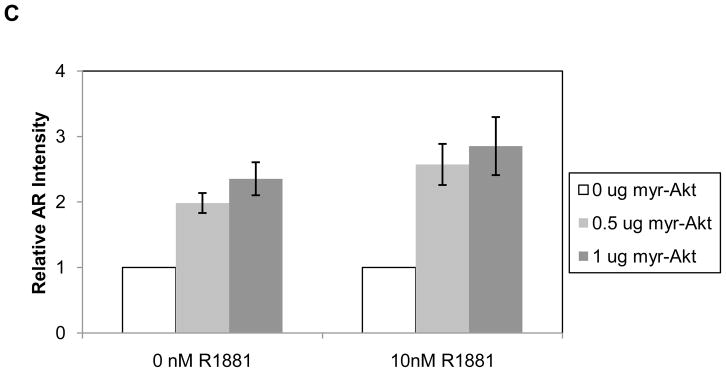
A) VCaP cells were serum and androgen starved in 0.05% cFBS or treated in 10% FBS overnight. Cells were then pretreated with 20μM Akti or vehicle for 30 minutes. After pretreatment, 10nM R1881 or vehicle was added for 2 hours. B) VCaP cells were transiently transfected with increasing amounts of myr-Akt1-HA. Cells were serum and androgen starved in 0.05% cFBS overnight then treated with either vehicle or 10nM R1881 for 2 hours. Vehicles were the same as described in Figure 1. Total protein was extracted and subjected to Western blot with antibodies against AR, phospho-AR S213, phospho-Akt S473, Akt (total), HA, and ERK-1 (loading control). C) Quantitation of relative AR protein levels in VCaP cells overexpressing myr-Akt. Data represents three independent experiments and error is represented by standard error of the mean.
Phosphorylation of AR at serine 213, a putative target of Akt, was also examined (Figure 2B). Ligand-dependent AR phosphorylation at serine 213 was previously shown to occur in prostate epithelial cells in vivo (Taneja et al., 2005), however, overexpression of Akt resulted in little, if any, AR serine 213 phosphorylation in VCaP cells. When comparing the impact of Akti on AR levels in LNCaP and LAPC4 versus VCaP (Figures 1 and 2), we cannot rule out that certain cells may be more susceptible to regulation of the AR pathway through Akt than others due to different genetic backgrounds of the cells. However, given the very different levels of P-Akt S473 in LNCaP and LAPC4 cells versus VCaP, there may not be enough Akt activity in VCaP cells to impact AR levels under the experimental conditions. Alternatively, Akti, which is preferential for Akt isoforms 1 and 2 (DeFeo-Jones et al., 2005; Lindsley et al., 2005), may not inhibit all of the Akt3 isoform that is present in VCaP cells. VCaP cells express all three isoforms of Akt whereas LNCaP and LAPC4 cells only express Akt 1 and 2 (Koseoglu et al., 2007) and Akt3 was not detected in either cell line (data not shown). Although Akt activity was assessed by examining the levels of P-Akt S473, it is possible that autophosphorylation at T72 and S246 or other putative phosphorylation sites (Chen et al., 2001; Conus et al., 2002; Li et al., 2006) contribute to Akt’s affect on AR levels. Overall, inhibition of Akt in cells expressing constitutively high levels of phospho-Akt results in decreased AR protein levels.
Myr-Akt1 expression in the prostate of transgenic animals
The results presented above indicate that inhibition of Akt kinase activity resulted in decreased levels of AR protein, suggesting crosstalk between these two pathways that is consistent with published findings (Xin et al., 2006). To determine if enhanced Akt activity impacts AR protein levels in vivo, we generated transgenic mice that overexpress constitutively active myristoylated Akt1, specifically in the prostate. Following pro-nuclear injection of a construct encoding the probasin ARR2 promoter (Zhang et al., 2000), HA epitope-tagged, myristoylated mouse Akt1 and a SV40 poly A sequence, founder animals were identified by Southern blot analysis (Figure 3A). Three founders identified by the asterisks in lanes 1, 5 and 6 (Figure 3A, left panel) were backcrossed into the C57BL/6 parental strain. Representative samples from transgenic F1 males are shown in Figure 3A, right panel. Mice heterozygous for ARR2-myr-Akt were bred to generate homozygous mice. Homozygocity for ARR2-myr-Akt was confirmed by Southern blot analysis, and these mice have been used for studies described below.
Figure 3. Southern blot analysis of founder ARR2-myr-Akt1 transgenic mice.

A) Southern blot of tail DNA from animals microinjected with ARR2-myr-Akt1-HA. Founder animals that have integrated the ARR2-myr-Akt DNA are shown by the asterisks. The genomic DNA was cleaved with KpnI and the expected sizes of the DNA fragments are diagramed in B. Southern blot of F1 progeny (A, right panel) shows expression of ARR2-myr-Akt1 only in the transgenic animals but not in wild-type (WT) animals. B) Diagram of the probasin promoter regulated, constitutively active Akt1 (myr-Akt1-HA) construct used to generate the ARR2-myr- Akt1 transgenic mice. The ARR2 probasin promoter is a composite of two androgen response regions (ARR) of the probasin promoter (ARR 1 (−244 to −96) placed 5′ to ARR 2 (−286 to +24)). DNA fragment sizes in kilobases (kb) resulting from KpnI cleavage are indicated. The isolated DNA shown in 3B is used as a probe. C) Protein lysates were made in TNN lysis buffer using ventral prostates of 3 month old ARR2-myr-Akt1 animals and WT controls. Lysates immunoblotted for HA (Top panel) using HA antibody conjugated to HRP. The positive control is overexpressed ARR2-myr-Akt-HA in 293 cells. The same membrane was then re-blotted (bottom panel) for ERK-1 as a loading control.
To verify expression of myr-Akt-HA protein, Western blot analysis was performed using lysates from wild type and transgenic animals (Figure 3C). The results indicate that as expected, the myr-Akt1 transgene was expressed in the ventral prostate of transgenic but not wild-type (WT) animals. The expression of P-Akt S473 and Akt1 was also examined in transgenic and WT prostates. P-Akt S473 and Akt1 expression increased approximately 40% in transgenic mice (data not shown).
Increased Akt activity results in elevated AR protein and mRNA levels
To determine the effect of increased Akt signaling on AR protein levels in vivo, AR levels were examined in age-matched (3.5 months) WT and transgenic animals expressing myristoylated Akt under the regulation of the probasin promoter. Four separate matched sets of tissue lysates consisting of pools of 3 prostates from either wild type or myr-Akt1 transgenic animals were immunoblotted for AR. The samples were also immunoblotted for the basal epithelial cell marker keratin 14 and tubulin (data not shown) as internal loading controls. Figure 4A shows that AR protein levels are markedly increased in the Akt transgenic compared to WT samples. A darker exposure of the AR immunoblot confirmed the presence of AR in WT mice (Figure 4A, middle panel). Equivalent levels of keratin 14 between the samples indicated comparable amounts of epithelial cells in the protein lysates.
Figure 4.

Androgen receptor protein is upregulated in ARR2-myr-Akt1 prostates compared to WT prostates. A) Pooled whole prostates from age-matched (3.5 months) myr-Akt1 or WT animals immunoblotted for AR (top panel), a longer exposure (exp.) of the same AR membrane (middle panel), and keratin 14 (bottom panel). B) AR mRNA is upregulated in ARR2-myr-Akt1 prostates compared to WT prostates. mRNA was extracted from age-matched (6 months) prostates from ARR2-myr-Akt1 and WT mice. Quantitative RT-PCR was performed measuring AR mRNA expression (arbitrary units). Samples are normalized to RPL19. Data is representative of three independent experiments and error bars represent standard deviation.
Upregulation of AR protein in response to overexpressed myr-Akt1 in the transgenic animals correlated with upregulation of AR mRNA. RNA from prostates of age-matched (6 months) ARR2-myr-Akt1 and WT animals was examined using quantitative RT-PCR. AR mRNA increased in transgenic animal compared to the WT. AR transcripts were normalized to RPL19 (Figure 4B). Normalization to epithelial cell markers keratin 14 or 18 showed similar results with upregulation of AR mRNA in the ARR2-myr-Akt1 mice (data not shown).
Overexpression of activated Akt leads to upregulation of senescence markers but not overt changes in cellular morphology
As detailed above, transgenic myr-Akt1 mice express increased levels of AR, a circumstance associated with development of recurrent prostate cancer. To determine if myr-Akt1 mice exhibited signs of hyperplasia, wild type and transgenic mice were sacrificed and examined for gross histological changes at 3.5, 6, 9, and 12 months. Prostates were dissected, fixed, and paraffin embedded for histological analysis. At necropsy, no observable tumors or changes in the mouse prostate were noted. Further, no discernable morphological differences between ARR2-myr-Akt1 prostates and age-matched wild type mouse prostates were evident following hematoxylin and eosin staining and examination of prostate tissue sections (Figure 5). Overexpression of ARR2-myr-Akt1 did not affect prostate cell size or growth because there was no difference in the weight or size of the prostate of the transgenic animals relative to wild type mice. Comparable levels of keratin 14 (Figure 4A) suggests that there was no loss of basal epithelial cells, consistent with the lack of a tumorigenic phenotype in the myr-Akt1 animals.
Figure 5. H&E staining of ARR2-myr-Akt1 and WT Prostates.

Hematoxylin and eosin staining of ARR2-myr-Akt and WT ventral prostates dissected at 3.5 months (A–B), 9 months (C–D), and 12 months (E–F). Panels A–F are shown at 200X magnification.
The fact that ARR2-myr-Akt1 did not have an impact on prostate cell growth or cause tumorigenesis led us to hypothesize that overexpression of myr-Akt1 induced oncogene-associated stress leading to cellular senescence in the adult prostate. Recent studies suggest a biological block to tumorigenesis inhibits the progression of preneoplastic lesions to neoplasia (Gorgoulis et al., 2005; Michaloglou et al., 2005). Similar observations have been made in mouse models in which oncogene-induced stress is found to be associated with signs of replication-induced stress and leads to cellular senescence as indicated by increased levels of γ-H2AX S139 and phospho-Chk2 (Bartkova et al., 2006). To determine if the ARR2-myr-Akt1 mice exhibited signaling changes indicative of cellular senescence, we examined levels of γ-H2AX and phospho-Chk2 Thr 68 in WT versus ARR2-myr-Akt1 mice. Prostates dissected from 3.5 month (Figure 6) and 6 and 9 months old mice (Supplemental Figure 1) were stained with antibodies against phospho-Chk2 and γ-H2AX. Prostate tissue from ARR2-myr-Akt1 animals at all time points exhibited more prevalent staining of nuclear phospho-Chk2 and γ-H2AX than that from WT animals, suggesting that expression of constitutively-active myr-Akt1 activated DNA damage response and senescence-inducing pathways even in the absence of any histological manifestations of PIN.
Figure 6. Senescence markers are upregulated in ARR2-myr-Akt1 prostates.
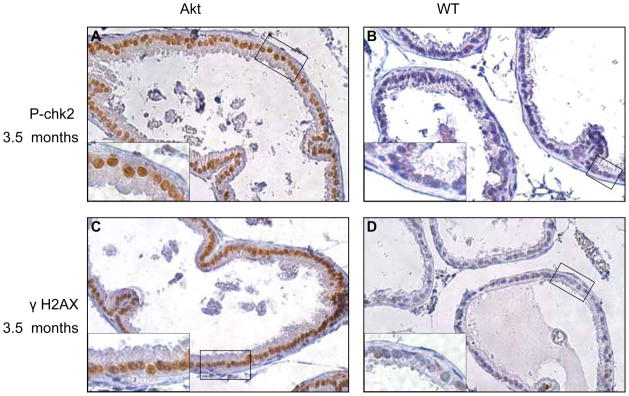
Immunohistochemistry was performed on 3.5 month old prostates from ARR2-myr-Akt1 and WT animals using phospho-Chk2 Thr68 (A and B) and γH2AX Ser139 (C and D) antibodies. Panels A–D are shown at 200X magnification. Insets show the same sample (boxed area) at 400X magnification and demonstrate enhanced nuclear staining in transgenic mice compared to WT controls.
Discussion
Results presented in this report indicate that an increase in Akt kinase activity correlates with increased levels of AR protein. These findings are relevant to human prostate cancers, since many have increased Akt activity due to PTEN mutation or enhanced growth factor receptor signaling. Interestingly, regulation of AR via Akt appears to occur primarily at the level of gene transcription since transgenic animals expressing constitutively-active myr-Akt1 have increased levels of AR mRNA as well as protein. While we do not know the mechanism of Akt-induced AR mRNA upregulation, we speculate that this might occur through Akt activation of NF- κB. Recent findings indicate that NF- κB interacts with the 5′ regulatory sequence of the AR gene to upregulate AR mRNA and protein levels (Dan et al., 2008). In addition, AR and NF- κB protein levels are strongly correlated in prostate cancer (Dan et al., 2008), supporting the idea that NF- κB may regulate AR expression during prostate cancer progression.
Although expression of the different isoforms of Akt are shown to correlate with cancerous lesions and clinical outcomes in prostate cancer (Le Page et al., 2006), the ARR2-myr-Akt1 transgenic mice described in this report did not display an obvious phenotype in contrast to previous reports showing that expression of activated Akt in the murine prostate induces highly penetrant prostatic intraepithelial neoplasia (PIN) in the ventral prostate (Majumder et al., 2003). It is unlikely that the difference is due to the genetic backgrounds since other studies also conducted experiments in a C57BL/6 background (Xu et al., 2007) similar to that used in our study. Our study differs from others in the promoter used (probasin-426 to +28 (Majumder et al., 2003) versus the ARR2 promoter containing two copies of the enhancer used here) and the inclusion of a polyadenylation sequence in our transgenic construct. Furthermore, it is likely that the significant increase in nuclear expression of γ-H2AX and phospho-Chk2 in our ARR2-myr-Akt1 animals are contributing to cellular senescence, thus blocking tumorigenesis. Still, the most likely explanation for the observed phenotypic variations between studies using similar transgenic mouse lines may be found in variations of myr-Akt1 expression levels due to the site of integration or the promoter used.
Previous studies have shown that the impact of Akt on AR differed in low passage versus high passage LNCaP cells and depended on the activation of Forkhead transcription factor, FOXO3a. In low passage LNCaP cells (i.e., passage numbers less than 35), AR and prostate specific antigen were shown to be upregulated due to FOXO3a activation after treatment with the PI 3-kinase inhibitor LY294002 (Yang et al., 2005). In addition, overexpression of constitutively active Akt (cAkt) in LNCaP cells at low passage numbers (less than 38) suppressed AR activity as assessed by MMTV-luciferase and AR protein expression when compared to high passage numbers (greater than 60), at which point MMTV-luciferase and AR expression was enhanced in response to overexpression of cAkt (Lin et al., 2003).
While studies presented in this report do not examine the impact of Akt on AR target gene transcription, we use lower passage number LNCaP cells (23–34) to show that an Akt inhibitor results in diminished AR expression, a result further supported by the observation that transgenic expression of myristoylated Akt results in increased AR protein levels. We speculate that differences between studies (Yang et al., 2005) may be due to the use of an Akt specific inhibitor to suppress endogenous Akt activity as opposed to the effect of overexpression of cAkt or inhibition of PI 3-kinase, upstream of Akt. Reduced expression of AR in response to Akt inhibition is likely due to the diminished pro-survival signaling (Manning and Cantley, 2007), altered cell cycle regulation (Liang and Slingerland, 2003), or increased degradation of AR. Indeed, proteasome inhibition with MG132 could partially rescue AR levels in the presence of Akti (data not shown).
Phosphorylation dependent degradation of AR has been reported in response to overexpression of cAkt and resulted in phosphorylation-dependent AR degradation (Lin et al., 2002). While we observed ligand-dependent phosphorylation of AR S213 (comparable to S210) in human prostate tissue and LAPC4 cells, we did not observe this in LNCaP cells. In fact, when we previously overexpressed the LNCaP AR T877A mutant in 293 cells, we observed robust phosphorylation of S213 in wild type AR, but greatly diminished phosphorylation of the mutant (Taneja et al., 2005). However, we have not ruled out the possibility that S213 is constitutively phosphorylated at low levels in LNCaP cells.
Regulation of AR in the LNCaP-AI subline appears to be independent of Akt. Interestingly, the androgen-independent sublines of LNCaP (AI and abl) responded differently to Akt inhibition. These cell lines have differing characteristics that may impact androgen- independent growth. Silencing of the cyclin-dependent kinase inhibitor p21WAF1 contributes to the androgen-independent phenotype of LNCaP-AI cells (Wang et al., 2001), whereas M-phase cell cycle genes such as UBE2C are upregulated in LNCaP-abl cells (Wang et al., 2009). In addition, other authors have presented evidence of gross differences in AR protein and mRNA regulation in androgen-dependent versus -independent cells, the latter expressing more stable AR protein and mRNA. For example, pulse chase experiments show that AR protein is 2–4 times more stable in cells derived from recurring prostate tumors than in LNCaP cells (Gregory et al., 2001). There are also differences in regulation of AR mRNA in androgen-dependent versus - independent cells: AR transcription is decreased in response to cytokines such as TNFα in LNCaP cells but not in androgen-independent cells (Ko et al., 2008).
Conventional anti-androgen treatments inhibit the activity of AR but activation of AR through other signaling molecules such as Akt may still lead to disease progression. Multiple studies have shown a correlation between phosphorylated Akt and prostate cancer progression and recurrence, making Akt an attractive therapeutic target. Unfortunately, our finding that AR protein levels are not decreased in all androgen-independent prostate cancer cells examined suggests that the AR pathway would be fully intact even in the presence of Akt inhibitors in some late stage prostate cancers. This is supported by studies showing that phase II clinical trials of androgen-independent or biochemically recurrent prostate cancer patients using the Akt inhibitor perifosine did not significantly improve clinical outcomes (Chee et al., 2007; Posadas et al., 2005). Thus, one might speculate that the window of opportunity for the clinical use of Akt inhibitors to treat prostate cancer may be restricted and that these agents may be useful to prevent progression of androgen-dependent disease to the anti-androgen resistant disease stage.
Supplementary Material
Acknowledgments
This work was supported by the American Cancer Society and NIH R01CA112226 (S.L.).
We thank members of the Logan laboratory (Paolo Mita, Julia Staverosky and Eric Schafler) for critical reading of the manuscript. We thank R. Reiter (University of California, Los Angeles) for LAPC4 cells, A. Ferrari (New York University School of Medicine) for the LNCaP-AI cells and Z. Culig (Innsbruck Medical University, Austria) for the LNCaP-abl cells.
Footnotes
Conflicts of Interest: None
References
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bookout AL, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl Recept Signal. 2003;1:e012. doi: 10.1621/nrs.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee KG, Longmate J, Quinn DI, Chatta G, Pinski J, Twardowski P, Pan CX, Cambio A, Evans CP, Gandara DR, et al. The AKT inhibitor perifosine in biochemically recurrent prostate cancer: a phase II California/Pittsburgh cancer consortium trial. Clin Genitourin Cancer. 2007;5:433–437. doi: 10.3816/CGC.2007.n.031. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, Chen H, Qiu Y. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001;276:31858–31862. doi: 10.1074/jbc.C100271200. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- Conus NM, Hannan KM, Cristiano BE, Hemmings BA, Pearson RB. Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt protein kinase. J Biol Chem. 2002;277:38021–38028. doi: 10.1074/jbc.M203387200. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, Bartsch G, Utermann G, Schneider MR, Parczyk K, et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer. 1999;81:242–251. doi: 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFeo-Jones D, Barnett SF, Fu S, Hancock PJ, Haskell KM, Leander KR, McAvoy E, Robinson RG, Duggan ME, Lindsley CW, et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol Cancer Ther. 2005;4:271–279. [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Gao M, Ossowski L, Ferrari AC. Activation of Rb and decline in androgen receptor protein precede retinoic acid-induced apoptosis in androgen-dependent LNCaP cells and their androgen-independent derivative. J Cell Physiol. 1999;179:336–346. doi: 10.1002/(SICI)1097-4652(199906)179:3<336::AID-JCP11>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–4319. [PubMed] [Google Scholar]
- Ko S, Shi L, Kim S, Song CS, Chatterjee B. Interplay of nuclear factor-kappaB and B-myb in the negative regulation of androgen receptor expression by tumor necrosis factor alpha. Mol Endocrinol. 2008;22:273–286. doi: 10.1210/me.2007-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koseoglu S, Lu Z, Kumar C, Kirschmeier P, Zou J. AKT1, AKT2 and AKT3-dependent cell survival is cell line-specific and knockdown of all three isoforms selectively induces apoptosis in 20 human tumor cell lines. Cancer Biol Ther. 2007;6:755–762. doi: 10.4161/cbt.6.5.3995. [DOI] [PubMed] [Google Scholar]
- Le Page C, Koumakpayi IH, Alam-Fahmy M, Mes-Masson AM, Saad F. Expression and localisation of Akt-1, Akt-2 and Akt-3 correlate with clinical outcome of prostate cancer patients. Br J Cancer. 2006;94:1906–1912. doi: 10.1038/sj.bjc.6603184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lu Y, Jin W, Liang K, Mills GB, Fan Z. Autophosphorylation of Akt at threonine 72 and serine 246. A potential mechanism of regulation of Akt kinase activity. J Biol Chem. 2006;281:13837–13843. doi: 10.1074/jbc.M602060200. [DOI] [PubMed] [Google Scholar]
- Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- Lin HK, Hu YC, Yang L, Altuwaijri S, Chen YT, Kang HY, Chang C. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. J Biol Chem. 2003;278:50902–50907. doi: 10.1074/jbc.M300676200. [DOI] [PubMed] [Google Scholar]
- Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. Embo J. 2002;21:4037–4048. doi: 10.1093/emboj/cdf406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, Guney I, Berger R, Herman P, Bikoff R, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14:146–155. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, Xue Q, Bikoff R, Ma H, Kantoff PW, Golub TR, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc Natl Acad Sci U S A. 2003;100:7841–7846. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Posadas EM, Gulley J, Arlen PM, Trout A, Parnes HL, Wright J, Lee MJ, Chung EJ, Trepel JB, Sparreboom A, et al. A phase II study of perifosine in androgen independent prostate cancer. Cancer Biol Ther. 2005;4:1133–1137. doi: 10.4161/cbt.4.10.2064. [DOI] [PubMed] [Google Scholar]
- Taneja SS, Ha S, Swenson NK, Huang HY, Lee P, Melamed J, Shapiro E, Garabedian MJ, Logan SK. Cell-specific regulation of androgen receptor phosphorylation in vivo. J Biol Chem. 2005;280:40916–40924. doi: 10.1074/jbc.M508442200. [DOI] [PubMed] [Google Scholar]
- Wang LG, Ossowski L, Ferrari AC. Overexpressed androgen receptor linked to p21WAF1 silencing may be responsible for androgen independence and resistance to apoptosis of a prostate cancer cell line. Cancer Res. 2001;61:7544–7551. [PubMed] [Google Scholar]
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc Natl Acad Sci U S A. 2006;103:7789–7794. doi: 10.1073/pnas.0602567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Majumder PK, Ross K, Shim Y, Golub TR, Loda M, Sellers WR. Identification of prostate cancer modifier pathways using parental strain expression mapping. Proc Natl Acad Sci U S A. 2007;104:17771–17776. doi: 10.1073/pnas.0708476104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Xie S, Jamaluddin MS, Altuwaijri S, Ni J, Kim E, Chen YT, Hu YC, Wang L, Chuang KH, et al. Induction of androgen receptor expression by phosphatidylinositol 3-kinase/Akt downstream substrate, FOXO3a, and their roles in apoptosis of LNCaP prostate cancer cells. J Biol Chem. 2005;280:33558–33565. doi: 10.1074/jbc.M504461200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Thomas TZ, Kasper S, Matusik RJ. A small composite probasin promoter confers high levels of prostate- specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology. 2000;141:4698–4710. doi: 10.1210/endo.141.12.7837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.