

Abstract
Polycystic ovary syndrome (PCOS) is one of the most common fertility disorders, affecting several million women worldwide. Women with PCOS manifest neuroendocrine, ovarian, and metabolic defects. A large number of animal models have evolved to understand the etiology of PCOS. These models provide support for the contributing role of excess steroids during development in programming the PCOS phenotype. However, considerable phenotypic variability is evident across animal models, depending on the quality of the steroid administered and the perinatal time of treatment relative to the developmental trajectory of the fetus/offspring. This review focuses on the reproductive and metabolic phenotypes of the various PCOS animal models that have evolved in the last decade to delineate the relative roles of androgens and estrogens in relation to the timing of exposure in programming the various dysfunctions that are part and parcel of the PCOS phenotype. Furthermore, the review addresses the contributory role of the postnatal metabolic environment in exaggerating the severity of the phenotype, the translational relevance of the various animal models to PCOS, and areas for future research.
Keywords: Infertility, PCOS, fetal programming, androgens, estrogens
More than 70 million people globally experience infertility.1 Among couples of childbearing age seeking medical help, in ~30 to 40% of the cases, it is exclusively a problem with the woman. Infertility disorders such as premature ovarian failure leading to early estrogen deficiency may lead to adverse consequences such as osteopenia, cardiovascular risk, and cognitive deficits. Because infertility can negatively impact quality of life and psychosocial well-being, approaches to prevent/overcome infertility must be developed.
Among fertility disorders, polycystic ovary syndrome (PCOS) is one of the most common. Economic burden of PCOS exceeds several billion dollars annually in the United States. A large percentage of women with PCOS do not respond to ovulation induction protocols.2 Even if successful ovulation is induced, conception rates are low and the percentage of pregnancies ending in spontaneous miscarriages is high.3,4 Women with PCOS are also at risk for ovarian hyperstimulation and multiple gestations.4–6 They are more likely to develop gestational diabetes and preeclampsia6 and show psychological disturbances.7,8 Overall, they have a lower degree of satisfaction about health and sexuality.7,8 About 70% of these women manifest insulin resistance,9 and insulin-lowering drugs reduce hyper-androgenism implicating a metabolic component in the etiology of PCOS.10–12 An increased risk of cardiovascular disease, dyslipidemia, hypertension, diabetes mellitus, and endometrial cancer in PCOS13,14 emphasizes the need not only to address the issues of infertility but also the long-term goals of preventing debilitating diseases and most importantly the transgenerational transfer of unwanted traits to the offspring. The etiology of PCOS is unknown and remains a topic of intense research.
Increasing evidence suggests that adult dysfunctions may result from abnormal programming of developing systems during intrauterine life.15 Some believe that androgen excess early in life may lead to the manifestation of PCOS in adulthood.16,17 In support, the PCOS phenotype is associated with conditions such as classical 21-hydroxylase deficiency in which the fetus has been exposed to high concentrations of sex steroids before birth.18 Several animal models have evolved to determine the impact of perinatal exposure to steroids on the development of adult reproductive and metabolic pathologies.19 Many of these animal models that manifested the PCOS phenotype involved perinatal treatment with testosterone (T). These perinatal T-treated models are often referred to as androgenized models, overlooking the ability of T to be aromatized to estrogen and then exerting its effects via estrogenic programming. Other models involve perinatal exposure to dihydrotestosterone (DHT), a nonaromatizable androgen, or estrogenic agents. This review focuses on animal models that have evolved in the last decade to (1) compare and contrast the reproductive and metabolic phenotypes of these animal models relative to women with PCOS and the nonhuman primate model for PCOS, (2) delineate the relative roles of androgens and estrogens in facilitating the various disruptions, (3) address the relative strengths and weaknesses of the different models, (4) pinpoint the translational significance of these animals to human PCOS, and (5) point to future directions to be taken.
DEVELOPMENTAL PROGRAMMING OF PCOS PHENOTYPE WITH PERINATAL T EXCESS
Studies assessing developmental effects of T focused on three species, Rhesus monkeys, sheep, and rats. Monkey and sheep studies have addressed the effects of T excess starting at two different gestational time points, early and late gestation. Rat studies have addressed exposure during prenatal and early postnatal periods (Table 120–71). These studies have found that developmental exposure to T excess leads to neuroendocrine, ovarian, and metabolic deficits (Fig. 1), the details of which are discussed next.
Table 1.
Comparison of Attributes of Prenatal Testosterone-Treated Monkeys, Sheep, and Rats and Postnatal Testosterone-Treated Rats with That of Women with Polycystic Ovary Syndrome
| PCOS Phenotype | Prenatal
|
Postnatal
|
|||||
|---|---|---|---|---|---|---|---|
| PCOS Women | Monkey | Sheep | RatSD | RatSD/W | |||
|
|
|
|
|
||||
| TP | TP | TF | TP | ||||
|
|
|
|
|
||||
| GD (40–60) to (55–120) | GD (110–115) to 139 | GD 30–90 | GD 60–90 | GD 16–19 | 3h PN | ||
| Hyperandrogenism | Yes20 | Yesfunc,33,34 | Yesfunc,34 | Yesfunc,47 | – | No,66; yes67 | No70,71 |
| LH excess | Yes21 | Yes34,35 | No34 | Yes48–50 | No49 | Yes66,67 | – |
| Oligo-anovulation | Yes20 | Yes35,36 | Yes35 | Yes51–53 | No51 | Yes67 | – |
| Infertility | Yes20 | Not tested,V,37 | Yes37 | Not tested,V,54 | Yes55 | Not testedV,66 | – |
| PCO morphology | Yes22 | Yes38 | Yes38 | Yes56,57 | – | Yes67 | – |
| Increased ovary weight/volume | Yes22 | Yes35 | Yes35 | Yes57 | – | – | – |
| Follicular persistence | ? | – | – | Yes52,53 | – | – | – |
| Enhanced follicular recruitment | YesA,23 | – | – | Yes57–59 | – | – | – |
| Increased intrafollicular androgen | Yes24 | No39 | No39 | – | – | – | – |
| Reduced oocyte competence | Yes22 | Yes39 | Yes39 | – | – | – | – |
| Disrupted E2 positive feedback | ? | No40 | – | Yes49,60 | Yes49 | Yes66 | – |
| Reduced E2 negative feedback | ? | Yes40 | – | Yes50 | – | – | – |
| Reduced P4 negative feedback | Yes25 | Yes41 | Yes41 | Yes61,62 | – | – | – |
| Increased GnRH sensitivity | Yes26 | Yes34 | – | Yes48 | – | No66 | – |
| Reduced insulin sensitivity | Yes9 | No42 | Yes42 | Yes63,64 | Yes64 | No68 | Yes70,71 |
| Pancreatic β-cell dysfunction | At risk27 | Yes42,43 | No42 | – | – | – | – |
| IUGR | YesB,28 | No44 | No44 | Yes54 | – | YesE,69 | – |
| Catch-up growth | YesB,29 | YesD,36 | – | Yes54 | No49 | – | – |
| Increased visceral fat | YesC,30 | Yes45 | – | – | – | Yes68 | Yes,70 No71 |
| Increased serum triglycerides | YesC,31 | – | – | – | – | Yes68 | Yes70 |
| Increased total cholesterol | YesC,31 | – | – | – | – | Yes68 | Yes70 |
| Increased free fatty acids | YesC,31 | Yes46 | – | – | – | – | No71 |
| Increased atherogenic index | YesC,31 | – | – | – | – | – | Yes70 |
| Hypertension | At risk32 | – | – | Yes65 | – | – | – |
based on cortical biopsies
Spanish cohort
in obese PCOS women
prior to menarche
prenatal treatment GD16 to 20
free
functional
propionate
Sprague-Dawley
virilized
Wistar Numbers indicate references.
PCOS, polycystic ovary syndrome; T, testosterone; GD, gestational day; LH, luteinizing hormone; PCO, polycystic ovary; E2, estradiol; P4, progesterone; GnRH, gonadotropin-releasing hormone; IUGR, intrauterine growth restriction.
Figure 1.
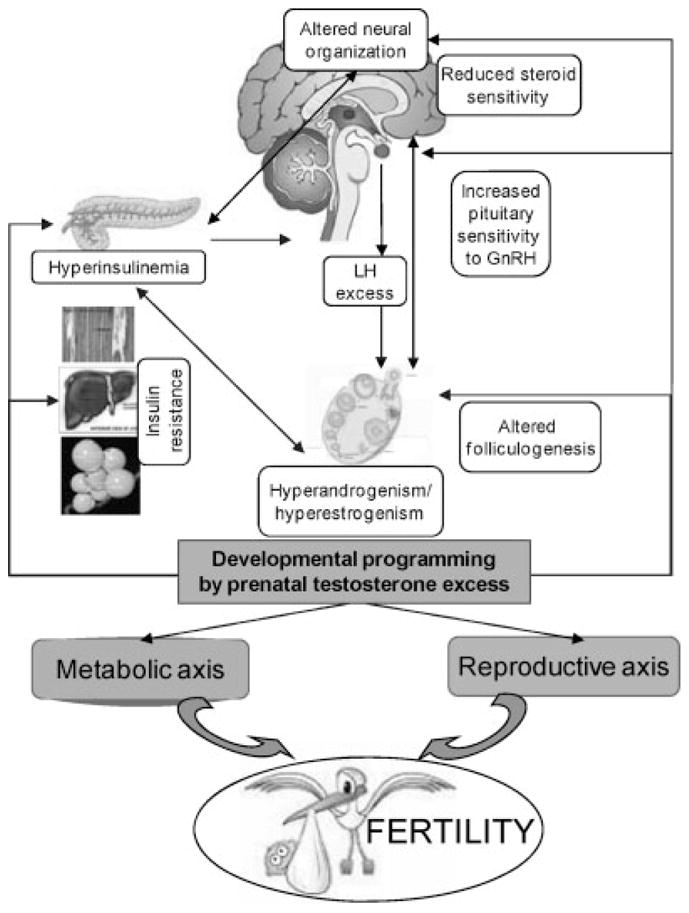
Schematic showing the impact of perinatal testosterone excess on neuroendocrine, ovarian, and metabolic programming and their contribution to infertility. GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone.
Neuroendocrine Studies
A common consequence of prenatal T excess is the induction of leuteinizing hormone (LH) excess in early-treated monkeys,34,35 early-treated sheep,48–50 and prenatal-treated rats.66,67 Detailed characterization of LH pulse dynamics performed in ovary-intact early-treated sheep found disruption of all three feedback systems, namely estradiol (E2)-negative,50 E2-positive,49,60 and progesterone (P4)-negative feedback. 61,62 A late shorter duration of treatment (gestational day [GD] 60 to 90) induced less severe disruptions at the E2- positive feedback level.49 Studies in early-treated monkeys (GD: 40 to 60 to 55 to 120) found reduced LH responsiveness to E2.34,40 Prenatal-treated rats (GD: 16–19)66 and early-treated sheep49 also manifest compro- mised E2 positive feedback responses. In-depth studies testing E2-negative and -positive feedback responses have not been undertaken in women with PCOS. Early-treated sheep61,62 and early- and late-treated monkeys41 manifest reduced sensitivity to P4-negative feedback, a feature seen in women with PCOS.25,26 More recent, neuroanatomical studies have found that kisspeptin/neurokinin-B/dynorphin neuronal population may be involved in altered negative feedback sensitivity.72 At the pituitary level, as in women with PCOS,25 pituitary sensitivity to gonadotropin-releasing hormone (GnRH) is increased in prenatal T-treated sheep48 and monkeys34,40 but not in rats.66 These differences may be a function of the study design; only studies in sheep,48 but not rats66 and monkeys,34 were undertaken after ablation of endogenous GnRH action.
Ovarian Studies
At the ovarian level, prenatal T excess leads to polycystic ovarian morphology with increased ovarian weight/volume in monkeys35,38 and sheep.56 Morphometric studies and serial ultrasonography studies undertaken in sheep provide evidence in support of increased ovarian follicular recruitment/depletion57 and persistence.52,53 An increase in antral follicle number following prenatal T excess was also evident in monkeys38 and rats.67 However, the measures in rats and monkeys38,67 as well as in women with PCOS23 are based on a single time point evaluation unlike serial ovarian stereology57/ultrasound52 undertaken at multiple developmental time points in sheep. It should also be recognized that rodents are polyovular and hence manifest polyfollicular morphology even when untreated. Furthermore, in addressing ovarian developmental programming, it is crucial to take into account the differences in the trajectory of ovarian differentiation. Sheep and subhuman primates are precocial with follicular differentiation completed in utero. In contrast, differentiation gets completed in rodent models only postnatally (Table 273–78). In-depth evaluations performed only with ovaries of sheep model of PCOS have revealed disruptions in androgen/estrogen receptor ratios,47 growth factor expression such as activin and follistatin,56 and insulin receptor signaling79 such as those seen in women with PCOS.80,81
Table 2.
Schematic Showing the Time (Days) of Appearance of Different Classes of Follicles in Humans, Monkeys, Sheep, Rats, and Mice
| Developmental Time Points | Human | Rhesus Monkey | Sheep | Rat | Mice | |
|---|---|---|---|---|---|---|
| Prenatal life | Implantation | 9 | 9 | 14 | 5.5 | 4 |
| Gonadal differentiation | 42–63 | 40 | 30 | 12.5 | 6 | |
| Start meiosis | 90 | 60 | 55 | 17 | 13 | |
| Primordial follicles | 112 | 100 | 75 | – | – | |
| Primary follicles | 130 | ? | 110 | – | – | |
| Antral follicles | 230 | 125 | 135 | – | – | |
| Birth | 270 | 170 | 147 | 22 | 20 | |
| Postnatal life | Primordial follicles | – | – | – | 1–2 | 2–5 |
| Primary follicles | – | – | – | 2–3 | 2–5 | |
| Antral follicles | – | – | – | 15 | 17 | |
Data for human, monkey, sheep, and mice adapted from Padmanabhan et al.73
Hyperandrogenemia
Studies conducted thus far document that prenatal T excess induces functional hyperandrogenism in monkeys manifested as enhanced responsiveness to human chorionic gonadotropin.33,34 Prenatal T-treated sheep also manifest functional hyperandrogenism reflected as increased ovarian47 and hypothalamic82 androgen receptor expression, and polyfollicular morphology.56,57 Studies in prenatal T-treated Sprague-Dawley rats are inconsistent in that hyperandrogenism was reported in one study67 but not the other.66 Both studies used the same regimen of T treatment both in terms of timing and dosage.
Cyclic Function and Fertility
Oligo-anovulation is a common feature of all three species (monkeys, sheep, and rodents) treated prenatally with T34–36,51–53,62,67 with the degree of disruption depending on the timing of treatment, with late-treated sheep and monkeys revealing lesser disruptions than the early-treated ones.34,51 Studies in monkeys, the only model where oocyte competence has been assessed, found that prenatal T excess reduces oocyte competence.39 Fertility tests following natural mating have been undertaken only with late-treated sheep (early-treated animals are virilized) and reveal a 60% reduction in pregnancy rates.55 Compromised fertility/fecundity is also a feature of women with PCOS.20,83
Cardiometabolic Studies
Developmentally, early-treated sheep manifested intra-uterine growth restriction (IUGR) and compensatory postnatal catch-up growth.54 An increase in postnatal growth rate was also evidenced in early-treated monkeys before menarche,36 although they did not manifest IUGR. Metabolic perturbations programmed by prenatal T excess include insulin resistance in late-treated monkeys,42 early- and late-treated sheep,63,64 and post-natal-treated rats70,71 but not prenatal-treated rats.68 Early-treated older monkeys have been reported to develop pancreatic β cell dysfunction.42,43 Increased visceral fat is another feature of early-treated older monkeys45 and prenatal-treated rats.68 Postnatal T treatment also increases fat mass in Wistar rats,70 although it has no effect in Sprague-Dawley rats.71 Both prenatal- and postnatal-treated rodent models manifest increased serum triglycerides and cholesterol68,70 suggestive of an extended critical period. Telemetry studies performed only in sheep found early treatment leads to hypertension.65 As such, prenatal T treatment has an impact on cardiometabolic aspects with the nature of disruptions differing between the species studied and possibly stemming from differences in timing of insult relative to organ differentiation.
Overall, the prenatal T-treated models manifest reproductive and metabolic features of PCOS consistent with the National Institutes of Health (NIH) 199084 (chronic anovulation and clinical and/or biochemical signs of hyperandrogenism), Rotterdam European Society of Human Reproduction and Embryology/American Society for Reproductive Medicine (ESHRE/ASRM) 200385 (oligo- and/or anovulation, clinical and/or biochemical signs of hyperandrogenism, and polycystic ovaries; two of three), Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) 200686 (oligo- and/or anovulation with clinical and/or biochemical signs of hyperandrogenism) criteria, and the cardiovascular disease risk AE-PCOS statement.87 Information is incomplete in the early postnatal T-treated rodent model70,71 to assess if they meet any of these criteria.
DEVELOPMENTAL PROGRAMMING OF PCOS PHENOTYPE WITH ANDROGEN EXCESS
The nonaromatizable androgen DHT was used as the programming agent in three prenatal models and two postnatal models (Table 3). The prenatal models include sheep (GD: 30 to 90), Sprague-Dawley rats (GD: 16 to 19), and mice (GD: 16 to 18), and the two postnatal models involve Wistar rats treated either 3 hours after birth (single dose) or 21 days after birth (duration: 90 days). Although the potential for estrogenic effect of DHT via conversion to 3β-diol and action through estrogen receptor-β exists,95 considering that the degree of such conversion in specific tissues/species remains unknown and is expected to be minimal, for the purpose of this review, DHT effects are discussed relative to its androgenic potential.
Table 3.
Comparison of Attributes of Prenatal Dihydrotestosterone-Treated Sheep and Mice, and Postnatal Dihydrotestosterone, Letrozole, and Estradiol Valerate–Treated Rats with That of Women with Polycystic Ovary Syndrome
| PCOS Phenotype | Prenatal | Postnatal | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| PCOS Women | Sheep | RatSD | Mouse | RatW | RatW | RatW | RatW | RatW | |
|
| |||||||||
| DHTP | DHTF | DHTP | DHTP | DHTP | Letrozole | Letrozole | EV | ||
|
| |||||||||
| GD 30–90 | GD 16–19 | GD 16–18 | 3h PN | 21d PN (90d) | 21d PN (90d) | 42d PN (21d) | 14 d PN | ||
| Hyperandrogenism | Yes20 | Nofunc,47 | No,66 Yes67 | Yes* | No70,89,90 | No91 | Yes91 | Yes92,93 | No94 |
| LH excess | Yes21 | Yes48 | Yes66,67 | Yes89 | – | – | – | Yes92,93 | No94 |
| Oligo-anovulation | Yes20 | No53 | Yes67 | Yes89,90 | – | Yes91 | Yes91 | Yes92 | Yes94 |
| Infertility | Yes20 | Not tested,V,53 | Not testedV,66 | – | – | – | – | – | – |
| PCO morphology | Yes22 | No57 | Yes67 | – | – | Yes91 | Yes91 | Yes92 | Yes94 |
| Increased ovary weight/volume | Yes22 | Yes, fetal57 | – | – | – | No91 | Yes91 | No92 | No94 |
| Follicular persistence | ? | No53 | – | – | – | – | – | – | – |
| Enhanced follicular recruitment | YesA,23 | Yes, fetal57 | – | – | – | – | – | – | – |
| Increased intrafollicular androgen | Yes24 | – | – | – | – | – | – | – | – |
| Reduced oocyte competence | Yes22 | – | – | – | – | – | – | – | – |
| Disrupted E2 positive feedback | ? | No88 | Yes66 | – | – | – | – | – | – |
| Reduced E2 negative feedback | ? | Yes88 | – | – | – | – | – | – | – |
| Reduced P4 negative feedback | Yes25 | – | – | – | – | – | – | – | – |
| Increased GnRH sensitivity | Yes26 | Yes48 | No66 | – | – | – | – | – | – |
| Reduced insulin sensitivity | Yes9 | Yes64 | – | No90,A | Yes70 | Yes91 | No91 | – | – |
| Pancreatic β-cell dysfunction | At risk27 | – | – | Yes90 | – | – | – | – | – |
| IUGR | YesB,28 | – | – | – | – | – | – | – | – |
| Catch-up growth | YesB,29 | – | – | – | – | – | – | – | – |
| Increased visceral fat | YesC,30 | – | – | No90 | No70 | Yes91 | No91 | – | – |
| Increased serum triglycerides | YesC,31 | – | – | – | No70 | No91 | No91 | – | – |
| Increased total cholesterol | YesC,31 | – | – | – | No70 | No91 | No91 | – | – |
| Increased free fatty acids | YesC,31 | – | – | – | – | No91 | No91 | – | – |
| Increased atherogenic index | YesC,31 | – | – | – | No70 | – | – | – | – |
| Hypertension | At risk32 | – | – | – | – | – | – | – | – |
Glucose intolerance present
free
propionate
Sprague-Dawley
virilized
Wistar
See text for the dual coding of Roland et al90 (“Developmental Programming of PCOS Phenotype with Androgen Excess,” subhead “Hyperandrogenism”).
PCOS, polycystic ovary syndrome; DHT, dihydrotestosterone; GD, gestational day; PN, postnatal; LH, luteinizing hormone; PCO, polycystic ovary; E2, estradiol; GnRH, gonadotropin-releasing hormone; IUGR, intrauterine growth restriction.
Neuroendocrine Studies
Detailed LH dynamics have been undertaken in sheep and rats and show that prenatal DHT treatment increases LH pulse frequency and amplitude.66,88 Single time point measures in mice also show that prenatal DHT treatment increases plasma LH levels.89 Detailed E2-negative feedback studies with prenatal DHT treat- ment have only been performed in sheep, and these show that E2-negative feedback responses are reduced,88 similar to that of prenatal T-treated sheep.50 E2-positive feedback is disrupted in DHT-treated rats66 but not sheep.88 At the pituitary level, prenatal DHT treatment, similar to findings with prenatal T, increased pituitary sensitivity to GnRH in sheep48 but not rats66 possibly due to the test being conducted without blocking endogenous GnRH input in rats.
Ovarian Studies
The effect of perinatal DHT treatment in the development of polycystic ovarian (PCO) morphology is species specific. Although both prenatal and postnatal DHT-treated rats display PCO morphology,67,91 this is not the case with sheep.57 Similar studies with prenatal DHT have not been undertaken in monkeys or mice. Ovarian morphometric and serial ultrasonography studies performed only in sheep support a transient increase in follicular recruitment57 but not follicular persistence.53
Hyperandrogenism
It remains to be resolved whether hyperandrogenism is a consistent feature of prenatal DHT-treated mice. Hyperandrogenism was reported as a consequence in one study conducted at 4 to 6 months of age.89 In the second study performed by the same group, hyperandrogenism was not evident at 5 months of age.90 Authors attributed the lack of hyperandrogenism in 5-month-old animals in the second study to the age when hyperandrogenism was examined (although there is overlap in age between this and the first study) or differences in the sensitivity of the T assay used (different assays were used in the two studies). The effect of prenatal DHT in Sprague-Dawley rats is also controversial, with one study manifesting hyperandrogenic status67 and another not.66 Hyper-androgenism is not a feature of postnatal DHT-treated rats.70,91 Prenatal DHT-treated sheep are functionally hyperandrogenic only during fetal life (manifested by increased androgen receptors in granulosa and stromal compartments) but not during adult life.47 These findings differ from the prenatal T-treated sheep, which shows evidence of hyperandrogenism both during fetal and adult life.47
Cyclic Function and Fertility
Cycle disruptions are evident in all models but differ in their attributes.53,67,89–91 The preovulatory E2 rise and LH surge dynamics studied only in prenatal DHT-treated sheep are not disrupted.88 Fertility tests have not been performed in any of the pre- or post-natal-treated models possibly due to their virilized phenotype.
Cardiometabolic Studies
Reduced insulin sensitivity is also a feature of prenatal DHT-treated sheep64 and the postnatal-treated rat models70,91 but not the prenatal DHT-treated mice,90 which display glucose intolerance.90 Increased visceral fat was a feature of late91 but not early70 postnatal DHT-treated rats or DHT-treated mice.90 No changes in lipid profiling were evident in both postnatal-treated rat models.70,91
The prenatal rat models show opposing findings with one meeting NIH, Rotterdam ESHRE/ASRM and the AE-PCOS criteria (cycle anomalies, PCO morphology, and hyperandrogenism)67 and the other showing only cycle disruptions (there is no evidence of hyperandrogenism and the ovarian phenotype has not been tested).66 The late postnatal rodent model91 fits only the Rotterdam ESHRE/ASRM criteria by virtue of the cycle anomalies and PCO morphology. The prenatal DHT-treated sheep model manifests only cycle disruptions53 but not hyperandrogenism or PCO morphology57 and therefore does not fit any of the PCOS criteria. The jury is still out on the prenatal DHT-treated mouse model in view of the discrepancy seen in the hyperandrogenic phenotype between the two studies.89,90 If hyperandrogenism is part of the consequence, the prenatal DHT-treated mouse model would meet the NIH, Rotterdam ESHRE/ASRM, as well as the AE-PCOS criteria.
DEVELOPMENTAL PROGRAMMING OF PCOS PHENOTYPE WITH ESTROGENS
Two different paradigms have been used to address the role of prenatal E2 programming. These include E2 valerate (EV) treatment beginning day 14 of neonatal life94 or administration of letrozole, a nonsteroidal aromatase inhibitor, to block conversion of androgen to estrogen (estrogen ablation approach) beginning either at postnatal day 21 for 3 months (early91) or at postnatal day 42 for 3 weeks (late92,93).
Neuroendocrine Studies
Detailed neuroendocrine investigations have not been performed with these models. LH excess (studies performed without controlling for cycle stage) is a feature of the late letrozole-treated model.92,93 This has not been studied in the early-treated model. In contrast, the EV model manifested low LH levels.94
Ovarian Studies, Cyclic Function, and Fertility
All three models display PCO morphology91,92,94,96 but disagree relative to ovarian weight (high in early letrozole,91 normal in late letrozole,92 and low in EV94).
Hyperandrogenism
Hyperandrogenism is a common feature of both letrozole models,91–93 whereas the EV-treated94 showed the opposite, namely hypoandrogenism.
Cyclic Function and Fertility
Cycle dysfunction is a common feature of the EV as well as the early- and late-treated letrozole models, although they differed in their attributes.91,92,94
Cardiometabolic Studies
Insulin sensitivity, visceral fat, and lipid profile were normal in the early letrozole-treated rats.91 Metabolic measures have not been studied in the other two animal models.
The EV model, which manifests cycle disruption and polycystic ovaries in the face of hypoandrogenism, meets the Rotterdam ESHRE/ASRM criteria of PCOS. Both letrozole models meet the NIH, Rotterdam ESHRE/ASRM as well as AE-PCOS criteria manifesting cyclic disruptions, hyperandrogenism, and PCO morphology. It should be noted that the adult phenotype of the two letrozole-treated models are similar in spite of differences in the timing of onset and duration of treatments. In the context of reprogramming, a limitation of the letrozole-treated model is that studies were performed immediately after stopping the treatment. As such, reported disruptions may be activational and dissipate after cessation of treatment.
ANDROGENIC VERSUS ESTROGENIC PROGRAMMING
The models discussed point to some aspects of the perinatal programming of the PCOS phenotype being driven by excess androgen and others by excess estrogen in a species-specific manner. In prenatal T-treated models, there is obvious potential for both androgenic and estrogenic programming. Sheep studies show that gestational T treatment increases both T and E2 concentrations in female fetuses,97 providing support that the resultant PCOS phenotype is likely the culmination of androgenic as well as estrogenic programming. Elevated fetal T levels but not estrogens were characteristics of gestational T-treated monkey fetuses.44 Similar information is lacking in the small animal models.
To discern whether each of the reproductive and metabolic disruptions previously discussed arise from androgenic or estrogenic effects, animal models that compare the quality of steroids spanning the same developmental time points provide the only valid comparisons. Four models fit this criteria: sheep treated from GD 30 to 90 with T or DHT, rats treated on GD 16 to 19 (prenatal) with T or DHT, rats treated 3 hours postnatal with T or DHT, and rats treated 21 day postnatal with DHT or letrozole (Table 4). Because the monkey model of PCOS involved only T treatment and the mouse model only DHT, such comparisons are not possible in these models.
Table 4.
Androgenic versus Estrogenic Programming of the Polycystic Ovary Syndrome Phenotype*
| PCOS Phenotype | Prenatal
|
Postnatal
|
||
|---|---|---|---|---|
| Sheep (GD 30–90) | Rat (GD 16–19) | Rat (3-hour PN) | Rat (21 days PN) | |
| Hyperandrogenism | Estrogenic | Inconsistent | Not disrupted | Inconsistent |
| LH excess | Androgenic | Androgenic | Not studied | Not studied |
| Oligo-anovulation | Estrogenic | Androgenic | Not studied | Androgenic |
| PCO morphology | Estrogenic | Androgenic | Not studied | Androgenic |
| Follicular persistence | Estrogenic | Not studied | Not studied | Not studied |
| Enhanced follicular recruitment | Androgenic | Not studied | Not studied | Not studied |
| Disrupted E2 positive feedback | Estrogenic | Androgenic | Not studied | Not studied |
| Reduced E2 negative feedback | Androgenic | Not studied | Not studied | Not studied |
| Increased GnRH sensitivity | Androgenic | Not disrupted | Not studied | Not studied |
| Reduced insulin sensitivity | Androgenic | Not studied | Androgenic | Inconsistent |
| Increased visceral fat | Not studied | Not studied | Inconsistent | Controversial |
| Abnormal lipid profile | Not studied | Not studied | Estrogenic | Not disrupted |
In GD 30 to 90 (prenatal) T versus DHT-treated sheep, GD 16 to 19 (prenatal) T versus DHT-treated rat, 3-hour PN T versus DHT-treated rat, and 21-day PN DHT versus letrozole-treated rat.
Assessment of androgenic or estrogenic regulation is based on outcomes described in Tables 1 and 3.PCOS, polycystic ovary syndrome; GD, gestational day; PN, postnatal; LH, luteinizing hormone; PCO, polycystic ovary; E2, estradiol; GnRH, gonadotropin-releasing hormone; DHT, dihydrotestosterone; T, testosterone.
Comparison of studies conducted with prenatal T- and DHT-treated sheep suggest that PCO morphology, follicular persistence, ovarian hyperandrogenism, oligo-anovulation, and the E2-positive feedback disruptions seen in adults are likely programmed by estrogenic actions, whereas LH excess, enhanced follicular recruitment, reduced sensitivity to E2-negative feedback, increased GnRH sensitivity, and reduced insulin sensitivity are programmed via androgens. Studies in prenatal T versus DHT rat models66,67,70 are in agreement with the sheep model48,63,64 relative to androgenic programming of LH excess and reduced insulin sensitivity. For comparison with women with PCOS and other models, discussion of the sheep model in this review has focused on the ovary-intact model. It needs to be recognized that dissection of androgenic and estrogenic programming of E2-positive and P4-negative feedback systems were delineated first using the ovar- iectomized E2-replaced prenatal T-treated model. 98,99
In contrast to findings in the sheep model,73 PCO morphology, oligo-anovulation, and E2 positive feedback disruptions in both prenatal- and postnatal-treated rats66,67,91 point to programming via androgens. Paradoxically, hyperandrogenism is an inconsistent finding between the two rat studies, which used identical paradigms in the same strain of rats.66,67 Similarly, androgenic programming achieved via DHT or ablation of estrogen with letrozole yielded inconsistent metabolic outcomes, the former being insulin resistant and having increased visceral fat but the latter not.91 Visceral adiposity and abnormal lipid profile in postnatal T- but not DHT-treated Wistar rats70 is supportive of estrogenic programming of these variables.
The inconsistencies seen between species are likely a function of the timing of treatment relative to timing of organ differentiation. However, inconsistencies in outcome such as seen in the DHT- and letrozole-treated models, both enforcing androgenic programming within the same strain of rats using similar exposure periods, suggest that the degree of steroid excess or imbalance in the estrogen-to-androgen ratio might be the underlying cause in the reprogramming of reproductive and metabolic dysfunction and development of the PCOS phenotype. Information on endogenous levels of various androgens and estrogens during the programming windows are required across species to sort out differences in outcomes.
METABOLIC AMPLIFICATION OF STEROIDAL PROGRAMMING
Evidence to date suggests that PCOS women have an increased propensity toward ovulatory dysfunction in the presence of increased adiposity.31 The prenatal T-treated monkeys19,45 and rats,68 similar to women with PCOS,31 manifested increased visceral adiposity. Obesity induced by overfeeding also exaggerated reproductive defects in the sheep model of PCOS culminating in anovulation,100 suggestive of metabolic amplification of disruptions. Increasing prevalence of childhood obesity101 might therefore provide a metabolic platform for uncovering or amplifying prenatally experienced developmental insults. Given the high prevalence of obesity and its comorbidities, diabetes, cardiovascular diseases, and metabolic syndrome, in the United States, more studies with various animal models are required to substantiate the detrimental effects of overfeeding/excess weight gain in the development of the PCOS phenotype.
ROLE OF HYPERINSULINEMIA IN THE DEVELOPMENT AND AMPLIFICATION OF THE PCOS PHENOTYPE
From a metabolic perspective, obesity and prenatal T excess both cause insulin resistance and compensatory hyperinsulinemia. A higher percentage of women with PCOS manifest insulin resistance and are at risk for developing type 2 diabetes.9 Lifestyle changes and weight loss that improve insulin sensitivity were found to improve ovulatory function in these women.102 A recent Cochrane review of 31 clinical trials found that insulin sensitizers enhance ovulation rates and improve menstrual patterns with success rates differing between studies,103 possibly due to the heterogeneity of the PCOS population being studied and the timing of initiation of treatment relative to when the pathology was established.
Studies conducted in prenatal T-treated sheep and Rhesus monkeys also point to beneficial effects of insulin sensitizer treatment.104,105 Treatment with rosiglitazone, an insulin sensitizer, begun during postpubertal life prevented further deterioration of reproductive function in prenatal T-treated sheep (cycles monitored over a 2-year period).104 Studies performed with an older cohort of prenatal T-treated monkeys also found that treatment with pioglitazone, another insulin sensitizer, improved cyclic function.105 In sheep, the beneficial effects of insulin sensitizer in improving reproductive function were evident at two levels: prevention from further deterioration of the reproductive axis and a reduction in the number of abnormally long cycles.104 In the older monkeys the beneficial effects of insulin sensitizer were evident as normalization of menstrual cycle length.105 Similar studies have not been undertaken with rat and mouse models.
Although improvement in reproductive function is clearly evident in prenatal T-treated sheep and monkey models,104,105 as is the case with PCOS women,103 the success rate has not been 100%, possibly because treatment was initiated after the pathology was established. In prenatal T-treated sheep, reproductive dysfunctions are evident postpubertally,51,52 whereas defects in insulin sensitivity are evident much before during neonatal life.63,64 Early insulin sensitizer treatment beginning when insulin sensitivity defects are manifested may prove to be more effective in achieving better success rates.
GENETIC VERSUS ENVIRONMENTAL INTERACTION IN PROGRAMMING THE PCOS PHENOTYPE
Clarification of underlying mechanisms by which developmental reprogramming of physiological function occurs is essential for targeting new strategies toward prevention. Both genetic and environmental factors have been implicated in the etiology of the PCOS phenotype.106 Familial clustering in first-degree relatives of PCOS subjects107 and higher prevalence of PCOS symptoms in monozygotic compared with dizygotic twins108 provide support for a genetic contribution. However, to date, no gene has been implicated in the development of a PCOS phenotype. But heterogeneity of phenotypic features in different PCOS families and even within the same family points to the importance of the environmental contribution. It is becoming increasingly apparent that environmental insults during development induce persistent changes in the epigenome leading to altered gene expression and increased risk of adult diseases.109 Interestingly, an epigenetic change, manifested as nonrandom X chromosome inactivation, has been reported in women with PCOS.110
Although maternal and environmental factors during development have been found to induce epigenetic alterations and reprogram the developmental ontogeny of the offspring, the interplay of epigenetics with genetics is likely the key determining factor in an individual’s susceptibility to pathology. The lower than 50% prevalence of inheritance in first-degree relatives does provide support for such gene by environment interactions.107 An understanding of the epigenetic mechanisms involved in models of PCOS would likely provide novel avenues for the prevention and treatment of PCOS and help reduce transgenerational susceptibility for acquiring the disrupted phenotype.
STRENGTHS OF DIFFERENT ANIMAL MODELS
All PCOS animal models discussed offer differing strengths. The highly compressed developmental time scale of developing rats and mice allows studies of transgenerational transfer of PCOS traits within a reasonable time frame. The transgenic approaches available in murine models are beneficial in pinpointing the site-specific role of suspected mediators. For instance, the green fluorescent protein–GnRH mouse has been a valuable resource in elucidating the direct effects of androgen and estrogen at the level of the GnRH neuron.89 The strengths of the sheep model of PCOS are that they are amenable to a wide variety of procedural manipulations including performance of detailed/repetitive hormonal profiling, noninvasive sequential monitoring of ovarian follicular dynamics via ultrasound, multiple neurotransmitter measures in the same animal (due to the large size of the brain), studies in natural settings with behavioral interactions intact, and its cost effectiveness. The subhuman primates are closer to humans from an evolutionary perspective and share similar placentation and hence would be an optimal model. However, the number of years taken to achieve reproductive maturity and the enormity of resources required restrict feasibility of studies spanning from the time of developmental insults to adult pathological outcomes in the same animal within a reasonable time frame.
While translating the findings from any of these animals to humans, it is important to interpret the findings relative to the developmental trajectory of the organ system being studied as to whether differentiation gets completed prenatally or postnatally and the similarity of regulatory mechanisms. For instance, sheep and subhuman primates complete their ovarian differentiation in utero, but it occurs ex utero in rats and mice (Table 2). Therefore, the ovarian reprogramming that occurs in utero in sheep, primates, and humans would be subject to influence from changes in both fetal and maternal milieus, which is not the case in the postnatal rodent models. Similarly, in understanding neuroendocrine disruptions, it should be recognized that progesterone blocks generation of the LH surge in sheep, monkeys, and humans, but it is a facilitator in rodents.111–113 In addressing studies focusing on the maternal-fetal interface, it should be taken into consideration that the placentation in sheep, rats, and mice differs from humans.
CLINICAL TRANSLATION AND PUBLIC HEATH RELEVANCE
The PCOS phenotype is associated with conditions such as classical 21-hydroxylase deficiency in which the fetus has been exposed to high amounts of sex steroids before birth,18 suggesting that androgen excess early in life may lead to manifestation of this phenotype in adulthood. Levels of T in 40% of human female fetuses are elevated to levels similar to that of male fetuses at 19 to 25 weeks of gestation.114 Interestingly, the gestational T-treated sheep female fetuses that manifest the PCOS phenotype are exposed to T at levels found in the male fetuses.97
Considering the experimental constraints in humans, animal models that manifest the PCOS phenotype are valuable resources for delineating the mechanisms contributing to the reproductive/metabolic disruptions seen in women with PCOS. More importantly, these models can serve as a testing ground for developing effective early prevention/treatment strategies to prevent/overcome reproductive/metabolic dysfunctions. The findings from these animal models may also have public health implications in the context of environmental exposures to steroid mimics. Human fetuses are subjected to abnormal steroidal programming via endocrine-disrupting chemicals in the environment such as bisphenol A and phthalates with estrogenic/antiandrogenic properties115 as well as during disease states.116
FUTURE DIRECTIONS
Future studies with animal models should capitalize on the identified strengths of various models to discern the early causal signals involved in the development and progression of PCOS. Studies should target time points during development that are comparable to time points of organ differentiation in humans and strive to discover the relative fetal and maternal contributions in programming the human PCOS phenotype. Because of the potential for such PCOS traits to be carried forward to subsequent generations, transgenerational studies that focus on causal mechanisms are very much needed to help segregate genetic/epigenetic interactions and differences in individual susceptibility. If prenatal steroid excess is indeed a contributing factor in the development of human PCOS syndrome, it is conceivable that differences in timing of developmental exposure to androgens/estrogens may account for the different PCOS phenotypes with subsequent lifestyle patterns playing a role in revealing or amplifying the severity of phenotype programmed early during development.
In parallel, clinical studies should target early gestational stages and gain information on developmental changes at the maternal level and when possible capitalize on amniocentesis and postmortem samples to assess fetal contribution. Term cord blood samples may not be optimal because much of the programming on the ovary and brain may have occurred early during gestation. These human studies should be expanded to analyze the relative contribution of both androgens and estrogens because T has the ability to be aromatized to estrogen and mediate estrogenic reprogramming. More importantly, studies should capitalize on the strengths of these animal models to develop prevention and treatment strategies aimed toward improving fertility and metabolic outcomes at the level of the individual.
Footnotes
The Developmental Origins of Health and Disease: Today’s Perspectives and Tomorrow’s Challenges; Guest Editor, Daniel B. Hardy, Ph.D.
References
- 1.Boivin J, Bunting L, Collins JA, Nygren KG. International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Hum Reprod. 2007;22(6):1506–1512. doi: 10.1093/humrep/dem046. [DOI] [PubMed] [Google Scholar]
- 2.Homburg R. Polycystic ovary syndrome: induction of ovulation. Baillieres Clin Endocrinol Metab. 1996;10(2):281–292. doi: 10.1016/s0950-351x(96)80127-3. [DOI] [PubMed] [Google Scholar]
- 3.Messinis IE. Ovulation induction: a mini review. Hum Reprod. 2005;20(10):2688–2697. doi: 10.1093/humrep/dei128. [DOI] [PubMed] [Google Scholar]
- 4.Hamilton-Fairley D, Franks S. Common problems in induction of ovulation. Baillieres Clin Obstet Gynaecol. 1990;4(3):609–625. doi: 10.1016/s0950-3552(05)80313-x. [DOI] [PubMed] [Google Scholar]
- 5.Tummon I, Gavrilova-Jordan L, Allemand MC, Session D. Polycystic ovaries and ovarian hyperstimulation syndrome: a systematic review. Acta Obstet Gynecol Scand. 2005;84(7):611–616. doi: 10.1111/j.0001-6349.2005.00788.x. [DOI] [PubMed] [Google Scholar]
- 6.Boomsma CM, Fauser BC, Macklon NS. Pregnancy complications in women with polycystic ovary syndrome. Semin Reprod Med. 2008;26(1):72–84. doi: 10.1055/s-2007-992927. [DOI] [PubMed] [Google Scholar]
- 7.Janssen OE, Hahn S, Tan S, Benson S, Elsenbruch S. Mood and sexual function in polycystic ovary syndrome. Semin Reprod Med. 2008;26(1):45–52. doi: 10.1055/s-2007-992924. [DOI] [PubMed] [Google Scholar]
- 8.Himelein MJ, Thatcher SS. Polycystic ovary syndrome and mental health: a review. Obstet Gynecol Surv. 2006;61(11):723–732. doi: 10.1097/01.ogx.0000243772.33357.84. [DOI] [PubMed] [Google Scholar]
- 9.Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18(6):774–800. doi: 10.1210/edrv.18.6.0318. [DOI] [PubMed] [Google Scholar]
- 10.Lord JM, Flight IH, Norman RJ. Insulin-sensitising drugs (metformin troglitazone rosiglitazone pioglitazone D-chiro-inositol) for polycystic ovary syndrome. Cochrane Database Syst Rev. 2003;3(3):CD003053. doi: 10.1002/14651858.CD003053. [DOI] [PubMed] [Google Scholar]
- 11.Essah PA, Nestler JE. The metabolic syndrome in polycystic ovary syndrome. J Endocrinol Invest. 2006;29(3):270–280. doi: 10.1007/BF03345554. [DOI] [PubMed] [Google Scholar]
- 12.Barber TM, McCarthy MI, Wass JA, Franks S. Obesity and polycystic ovary syndrome. Clin Endocrinol (Oxf) 2006;65(2):137–145. doi: 10.1111/j.1365-2265.2006.02587.x. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman LK, Ehrmann DA. Cardiometabolic features of polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab. 2008;4(4):215–222. doi: 10.1038/ncpendmet0755. [DOI] [PubMed] [Google Scholar]
- 14.Navaratnarajah R, Pillay OC, Hardiman P. Polycystic ovary syndrome and endometrial cancer. Semin Reprod Med. 2008;26(1):62–71. doi: 10.1055/s-2007-992926. [DOI] [PubMed] [Google Scholar]
- 15.Barker DJP. Programming the baby. In: Gillman MW, Barker DJP, editors. Mothers, Babies, and Disease in Later Life. London, United Kingdom: BMJ Publishing Group; 1994. pp. 14–36. [Google Scholar]
- 16.Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord. 2007;8(2):127–141. doi: 10.1007/s11154-007-9046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies MJ, Norman RJ. Programming and reproductive functioning. Trends Endocrinol Metab. 2002;13(9):386–392. doi: 10.1016/s1043-2760(02)00691-4. [DOI] [PubMed] [Google Scholar]
- 18.Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuro-endocrine function in women. J Clin Endocrinol Metab. 1994;79(5):1328–1333. doi: 10.1210/jcem.79.5.7962325. [DOI] [PubMed] [Google Scholar]
- 19.Abbott DH, Dumesic DA, Levine JE, Dunaif A, Padmanabhan V. Animal models and fetal programming of PCOS. In: Azziz R, Nestler JE, Dewailly D, editors. Contemporary Endocrinology: Androgen Excess Disorders in Women: Polycystic Ovary Syndrome and Other Disorders. Totowa, NJ: Humana Press; 2006. pp. 259–272. [Google Scholar]
- 20.Rosenfield RL. Current concepts of polycystic ovary syndrome. Baillieres Clin Obstet Gynaecol. 1997;11(2):307–333. doi: 10.1016/s0950-3552(97)80039-9. [DOI] [PubMed] [Google Scholar]
- 21.Rebar R, Judd HL, Yen SS, Rakoff J, Vandenberg G, Naftolin F. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest. 1976;57(5):1320–1329. doi: 10.1172/JCI108400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franks S, Roberts R, Hardy K. Gonadotrophin regimens and oocyte quality in women with polycystic ovaries. Reprod Biomed Online. 2003;6(2):181–184. doi: 10.1016/s1472-6483(10)61708-7. [DOI] [PubMed] [Google Scholar]
- 23.Webber LJ, Stubbs S, Stark J, et al. Formation and early development of follicles in the polycystic ovary. Lancet. 2003;362(9389):1017–1021. doi: 10.1016/s0140-6736(03)14410-8. [DOI] [PubMed] [Google Scholar]
- 24.Foong SC, Abbott DH, Zschunke MA, Lesnick TG, Phy JL, Dumesic DA. Follicle luteinization in hyperandrogenic follicles of polycystic ovary syndrome patients undergoing gonadotropin therapy for in vitro fertilization. J Clin Endocrinol Metab. 2006;91(6):2327–2333. doi: 10.1210/jc.2005-2142. [DOI] [PubMed] [Google Scholar]
- 25.Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 1998;83(2):582–590. doi: 10.1210/jcem.83.2.4604. [DOI] [PubMed] [Google Scholar]
- 26.Patel K, Coffler MS, Dahan MH, Malcom PJ, Deutsch R, Chang RJ. Relationship of GnRH-stimulated LH release to episodic LH secretion and baseline endocrine-metabolic measures in women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 2004;60(1):67–74. doi: 10.1111/j.1365-2265.2004.01945.x. [DOI] [PubMed] [Google Scholar]
- 27.Ehrmann DA. β-cell dysfunction, glucose intolerance, and diabetes in the polycystic syndrome. In: Azziz R, Nestler JE, Dewailly D, editors. Androgen Excess Disorders in Women. Totowa, NJ: Humana Press; 2007. pp. 319–324. [Google Scholar]
- 28.Ibáñez L, Potau N, Francois I, de Zegher F. Precocious pubarche, hyperinsulinism, and ovarian hyperandrogenism in girls: relation to reduced fetal growth. J Clin Endocrinol Metab. 1998;83(10):3558–3562. doi: 10.1210/jcem.83.10.5205. [DOI] [PubMed] [Google Scholar]
- 29.de Zegher F, Ibáñez L. Prenatal growth restraint followed by catch-up of weight: a hyperinsulinemic pathway to polycystic ovary syndrome. Fertil Steril. 2006;86(Suppl 1):S4–S5. doi: 10.1016/j.fertnstert.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Diamanti-Kandarakis E. Role of obesity and adiposity in polycystic ovary syndrome. Int J Obes (Lond) 2007;31(Suppl 2):S8–S13. doi: 10.1038/sj.ijo.0803730. discussion S31–S32. [DOI] [PubMed] [Google Scholar]
- 31.Pasquali R, Gambineri A. The endocrine impact of obesity and body habitus in the polycystic syndrome. In: Azziz R, Nestler JE, Dewailly D, editors. Androgen Excess Disorders in Women. Totowa NJ: Humana Press; 2007. pp. 283–291. [Google Scholar]
- 32.Essah PA, Nestler JE. The metabolic syndrome in polycystic ovary syndrome. J Endocrinol Invest. 2006;29(3):270–280. doi: 10.1007/BF03345554. [DOI] [PubMed] [Google Scholar]
- 33.Eisner JR, Barnett MA, Dumesic DA, Abbott DH. Ovarian hyperandrogenism in adult female rhesus monkeys exposed to prenatal androgen excess. Fertil Steril. 2002;77(1):167–172. doi: 10.1016/s0015-0282(01)02947-8. [DOI] [PubMed] [Google Scholar]
- 34.Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update. 2005;11(4):357–374. doi: 10.1093/humupd/dmi013. [DOI] [PubMed] [Google Scholar]
- 35.Dumesic DA, Abbott DH, Eisner JR, Goy RW. Prenatal exposure of female rhesus monkeys to testosterone propionate increases serum luteinizing hormone levels in adulthood. Fertil Steril. 1997;67(1):155–163. doi: 10.1016/s0015-0282(97)81873-0. [DOI] [PubMed] [Google Scholar]
- 36.Goy RW, Robinson JA. Prenatal exposure of rhesus monkeys to patent androgens: morphological, behavioral, and physiological consequences. Banbury Rep. 1982;11:355–378. [Google Scholar]
- 37.Abbott DH, Dumesic DA, Eisner JR, Kemnitz JW, Goy RW. The prenatally androgenized female rhesus monkey as a model for polycystic ovarian syndrome. In: Azziz R, Nestler JE, Dewailly D, editors. Androgen Excess Disorders in Women. Philadelphia, PA: Lippincott-Raven Press; 1997. pp. 369–382. [Google Scholar]
- 38.Abbott DH, Eisner JR, Colman RJ, Kemnitz J, Dumesic DA. Prenatal androgen excess programs for PCOS in female rhesus monkeys. In: Chang RJ, Dunaif A, Hiendel J, editors. Polycystic Ovary Syndrome. New York, NY: Marcel Dekker; 2002. pp. 119–133. [Google Scholar]
- 39.Dumesic DA, Schramm RD, Peterson E, Paprocki AM, Zhou R, Abbott DH. Impaired developmental competence of oocytes in adult prenatally androgenized female rhesus monkeys undergoing gonadotropin stimulation for in vitro fertilization. J Clin Endocrinol Metab. 2002;87(3):1111–1119. doi: 10.1210/jcem.87.3.8287. [DOI] [PubMed] [Google Scholar]
- 40.Steiner RA, Clifton DK, Spies HG, Resko JA. Sexual differentiation and feedback control of luteinizing hormone secretion in the rhesus monkey. Biol Reprod. 1976;15(2):206–212. doi: 10.1095/biolreprod15.2.206. [DOI] [PubMed] [Google Scholar]
- 41.Levine JE, Terasawa E, Hoffman SM, Dobbert MJW, Foecking EM, Abbott DH. Luteinizing hormone (LH) hypersecretion and diminished LH responses to RU486 in a non human primate model for polycystic ovary syndrome (PCOS). Paper presented at: Annual Meeting of the Endocrine Society; June 4, 2005; San Diego, CA. [Google Scholar]
- 42.Eisner JR, Dumesic DA, Kemnitz JW, Abbott DH. Timing of prenatal androgen excess determines differential impairment in insulin secretion and action in adult female rhesus monkeys. J Clin Endocrinol Metab. 2000;85(3):1206–1210. doi: 10.1210/jcem.85.3.6453. [DOI] [PubMed] [Google Scholar]
- 43.Abbott DH, Bruns CM, Barnett DK, et al. Metabolic and reproductive consequences of prenatal testosterone exposure. Paper presented at: Annual Meeting of the Endocrine Society; June 19, 2003; Philadelphia, PA. [Google Scholar]
- 44.Abbott DH, Barnett DK, Levine JE, et al. Endocrine antecedents of polycystic ovary syndrome in fetal and infant prenatally androgenized female rhesus monkeys. Biol Reprod. 2008;79(1):154–163. doi: 10.1095/biolreprod.108.067702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eisner JR, Dumesic DA, Kemnitz JW, Colman RJ, Abbott DH. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes Res. 2003;11(2):279–286. doi: 10.1038/oby.2003.42. [DOI] [PubMed] [Google Scholar]
- 46.Abbott DH, Eisner JR, Goodfriend TL, et al. Leptin and total free fatty acids are elevated in the circulation of prenatally androgenized female rhesus monkeys. Paper presented at: Annual Meeting of the Endocrine Society; June 18, 2002; San Francisco, CA. [Google Scholar]
- 47.Ortega HH, Salvetti NR, Padmanabhan V. Developmental programming: prenatal androgen excess disrupts ovarian steroid receptor balance. Reproduction. 2009;137(5):865–877. doi: 10.1530/REP-08-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manikkam M, Thompson RC, Herkimer C, et al. Developmental programming: impact of prenatal testosterone excess on pre- and postnatal gonadotropin regulation in sheep. Biol Reprod. 2008;78(4):648–660. doi: 10.1095/biolreprod.107.063347. [DOI] [PubMed] [Google Scholar]
- 49.Sharma TP, Herkimer C, West C, et al. Fetal programming: prenatal androgen disrupts positive feedback actions of estradiol but does not affect timing of puberty in female sheep. Biol Reprod. 2002;66(4):924–933. doi: 10.1095/biolreprod66.4.924. [DOI] [PubMed] [Google Scholar]
- 50.Sarma HN, Manikkam M, Herkimer C, Dell’Orco J, Foster DL, Padmanabhan V. Fetal programming: excess prenatal testosterone reduces postnatal LH, but not FSH responsiveness to estradiol negative feedback in the female. Endocrinology. 2005;146:4281–4291. doi: 10.1210/en.2005-0322. [DOI] [PubMed] [Google Scholar]
- 51.Birch RA, Padmanabhan V, Foster DL, Unsworth WP, Robinson JE. Prenatal programming of reproductive neuroendocrine function: fetal androgen exposure produces progressive disruption of reproductive cycles in sheep. Endocrinology. 2003;144(4):1426–1434. doi: 10.1210/en.2002-220965. [DOI] [PubMed] [Google Scholar]
- 52.Manikkam M, Steckler TL, Welch KB, Inskeep EK, Padmanabhan V. Fetal programming: prenatal testosterone treatment leads to follicular persistence/luteal defects; partial restoration of ovarian function by cyclic progesterone treatment. Endocrinology. 2006;147(4):1997–2007. doi: 10.1210/en.2005-1338. [DOI] [PubMed] [Google Scholar]
- 53.Steckler T, Manikkam M, Inskeep EK, Padmanabhan V. Developmental programming: follicular persistence in prenatal testosterone-treated sheep is not programmed by androgenic actions of testosterone. Endocrinology. 2007;148(7):3532–3540. doi: 10.1210/en.2007-0339. [DOI] [PubMed] [Google Scholar]
- 54.Manikkam M, Crespi EJ, Doop DD, et al. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology. 2004;145(2):790–798. doi: 10.1210/en.2003-0478. [DOI] [PubMed] [Google Scholar]
- 55.Steckler TL, Roberts EK, Doop DD, Lee TM, Padmanabhan V. Developmental programming in sheep: administration of testosterone during 60–90 days of pregnancy reduces breeding success and pregnancy outcome. Theriogenology. 2007;67(3):459–467. doi: 10.1016/j.theriogenology.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 56.West C, Foster DL, Evans NP, Robinson J, Padmanabhan V. Intra-follicular activin availability is altered in prenatally-androgenized lambs. Mol Cell Endocrinol. 2001;185(1–2):51–59. doi: 10.1016/s0303-7207(01)00632-3. [DOI] [PubMed] [Google Scholar]
- 57.Smith P, Steckler TL, Veiga-Lopez A, Padmanabhan V. Developmental programming: differential effects of prenatal testosterone and dihydrotestosterone on follicular recruitment, depletion of follicular reserve, and ovarian morphology in sheep. Biol Reprod. 2009;80(4):726–736. doi: 10.1095/biolreprod.108.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forsdike RA, Hardy K, Bull L, et al. Disordered follicle development in ovaries of prenatally androgenized ewes. J Endocrinol. 2007;192(2):421–428. doi: 10.1677/joe.1.07097. [DOI] [PubMed] [Google Scholar]
- 59.Steckler T, Wang J, Bartol FF, Roy SK, Padmanabhan V. Fetal programming: prenatal testosterone treatment causes intrauterine growth retardation, reduces ovarian reserve and increases ovarian follicular recruitment. Endocrinology. 2005;146(7):3185–3193. doi: 10.1210/en.2004-1444. [DOI] [PubMed] [Google Scholar]
- 60.Unsworth WP, Taylor JA, Robinson JE. Prenatal programming of reproductive neuroendocrine function: the effect of prenatal androgens on the development of estrogen positive feedback and ovarian cycles in the ewe. Biol Reprod. 2005;72(3):619–627. doi: 10.1095/biolreprod.104.035691. [DOI] [PubMed] [Google Scholar]
- 61.Robinson JE, Forsdike RA, Taylor JA. In utero exposure of female lambs to testosterone reduces the sensitivity of the gonadotropin-releasing hormone neuronal network to inhibition by progesterone. Endocrinology. 1999;140(12):5797–5805. doi: 10.1210/endo.140.12.7205. [DOI] [PubMed] [Google Scholar]
- 62.Veiga-Lopez A, Ye W, Phillips DJ, Herkimer C, Knight PG, Padmanabhan V. Developmental programming: deficits in reproductive hormone dynamics and ovulatory outcomes in prenatal, testosterone-treated sheep. Biol Reprod. 2008;78(4):636–647. doi: 10.1095/biolreprod.107.065904. [DOI] [PubMed] [Google Scholar]
- 63.Recabarren SE, Padmanabhan V, Codner E, et al. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. Am J Physiol Endocrinol Metab. 2005;289(5):E801–E806. doi: 10.1152/ajpendo.00107.2005. [DOI] [PubMed] [Google Scholar]
- 64.Padmanabhan V, Veiga-Lopez A, Abbott DH, Recabarren SE, Herkimer C. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology. 2010;151(2):595–605. doi: 10.1210/en.2009-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.King AJ, Olivier NB, Mohankumar PS, Lee JS, Padmanabhan V, Fink GD. Hypertension caused by prenatal testosterone excess in female sheep. Am J Physiol Endo-crinol Metab. 2007;292(6):E1837–E1841. doi: 10.1152/ajpendo.00668.2006. [DOI] [PubMed] [Google Scholar]
- 66.Foecking EM, Szabo M, Schwartz NB, Levine JE. Neuroendocrine consequences of prenatal androgen exposure in the female rat: absence of luteinizing hormone surges, suppression of progesterone receptor gene expression, and acceleration of the gonadotropin-releasing hormone pulse generator. Biol Reprod. 2005;72(6):1475–1483. doi: 10.1095/biolreprod.105.039800. [DOI] [PubMed] [Google Scholar]
- 67.Wu XY, Li ZL, Wu CY, et al. Endocrine traits of polycystic ovary syndrome in prenatally androgenized female Sprague-Dawley rats. Endocr J. 2010;57(3):201–209. doi: 10.1507/endocrj.k09e-205. [DOI] [PubMed] [Google Scholar]
- 68.Demissie M, Lazic M, Foecking EM, Aird F, Dunaif A, Levine JE. Transient prenatal androgen exposure produces metabolic syndrome in adult female rats. Am J Physiol Endocrinol Metab. 2008;295(2):E262–E268. doi: 10.1152/ajpendo.90208.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Slob AK, den Hamer R, Woutersen PJ, van der Werff ten Bosch JJ. Prenatal testosterone propionate and postnatal ovarian activity in the rat. Acta Endocrinol (Copenh) 1983;103(3):420–427. doi: 10.1530/acta.0.1030420. [DOI] [PubMed] [Google Scholar]
- 70.Alexanderson C, Eriksson E, Stener-Victorin E, et al. Postnatal testosterone exposure results in insulin resistance, enlarged mesenteric adipocytes, and an atherogenic lipid profile in adult female rats: comparisons with estradiol and dihydrotestosterone. Endocrinology. 2007;148(11):5369–5376. doi: 10.1210/en.2007-0305. [DOI] [PubMed] [Google Scholar]
- 71.Nilsson C, Niklasson M, Eriksson E, Björntorp P, Holmäng A. Imprinting of female offspring with testosterone results in insulin resistance and changes in body fat distribution at adult age in rats. J Clin Invest. 1998;101(1):74–78. doi: 10.1172/JCI1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cheng G, Coolen LM, Padmanabhan V, Goodman RL, Lehman MN. The kisspeptin/neurokinin B/dynorphin (KNDy) cell population of the arcuate nucleus: sex differences and effects of prenatal testosterone in sheep. Endocrinology. 2010;151(1):301–311. doi: 10.1210/en.2009-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Padmanabhan V, Veiga-Lopez A, Abbott DH, Dumesic DA. Developmental programming of ovarian dysfunction. In: Gonzalez-Bulnes A, editor. Novel Concepts in Ovarian Endocrinology. Kerala, India: Research Signpost Editors; 2007. pp. 328–352. [Google Scholar]
- 74.Kennedy TG, Gillio-Meina C, Phang SH. Prostaglandins and the initiation of blastocyst implantation and decidualization. Reproduction. 2007;134(5):635–643. doi: 10.1530/REP-07-0328. [DOI] [PubMed] [Google Scholar]
- 75.Pelliniemi LJ, Fröjdman K. Structural and regulatory macromolecules in sex differentiation of gonads. J Exp Zool. 2001;290(5):523–528. doi: 10.1002/jez.1096. [DOI] [PubMed] [Google Scholar]
- 76.Hemsworth BN, Jackson H. Effect of busulphan on the developing ovary in the rat. J Reprod Fertil. 1963;6:229–233. doi: 10.1530/jrf.0.0060229. [DOI] [PubMed] [Google Scholar]
- 77.Rajah R, Glaser EM, Hirshfield AN. The changing architecture of the neonatal rat ovary during histogenesis. Dev Dyn. 1992;194(3):177–192. doi: 10.1002/aja.1001940303. [DOI] [PubMed] [Google Scholar]
- 78.Malamed S, Gibney JA, Ojeda SR. Ovarian innervation develops before initiation of folliculogenesis in the rat. Cell Tissue Res. 1992;270(1):87–93. doi: 10.1007/BF00381883. [DOI] [PubMed] [Google Scholar]
- 79.Ortega HH, Rey F, Velazquez MM, Padmanabhan V. Developmental programming: effect of prenatal steroid excess on intraovarian components of insulin signaling pathway and related proteins in sheep. Biol Reprod. 2010;82(6):1065–1075. doi: 10.1095/biolreprod.109.082719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Welt CK, Taylor AE, Fox J, Messerlian GM, Adams JM, Schneyer AL. Follicular arrest in polycystic ovary syndrome is associated with deficient inhibin A and B biosynthesis. J Clin Endocrinol Metab. 2005;90(10):5582–5587. doi: 10.1210/jc.2005-0695. [DOI] [PubMed] [Google Scholar]
- 81.Fujiwara T, Sidis Y, Welt C, et al. Dynamics of inhibin subunit and follistatin mRNA during development of normal and polycystic ovary syndrome follicles. J Clin Endocrinol Metab. 2001;86(9):4206–4215. doi: 10.1210/jcem.86.9.7798. [DOI] [PubMed] [Google Scholar]
- 82.Brown E, Lee T, Padmanabhan V, Lehman MN, Coolen LM. The ventral tegmental area dopamine system is masculinized by prenatal testosterone via androgen receptor action in female sheep. Paper presented at: Annual Meeting of Society for Behavioral Neuroendocrinology; July 18, 2010; Toronto, ON, Canada. [Google Scholar]
- 83.Fauser BC, Diedrich K, Devroey P Evian Annual Reproduction Workshop Group 2007. Predictors of ovarian response: progress towards individualized treatment in ovulation induction and ovarian stimulation. Hum Reprod Update. 2008;14(1):1–14. doi: 10.1093/humupd/dmm034. [DOI] [PubMed] [Google Scholar]
- 84.Zawadki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens JR, Haseltine FP, Merriam GR, editors. Polycystic Ovary Syndrome. Boston, MA: Blackwell Scientific; 1992. pp. 377–384. [Google Scholar]
- 85.Rotterdam ESHRE/ASRM-Sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS) Hum Reprod. 2004;19(1):41–47. doi: 10.1093/humrep/deh098. [DOI] [PubMed] [Google Scholar]
- 86.Azziz R, Carmina E, Dewailly D, et al. Androgen Excess Society. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 2006;91(11):4237–4245. doi: 10.1210/jc.2006-0178. [DOI] [PubMed] [Google Scholar]
- 87.Wild RA, Carmina E, Diamanti-Kandarakis E, et al. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J Clin Endocrinol Metab. 2010;95(5):2038–2049. doi: 10.1210/jc.2009-2724. [DOI] [PubMed] [Google Scholar]
- 88.Veiga-Lopez A, Astapova OI, Aizenberg EF, Lee JS, Padmanabhan V. Developmental programming: contribution of prenatal androgen and estrogen to estradiol feedback systems and periovulatory hormonal dynamics in sheep. Biol Reprod. 2009;80(4):718–725. doi: 10.1095/biolreprod.108.074781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci U S A. 2004;101(18):7129–7134. doi: 10.1073/pnas.0308058101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roland AV, Nunemaker CS, Keller SR, Moenter SM. Prenatal androgen exposure programs metabolic dysfunction in female mice. J Endocrinol. 2010;207(2):213–223. doi: 10.1677/JOE-10-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mannerås L, Cajander S, Holmäng A, et al. A new rat model exhibiting both ovarian and metabolic characteristics of polycystic ovary syndrome. Endocrinology. 2007;148(8):3781–3791. doi: 10.1210/en.2007-0168. [DOI] [PubMed] [Google Scholar]
- 92.Kafali H, Iriadam M, Ozardali I, Demir N. Letrozole-induced polycystic ovaries in the rat: a new model for cystic ovarian disease. Arch Med Res. 2004;35(2):103– 108. doi: 10.1016/j.arcmed.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 93.Baravalle C, Salvetti NR, Mira GA, Pezzone N, Ortega HH. Microscopic characterization of follicular structures in letrozole-induced polycystic ovarian syndrome in the rat. Arch Med Res. 2006;37(7):830–839. doi: 10.1016/j.arcmed.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 94.Rosa-E-Silva A, Guimaraes MA, Padmanabhan V, Lara HE. Prepubertal administration of estradiol valerate disrupts cyclicity and leads to cystic ovarian morphology during adult life in the rat: role of sympathetic innervation. Endocrinology. 2003;144(10):4289–4297. doi: 10.1210/en.2003-0146. [DOI] [PubMed] [Google Scholar]
- 95.Handa RJ, Pak TR, Kudwa AE, Lund TD, Hinds L. An alternate pathway for androgen regulation of brain function: activation of estrogen receptor beta by the metabolite of dihydrotestosterone, 5alpha-androstane-3beta,17beta-diol. Horm Behav. 2008;53(5):741–752. doi: 10.1016/j.yhbeh.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sotomayor-Zárate R, Dorfman M, Paredes A, Lara HE. Neonatal exposure to estradiol valerate programs ovarian sympathetic innervation and follicular development in the adult rat. Biol Reprod. 2008;78(4):673–680. doi: 10.1095/biolreprod.107.063974. [DOI] [PubMed] [Google Scholar]
- 97.Veiga-Lopez A, Steckler TL, Abbott DH, et al. Developmental programming: impact of excess prenatal testosterone on intra-uterine fetal endocrine milieu and growth in sheep. Biol Reprod. 2011;84(1):87–96. doi: 10.1095/biolreprod.110.086686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wood RI, Foster DL. Sexual differentiation of reproductive neuroendocrine function in sheep. Rev Reprod. 1998;3(2):130–140. doi: 10.1530/ror.0.0030130. [DOI] [PubMed] [Google Scholar]
- 99.Foster DL, Padmanabhan V, Wood RI, Robinson JE. Sexual differentiation of the neuroendocrine control of gonadotrophin secretion: concepts derived from sheep models. Reprod Suppl. 2002;59:83–99. [PubMed] [Google Scholar]
- 100.Steckler TL, Herkimer C, Dumesic DA, Padmanabhan V. Developmental programming: excess weight gain amplifies the effects of prenatal testosterone excess on reproductive cyclicity—implication for polycystic ovary syndrome. Endo-crinology. 2009;150(3):1456–1465. doi: 10.1210/en.2008-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ludwig DS. Childhood obesity—the shape of things to come. N Engl J Med. 2007;357(23):2325–2327. doi: 10.1056/NEJMp0706538. [DOI] [PubMed] [Google Scholar]
- 102.Moran LJ, Brinkworth GD, Norman RJ. Dietary therapy in polycystic ovary syndrome. Semin Reprod Med. 2008;26(1):85–92. doi: 10.1055/s-2007-992928. [DOI] [PubMed] [Google Scholar]
- 103.Tang T, Lord JM, Norman RJ, Yasmin E, Balen AH. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst Rev. 2010;(1):CD003053. doi: 10.1002/14651858.CD003053.pub4. [DOI] [PubMed] [Google Scholar]
- 104.Veiga-Lopez A, Lee JS, Padmanabhan V. Developmental programming: insulin sensitizer treatment improves reproductive function in prenatal testosterone-treated female sheep. Endocrinology. 2010;151(8):4007–4017. doi: 10.1210/en.2010-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou R, Bruns CM, Bird IM, et al. Pioglitazone improves insulin action and normalizes menstrual cycles in a majority of prenatally androgenized female rhesus monkeys. Reprod Toxicol. 2007;23(3):438–448. doi: 10.1016/j.reprotox.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dasgupta S, Reddy BM. Present status of understanding on the genetic etiology of polycystic ovary syndrome. J Postgrad Med. 2008;54(2):115–125. doi: 10.4103/0022-3859.40778. [DOI] [PubMed] [Google Scholar]
- 107.Yildiz BO, Yarali H, Oguz H, Bayraktar M. Glucose intolerance, insulin resistance, and hyperandrogenemia in first degree relatives of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88(5):2031–2036. doi: 10.1210/jc.2002-021499. [DOI] [PubMed] [Google Scholar]
- 108.Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab. 2006;91(6):2100–2104. doi: 10.1210/jc.2005-1494. [DOI] [PubMed] [Google Scholar]
- 109.Waterland RA. Is epigenetics an important link between early life events and adult disease? Horm Res. 2009;71(Suppl 1):13–16. doi: 10.1159/000178030. [DOI] [PubMed] [Google Scholar]
- 110.Hickey TE, Legro RS, Norman RJ. Epigenetic modification of the X chromosome influences susceptibility to polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(7):2789–2791. doi: 10.1210/jc.2006-0069. [DOI] [PubMed] [Google Scholar]
- 111.Gemzell-Danielsson K, Marions L. Mechanisms of action of mifepristone and levonorgestrel when used for emergency contraception. Hum Reprod Update. 2004;10(4):341–348. doi: 10.1093/humupd/dmh027. [DOI] [PubMed] [Google Scholar]
- 112.Kasa-Vubu JZ, Dahl GE, Evans NP, et al. Progesterone blocks the estradiol-induced gonadotropin discharge in the ewe by inhibiting the surge of gonadotropin-releasing hormone. Endocrinology. 1992;131(1):208–212. doi: 10.1210/endo.131.1.1611998. [DOI] [PubMed] [Google Scholar]
- 113.Levine JE. New concepts of the neuroendocrine regulation of gonadotropin surges in rats. Biol Reprod. 1997;56(2):293–302. doi: 10.1095/biolreprod56.2.293. [DOI] [PubMed] [Google Scholar]
- 114.Beck-Peccoz P, Padmanabhan V, Baggiani AM, et al. Maturation of hypothalamic-pituitary-gonadal function in normal human fetuses: circulating levels of gonadotropins, their common alpha-subunit and free testosterone, and discrepancy between immunological and biological activities of circulating follicle-stimulating hormone. J Clin Endo-crinol Metab. 1991;73(3):525–532. doi: 10.1210/jcem-73-3-525. [DOI] [PubMed] [Google Scholar]
- 115.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30(4):293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sir-Petermann T, Codner E, Pérez V, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endo-crinol Metab. 2009;94(6):1923–1930. doi: 10.1210/jc.2008-2836. [DOI] [PMC free article] [PubMed] [Google Scholar]