

Abstract
Individual differences in affective and social processes may arise from variability in amygdala-medial prefrontal (mPFC) circuitry and related genetic heterogeneity. To explore this possibility in humans, we examined the structural correlates of trait negative affect in a sample of 1050 healthy young adults with no history of psychiatric illness. Analyses revealed that heightened negative affect was associated with increased amygdala volume and reduced thickness in a left mPFC region encompassing the subgenual and rostral anterior cingulate cortex. The most extreme individuals displayed an inverse correlation between amygdala volume and mPFC thickness, suggesting that imbalance between these structures is linked to negative affect in the general population. Subgroups of participants were further evaluated on social (n = 206) and emotional (n = 533) functions. Individuals with decreased mPFC thickness exhibited the poorest social cognition and were least able to correctly identify facial emotion. Given prior links between disrupted amygdala–mPFC circuitry and the presence of major depressive disorder (MDD), we explored whether the individual differences in anatomy observed here in healthy young adults were associated with polygenic risk for MDD (n = 438) using risk scores derived from a large genome-wide association analysis (n = 18,759). Analyses revealed associations between increasing polygenic burden for MDD and reduced cortical thickness in the left mPFC. These collective findings suggest that, within the healthy population, there is significant variability in amygdala–mPFC circuitry that is associated with poor functioning across affective and social domains. Individual differences in this circuitry may arise, in part, from common genetic variability that contributes to risk for MDD.
Introduction
Distinct behavioral phenotypes may directly or indirectly rely upon common brain systems. A well characterized amygdala–medial prefrontal cortex (mPFC) circuit contributes to both emotional (Davis and Whalen, 2001; Devinsky et al., 1995; Milad and Quirk, 2012; Phelps and LeDoux, 2005) and social processes (Adolphs, 2001; Amodio and Frith, 2006; Phelps, 2006; Van Overwalle, 2009). Dysregulated affect and impaired social cognition co-occur within disorders marked by abnormalities of the amygdala and mPFC [e.g., major depressive disorder (MDD)] (Curran et al., 1993; Drevets et al., 1997; Hajek et al., 2008; Mayberg, 1997; Price and Drevets, 2010; Savitz and Drevets, 2009b). A leading hypothesis is that variation in amygdala–mPFC circuitry, present within the general population, sets the stage for illness onset through its influence on both affective and social traits (Mayberg, 1997; Price and Drevets, 2010).
Consistent with a role in the modulation of affective and visceral functions, the mPFC and amygdala share reciprocal connections (Milad and Quirk, 2012; Phelps and LeDoux, 2005; Price and Drevets, 2010). In healthy populations, the strength of intrinsic (Kim et al., 2011a) and task-evoked (Cremers et al., 2010; Pezawas et al., 2005) amygdala–mPFC coupling decreases, and white-matter is altered (Kim and Whalen, 2009) in individuals with high levels of anxiety or related behavioral profiles. Coordinated and reciprocal amygdala–mPFC circuits are essential for adaptive affect regulation (Etkin, 2010; Kim et al., 2011b; Phelps and LeDoux, 2005), and dysfunction within the extended amygdala–mPFC network has been hypothesized to contribute to psychiatric illness including MDD (Mayberg, 1997; Price and Drevets, 2010).
Suggesting genetic contributions to individual differences in amygdala–mPFC circuitry, MDD is heritable (Sullivan et al., 2000) and thought to be influenced by a large number of common allelic variations of very small effect. Although polygenic vulnerabilities have been identified (Ripke et al., 2012), their neurobiological influence has yet to be established. Subtle alterations within the amygdala and mPFC have been observed in healthy populations carrying individual genetic variants associated with illness risk (Canli et al., 2005; Hariri et al., 2002; Pezawas et al., 2005; Savitz and Drevets, 2009b). Collectively, these results suggest that polygenic vulnerability for the onset of MDD could be reflected in the structural integrity of the amygdala and/or mPFC even within the general population, and, further, that such variation may influence multiple affective and social functions dependent upon amygdala–mPFC circuitry.
The goal of the present study was to explore whether individual differences in amygdala–mPFC circuitry relate to negative affect, social functioning, and polygenic MDD risk. Anatomical variability within the amygdala and mPFC was examined in a large cohort of treatment naive and psychiatrically healthy young adults. Because of their respective roles in affective reactivity and regulation, we hypothesized increased amygdala volume and reduced mPFC thickness in those individuals with heightened trait negative affect and impaired social functioning. Given the critical role the amygdala–mPFC circuit is theorized to play in the onset of affective illnesses, we hypothesized that the anatomical regions linked to affective and social traits would associate with polygenic vulnerability for MDD.
Materials and Methods
Participants.
Native English-speaking young adults (ages 18 to 35) with normal or corrected-to-normal vision were recruited from Harvard University, Massachusetts General Hospital, and the surrounding Boston communities. To prevent possible spurious effects resulting from population admixture and cultural biases in self-reported affect (Markus and Kitayama, 1991), analyses were restricted to non-Hispanic Caucasians of European ancestry. History of psychiatric illness and medication usage was assessed during an initial structured phone screen. On the day of MRI data collection, participants were supervised during the completion of an additional questionnaire concerning their physical health, past and present history of psychiatric illness, medication usage, and family history of psychiatric illness. Participants were excluded if their self-reported health information indicated a history of head trauma, current/past Axis I pathology or neurological disorder, current/past psychotropic medication usage, acute physical illness, and/or loss of consciousness. Genotyping was accomplished using a saliva sample (Oragene, DNA Genotek). Following the collection of their MRI data, participants completed an online self-report battery (see below) and were excluded if their data suggested noncompliance. Participants were considered noncompliant if they failed to answer more than eight questions, admitted to seeking outside assistance during the completion of the battery, or if they did not complete the online assessment. Participants provided written informed consent in accordance with guidelines set by the Partners Health Care Institutional Review Board or the Harvard University Committee on the Use of Human Subjects in Research.
Between October 2008 and February 2011, participants were recruited for the analyses of trait negative affect. Data for these analyses consisted of 1050 participants (age: 21.37 ± 2.95; female: 54.10%; right handed: 92.20%; years of education: 14.70 ± 1.95; estimated IQ 113.21 ± 9.17). In total, 18% of the sample reported a family history of psychiatric illness [depression (10.8%), anxiety disorders (4.6%), attention deficit/hyperactivity disorder (ADHD; 3.7%), schizophrenia or other psychotic disorders (0.7%), bipolar disorder (1.8%), alcohol use disorders (5.4%), substance use disorders (2.7%), personality disorders (0.7%), and psychiatric illnesses where the diagnostic details were unknown (1.2%)].
From September 2009 through July 2011, participants were recruited for the analyses of social functioning and emotion perception. Data for the analyses of self-reported social functioning consisted of 206 participants (age: 20.49 ± 2.55; female: 68.10%; right handed: 90.80%; years of education: 14.09 ± 1.84; estimated IQ 112.72 ± 8.10) of which 178 overlapped with the initial sample. The dataset for the emotional perception analyses consisted of 533 participants (age: 21.11 ± 2.82; female: 54.20%; right handed: 93.10%; years of education: 14.57 ± 1.99; estimated IQ 113.13 ± 8.81) of which 456 overlapped with the initial sample. Self-reported social functioning was collected on 138 of the 533 participants who completed the emotional perception task.
Participants were recruited for analyses of the relations between brain structure and polygenic risk for MDD from November 2008 through March 2010. Data consisted of 470 participants (age: 21.45 ± 3.16; female: 56.85%; right handed: 86.80%; years of education: 14.71 ± 1.88; estimated IQ 113.45 ± 9.13) of which 400 overlapped with the initial sample. Participants included in the polygenic analyses did not significantly differ from the initial sample in their self-reported negative affect, social functioning, or emotion perception task performance (t < 0.76, p > 0.49).
MRI data acquisition.
All imaging data were collected on matched 3T Tim Trio scanners (Siemens) at Harvard University and Massachusetts General Hospital using the vendor-supplied 12-channel phased-array head coil. Structural data included a high-resolution multi-echo T1-weighted magnetization-prepared gradient-echo image (multi-echo MP-RAGE) using the following parameters: TR = 2200 ms, TI = 1100 ms, TE = 1.54 ms for image 1 to 7.01 ms for image 4, FA = 7°, 1.2 × 1.2 × 1.2 mm, and FOV = 230. Software upgrades (VB13, VB15, VB17) occurred during data collection. Reported results are after partialing out variance associated with site (Harvard Bay 1, MGH Bay 4, MGH Bay 8, etc.) and software upgrade.
Online self-report battery.
Following MRI data collection the participants were provided a card with a random de-identified code and a web address to conduct online personality and cognitive measures. The battery was hosted on a secure internal server and presented through the LimeSurvey user interface (http://www.limesurvey.org/). The present analyses incorporated five self-report measures associated with the experience of negative affect (Barrett and Bliss-Moreau, 2009; Watson and Tellegen, 1985; Watson et al., 1999). These scales included the trait form of the Spielberger State/Trait Anxiety Inventory (STAI), which assesses feelings of apprehension, tension, nervousness, and worry (Spielberger and Gorsuch, 1970); the neuroticism scale from the NEO five-factor inventory, which captures a tendency to experience negative emotions, such as anger, anxiety, or depression and has been associated with anxiety, hostility, depression, self-consciousness, impulsiveness, and vulnerability to stress (Costa and McCrae, 1992); the behavioral inhibition component of the Behavioral Inhibition/Behavioral Activation Scale (BIS/BAS) that indexes feelings of fear, anxiety, and frustration as well as sadness in response to the experience of these cues (Carver and White, 1994); the total mood disturbance score from the Profile of Mood States (POMS), which evaluates feelings of anger, depression, fatigue, and tension (McNair et al., 1971); and the harm avoidance scale from the Temperament and Character Inventory (TCI) (Cloninger, 1987), which assesses temperamental predisposition to anticipatory worry, fear of uncertainty, shyness, and fatigability. In addition to sharing significant conceptual overlap the scales displayed strong cross-measure correlations (Table 1; Cronbach's α = 0.84). The trait negative affect composite score was calculated as the average of the Z-scores for each individual scale.
Table 1.
Negative affect composite score cross-scale correlations
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| 1. STAI—Trait | — | ||||
| 2. NEO Neuroticism | 0.80 | — | |||
| 3. Behavioral Inhibition Scale | 0.48 | 0.60 | — | ||
| 4. POMS Total Mood Disturbance | 0.71 | 0.69 | 0.34 | — | |
| 5. TCI Harm Avoidance | 0.67 | 0.73 | 0.61 | 0.54 | — |
Reported values represent Pearson r values; all p values <0.001.
Social proficiency was assessed through three scales of current and retrospective impairments in social functioning, including measures of social detachment, decreased social motivation, and social avoidance. These included the aloof-introverted subscale from the Interpersonal Adjective Scales (IAS), which indexes the self-descriptive accuracy of adjectives such as dissocial, unsociable, introverted, and distant (Wiggins et al., 1988); the aloof personality component of the Broad Autism Phenotype Questionnaire (BAPQ), which measures lack of interest in or enjoyment of social interactions (Hurley et al., 2007); and the social/school factor from the Retrospective Self-Report of Inhibition (RSRI), a recall measure of temperamental restraint across childhood social situations (Reznick et al., 1992). The social functioning composite score was calculated as the average of the Z-scores for each individual scale (r = 0.50 to 0.75; Cronbach's α = 0.80).
Accuracy of emotion perception was assessed through the online completion of a modified version of the Penn Emotion Recognition Task (Carter et al., 2009). Participants were presented with a series of faces and asked to identify if the expression was neutral or one of four emotions (happiness, anger, fear, and sadness).
Genetic data.
Genotyping was performed at the Broad Institute using the Illumina Infinium OMNI 1 quad chip (1,140,419 SNPs). The following quality control (QC) steps were performed using PLINK v1.07 (Purcell et al., 2007). Individuals were excluded for missing genotype rates >5% (n = 12), sex inconsistency based on the X chromosome inbreeding coefficient estimates (n = 1), and excessive identity-by-descent estimates (Pi-hat > 0.125) indicating genotypic relatedness (n = 11). To account for population substratification, multidimensional scaling (MDS) analyses were conducted with the HapMap phase 3 reference data (Altshuler et al., 2010b). Visual comparison of the first two MDS factors resulted in the exclusion of four participants as population outliers. SNPs with a minor allele frequency <0.01, missing rates >0.05, and suggestive deviation from Hardy–Weinberg Equilibrium (p < 10−6) were removed. SNPs were also removed if allele information was incompatible with reference Hapmap CEU data, strand information was ambiguous, or if there existed nonrandom missingness based on the PLINK haplotype test. After QC, data for 442 participants were retained with 763,104 SNPs. Of these, 438 participants passed the behavioral inclusion criteria (see Participants, above). Total genotyping rate for the final sample was 99.50%. Imputation increases genomic coverage providing additional support for genotyped-allele-based analysis (de Bakker et al., 2008). Non-genotyped SNPs were imputed using Beagle v3.3 (Browning and Browning, 2009) with the 1000 Genome Project CEU-TSI data as the reference sample (Altshuler et al., 2010a). After filtering out SNPs with a low imputation quality score (i.e., R2<0.8), imputed data included the dosage information (i.e., estimated counts of reference alleles ranging from 0.0 to 2.0) of 5,597,521 SNPs.
The genome-wide polygenic analyses, proposed by Purcell et al. (2009) consider the proportion of variation in disease risk explained through the additive effect of thousands of common variants. A subset of SNPs that are nominally associated with a target disease are identified in a discovery genome-wide association study (GWAS) case-control dataset using nominal p value thresholds (for example, association p < 0.1, p < 0.2, …, p < 0.5). The selected SNPs are then used to quantify the genetic disease risk of individuals in an independent dataset. Risk scores consist of the sum of the number of reference alleles carried by each individual, weighted by their estimated effect sizes obtained from the discovery GWAS study.
Polygenic depression risk scores were generated using data available through the Psychiatric Genomics Consortium (PGC; N = 9240 MDD cases/9519 controls) (Ripke, 2012). Abnormalities within the structure and function of the mPFC network have been established in a range of psychiatric disorders in addition to MDD [e.g., bipolar disorder (Drevets et al., 1997; Phillips and Vieta, 2007; Price and Drevets, 2010; Savitz and Drevets, 2009a; Strakowski et al., 1999), schizophrenia (Benes, 2000; Koo et al., 2008; Shenton et al., 2001; Williams et al., 2004), and ADHD (Castellanos and Proal, 2012; Seidman et al., 2006)]. To examine the specificity of polygenic effects on amygdala/mPFC structure, additional polygenic risk scores were derived from PGC GWASs of bipolar disorder (Sklar et al., 2011), ADHD (Neale et al., 2010), schizophrenia (Ripke et al., 2011), as well as nonpsychiatric illnesses including coronary artery disease, Crohn's disease, hypertension, rheumatoid arthritis, type I diabetes (Burton et al., 2007), and type II diabetes (Voight et al., 2010). For each discovery GWAS dataset, p value thresholds of 0.1, 0.2, 0.3, 0.4, and 0.5 were used to select a subset of disease-associated SNPs. Table 2 lists the summary statistics for the four psychiatric and six nonpsychiatric GWAS datasets used as discovery samples. Additional analyses examined the stability of the effects of interest across polygenic scores derived from the imputation-based dosage data.
Table 2.
Summary statistics for the discovery GWAS case control datasets
| Category | Disorder | Data Source | Sample Size |
SNP # | |
|---|---|---|---|---|---|
| Case | Control | ||||
| Psychiatric illness | Major depressive disorder | PGC | 9240 | 9519 | 1,235,033 |
| ADHD | PGC | 2887 | 2635 | 1,234,271 | |
| Bipolar disorder | PGC | 6990 | 4820 | 1,237,862 | |
| Schizophrenia | PGC | 9379 | 7736 | 1,241,747 | |
| Nonpsychiatric illness | Coronary artery disease | WTCCC | 1926 | 2935 | 404,508 |
| Crohn's disease | WTCCC | 1748 | 2935 | 404,699 | |
| Hypertension | WTCCC | 1952 | 2935 | 404,339 | |
| Rheumatoid arthritis | WTCCC | 1860 | 2935 | 404,469 | |
| Type 1 diabetes | WTCCC | 1963 | 2935 | 404,477 | |
| Type 2 diabetes | Diagram | 8130 | 38,987 | 2,255,856 | |
PGC (https://pgc.unc.edu/index.php); WTCCC, Wellcome Trust Case Control Consortium (http://www.wtccc.org.uk/); Diagram, Diabetes Genetics Replication And Meta-Analysis Consortium (http://www.well.ox.ac.uk/DIAGRAM/index.html).
Structural MRI data preprocessing.
Data were analyzed using FreeSurfer version 4.5.0 software (http://surfer.nmr.mgh.harvard.edu). FreeSurfer provides automated algorithms for the volumetric segmentation of subcortical structures and estimation of cortical thickness (Fischl and Dale, 2000; Fischl et al., 2002). The FreeSurfer pipeline allows users to analyze estimated cortical thickness independent of cortical volume. Cortical thickness and surface area, which sum to reflect total gray matter volume, are genetically and phenotypically distinct measures (Panizzon et al., 2009; Winkler et al., 2010). Accordingly, thickness estimates were selected as the dependent variable of interest for analyses of cortical anatomy. Cortical thickness was calculated as the closest distance from the gray/white boundary to the gray/CSF boundary at each vertex on the tessellated surface (Fischl and Dale, 2000). Following surface-based registration, and before the analysis of cortical thickness, a 22 mm FWHM smoothing kernel was applied to each participant's data. For the purpose of visualization, resulting maps were displayed on the inflated PALS cortical surface using Caret software (Van Essen, 2005). Estimated intracranial volume (ICV) was calculated using the approach of Buckner et al. (2004) as implemented in FreeSurfer 4.5.0.
Structural phenotype reliability.
The accuracy of FreeSurfer-derived structural estimates have been validated against histological analysis (Rosas et al., 2002) and manual measurements (Kuperberg et al., 2003). ICV has also been validated against manual measurement (Buckner et al., 2004). For the present study we took the additional step of directly assessing the reliability of the volume or thickness of each phenotype of interest (ICV, amygdala, hippocampus, mPFC). To accomplish this, a supplementary dataset (n = 91) was acquired over the course of the primary collection effort. Data were collected from these participants on two independent days (mean = 90 d apart; min = 2; max = 624). Each independent session was processed through the automated FreeSurfer pipeline separately. These data contain 46 participants whose initial runs are also included in the primary analyses. Pearson correlations were used to compare the two visits. Analyses revealed high test–retest reliability of each of our phenotypes of interest (r = 0.76 to 0.99; Fig. 1). Observed regional variation in reliability likely reflects scan–rescan shifts in head positioning and/or motion, which may disproportionately impact small structures (e.g., amygdala relative to ICV) and estimates of cortical thickness (Han et al., 2006; Jovicich et al., 2009; Morey et al., 2010).
Figure 1.

Anatomical measures are reliable. Scatter plots represent values across two separate scanning sessions for the critical volumetric and cortical thickness estimates. A–D, Estimated intracranial volume (A), left amygdala volume (B), left hippocampal volume (C), left medial prefrontal cortex thickness (D). Individual circles represent estimated volume and thickness measurements for each participant (n = 91). Reported r values reflect Pearson correlations of values from Visit 1 and Visit 2.
Statistical analyses.
A substantial literature supports the role of the amygdala in the regulation of emotional responses. Analyses of subcortical anatomical variability targeted this structure (Fig. 2A). Block linear regressions were conducted separately for both the left and right amygdala. These analyses partialed out the variance associated with site, console software version, estimated IQ, age, sex, and ICV and then examined the relation between amygdala volume and negative affect. Follow-up analyses were conducted on the remaining subcortical structures to examine the specificity of these effects.
Figure 2.

Amygdala and medial prefrontal cortex are associated with negative affect. A, Representative segmentations of the left amygdala and right amygdala. The scatter plot displays the correlation between trait negative affect and left amygdala volume. B, The targeted medial prefrontal subregion defined using FreeSurfer is displayed. The region includes portions of the left rostral anterior cingulate cortex and the subgenual anterior cingulate cortex (see Materials and Methods, Statistical analysis). Map effects do not limit exploration to the medial prefrontal cortex but also converged on the same general region, reinforcing that it was appropriate to target this subregion for hypothesis-driven analyses. The scatter plot is displayed as in A.
Given the a priori hypothesis regarding opposing effects in the amygdala and the mPFC, surface-based analyses were conducted on the FreeSurfer parcelation of the region labeled by Desikan et al. (2006) as the rostral anterior cingulate (rACC; Fig. 2B). This region was defined by Desikan et al. to include the rostral extent of the cingulate sulcus with the caudal boundary being the genu of the corpus callosum, encompassing portions of rACC and perigenual ACC. As above, block linear regressions were conducted by first partialing out the variance associated with site, console software version, estimated IQ, age, and sex and then by examining the relation between thickness estimates and the negative affect composite score. Follow-up surface-based analyses were then conducted to assess the specificity of the observed effects (to facilitate comprehensive display, surface effects are plotted with the threshold p < 0.005 uncorrected for multiple comparisons).
Analyses of social functioning, emotional perception, and polygenic vulnerability focused on the regions emerging from the analyses of cortical thickness and negative affect (Fig. 3D). Block linear regressions first partialed out the variance associated with site, console software version, estimated IQ, age, and sex and then examined the relation between thickness estimates and the variable of interest. Median response time was partialed out during analyses of errors in emotional perception. When considering polygenic risk scores analyses, we took the additional step of partialing out the variance associated with potential population substratification (the first four MDS factors, C1–C4), and number of nonmissing SNPs.
Figure 3.

Trait negative affect shows opposing effects on the amygdala and medial prefrontal cortex. A–C, Coronal slices display the correlation strengths between the volumes of each subcortical structure (based on automated segmentations; Fischl et al., 2002) and trait negative affect across the entire sample (A), as well as for the female (B) and male (C) participants. Correlations partial out variance associated with collection site, scanner software, estimated IQ, age, sex, and estimated intracranial volume (Buckner et al., 2004). D–F, Surface-based renderings of the left midline reflect the strength of the correlation between each vertex and negative affect across the entire sample (D), as well as for the female (E) and male (F) participants. Reported correlations are after partialing out the variance associated with collection site, scanner software, estimated IQ, age, and sex. Cortical thickness was estimated using the procedures of Fischl and Dale (2000) and then displayed on the inflated surface (Van Essen, 2005). Display threshold is set at p < 0.005 to allow complete visualization of the effect pattern. Color bars reflect Pearson correlations.
Results
Increased amygdala volume is associated with negative affect
Given its hypothesized role in anxiety and affective illnesses, we began our analyses focusing on the relation between the amygdala and trait-negative affect. After partialing out the variance associated with site, console software version, estimated IQ, age, sex, and ICV, left amygdala volume accounted for a significant percentage of the variance in negative affect (F(1,1042) = 19.67; p < 0.001; r = 0.14; Figs. 2A, 3A). This effect was echoed in the right amygdala (F(1,1042) = 7.69; p < 0.01; r = 0.09) and the relation between amygdala volume and the composite score of negative affect did not significantly differ by hemisphere (Z = 1.17, p = 0.24). Follow-up analyses examined the specificity of this effect across each subcortical structure. Heightened negative affect was associated with increased left (F(1,1042) = 12.56; p < 0.001; r = 0.11) and right hippocampal volumes (F(1,1042) = 6.60; p < 0.01; r = 0.08). The remaining subcortical structures (caudate, globus pallidus, nucleus accumbens, putamen, thalamus) were not significantly associated with negative affect (F < 3.25; p > 0.07; r < 0.06; Fig. 3A). Consistent with these analyses, correlations between amygdala and hippocampus volumes (r > 0.42) were significantly stronger than their respective relations with other sub-cortical regions (r < 0.29; Z < 3.41; p < 0.001).
Decreased medial prefrontal thickness is associated with negative affect
We next explored cortical effects. In line with the hypothesized role of the mPFC in the downregulation of amygdala activity, reduced left mPFC thickness was associated with increased negative affect (F(1,1043) = 8.28; p < 0.005; r = −0.09; Fig. 2B). No effects emerged on the right (F(1,1043) = 0.16; p = 0.69; r < −0.01). When considering age (F(1,1048) = 10.02; p < 0.005; r = −0.10), sex (F(1,1047) = 18.37; p < 0.001; r = 0.13), estimated IQ (F(1,1046) = 1.08; p = 0.30; r = 0.03), console software version (F(1,1045) < 0.01; p = 0.99; r < −0.01), and collection site (F(1,1044) = 4.89; p < 0.05; r = −0.07), left amygdala volume (after partialing out ICV) and mPFC thickness accounted for 3% of the remaining variance in self-reported negative affect (F(1,1042) = 13.67; p < 0.001; r = 0.16).
Given the opposing relation of each region with the negative affect composite score, the structural estimates for the amygdala and mPFC were entered sequentially in two regression analyses. After partialing out the variance associated with mPFC thickness, the association between negative affect and the amygdala remained (F(1,1042) = 18.92; p < 0.001; r = 0.13), as did the inverse relation between negative affect and mPFC thickness after partialing out variance associated with amygdala volume (F(1,1042) = 7.52; p < 0.01; r = −0.09).
As the effect in the mPFC was established within an a priori defined region, follow-up whole surface analyses established that the effect was anatomically preferential, largely confined at our significance threshold to the rACC extending into the subgenual ACC (sgACC) (Fig. 3D).
Cross-scale consistency
To establish that the effects of any one scale did not drive the observed relation between anatomical variability and trait negative affect, we duplicated the volumetric and cortical thickness regression analyses for each of five included self-report measures of affect, anxiety, and personality. When considering left amygdala volume, increased scores on each scale were associated with increased volume (STAI trait: F(1,1042) = 13.31; p < 0.001; r = 0.12; NEO neuroticism: F(1,1042) = 13.98; p < 0.001; r = 0.12; BIS: F(1,1042) = 9.71; p < 0.005; r = 0.09; POMS total mood disturbance: F(1,1042) = 10.61; p < 0.001; r = 0.11; TCI harm avoidance: F(1,1042) = 21.51; p < 0.001; r = 0.14). Increasing right amygdala volume was associated with increased scores on four of the scales (NEO neuroticism: F(1,1042) = 5.13; p < 0.05; r = 0.07; BIS: F(1,1042) = 3.86; p < 0.05; r = 0.06; POMS total mood disturbance: F(1,1042) = 4.64; p < 0.05; r = 0.07; TCI harm avoidance: F(1,1042) = 11.46; p < 0.001; r = 0.10). STAI trait anxiety displayed a trend in the same direction (F(1,1042) = 3.23; p = 0.07; r = 0.06) and this relation did not significantly differ from the other associations (Z < 0.92; p > 0.36).
The opposing effects in the mPFC were also evident in four of the scales (STAI trait: F(1,1043) = 6.81; p < 0.01; r = −0.08; NEO neuroticism: F(1,1043) = 4.90; p < 0.05; r = −0.07; POMS total mood disturbance: F(1,1043) = 7.86; p < 0.005; r = −0.09; TCI harm avoidance: F(1,1043) = 6.80; p < 0.01; r = −0.08). Although the behavioral inhibition scale did not display a significant relation with mPFC thickness, the observed effect was in the same direction (F(1,1043) = 2.98; p = 0.08; r = −0.05) and did not significantly differ from the other relations (Z < 0.92; p > 0.36).
Individual differences are present in both men and women
As anxiety and depressive disorders are more common among women (Nolen-Hoeksema and Girgus, 1994), we examined whether there is a sex difference in the amygdala–mPFC association with negative affect. Women displayed increased negative affect (0.10 ± 0.86 vs −0.12 ± 0.80; t(1048) = 4.29; p < 0.001) and left mPFC thickness (3.15 ± 0.27 vs 3.07 ± 0.24; t(1048) = 5.23; p < 0.001) relative to men. No differences emerged when considering amygdala volumes (after partialing out ICV; t < 1.28; p > 0.20).
Critically, left amygdala volume was similarly associated with the negative affect composite score for both women (F(1,561) = 10.55; p < 0.001; r = 0.14) and men (F(1,475) = 7.64; p < 0.01; r = 0.13). Among women, the left hippocampus (F(1,561) = 6.56; p < 0.01; r = 0.11) demonstrated a significant relation with negative affect, while a trend emerged in the right amygdala (F(1,561) = 2.80; p = 0.09; r = 0.07). No other subcortical structures approached significance (F < 2.45; p > 0.12; r < 0.07; Fig. 3B). Among men, negative affect was associated with right amygdala (F(1,475) = 4.11; p < 0.05; r = 0.09) and hippocampal volumes (left: F(1,475) = 5.17; p < 0.05; r = 0.10; right: F(1,475) = 4.44; p < 0.05; r = 0.10). No other subcortical structures approached significance (F < 1.02; p > 0.31; r < 0.05; Fig. 3C). The inverse relation between left mPFC thickness and negative affect was observed and of similar magnitude for both sexes (women, F(1,562) = 5.60; p < 0.05; r = −0.10; men, F(1,476) = 4.50; p < 0.05; r = −0.10; Fig. 3E,F).
Extreme negative affect reveals an inverse relation between the structure of the amygdala and medial prefrontal cortex
Impairments in emotional experience are hypothesized to result from a breakdown in the dynamic interactions between opposing subcortical and cortical structures (Mayberg, 1997; Price and Drevets, 2010). To search for indirect evidence suggesting weakened and/or imbalanced coupling, the full sample was rank ordered by their negative affect scores. A rolling correlation was then calculated between amygdala volume and mPFC thickness across the full range of negative affect. The rolling average revealed opposing relations between negative affect and the structure of the left amygdala and cortex (Fig. 4A). While there was an absence of a cross-structure relation over much of the sample, a robust negative correlation between amygdala volume and mPFC thickness emerged in individuals reporting the most severe negative affect (Fig. 4B,C).
Figure 4.
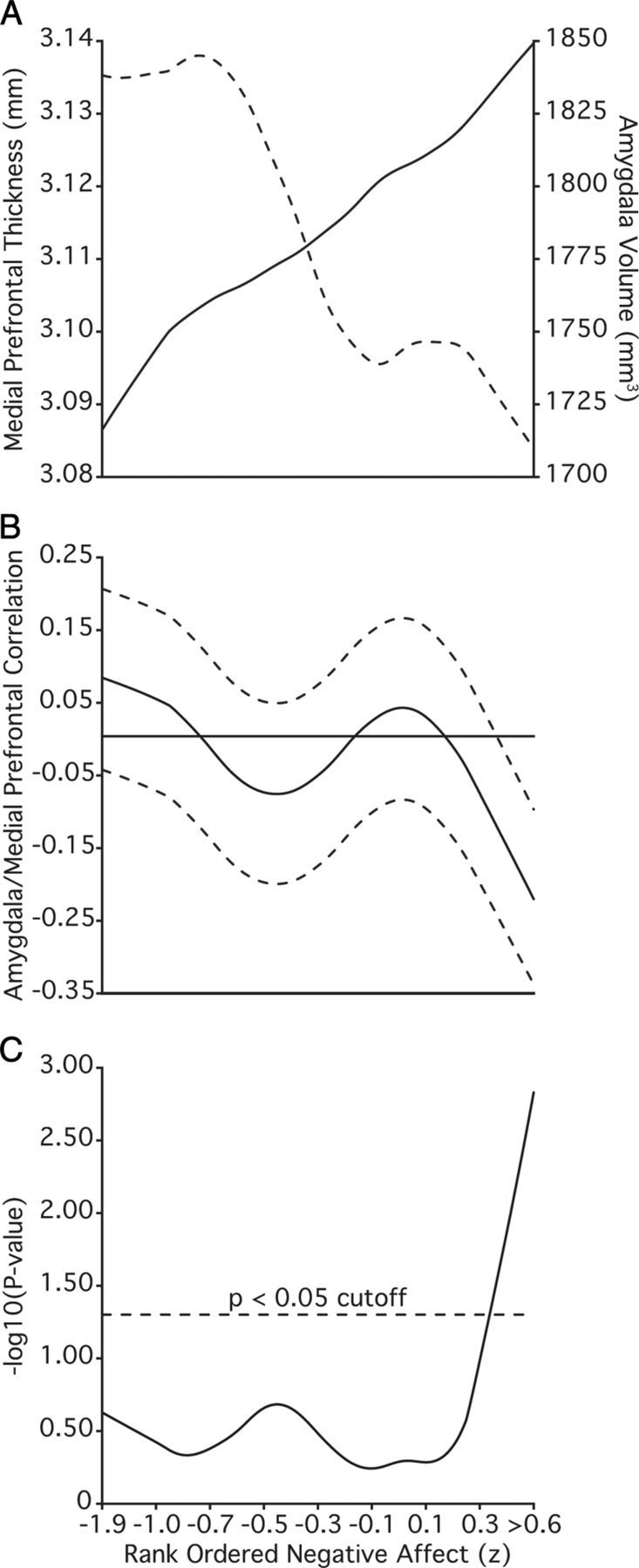
Rank ordering of individuals by negative affect reveals an inverse correlation between amygdala volume and medial prefrontal thickness. Data rank ordered by negative affect, averaged (n − n + 249), and smoothed with a lowess filter (span = 250). Reported values are after partialing out the variance associated intracranial volume from the left amygdala as well as collection site, scanner software, estimated IQ, age, and sex from both structures. A, Rolling average of left amygdala volume (solid line) and left medial prefrontal thickness (dotted line). B, Rolling Pearson correlation between amygdala volume and medial prefrontal thickness. Dotted lines reflect 95% confidence interval. C, Resulting p values (−log10). Individuals reporting the most severe symptoms show the strongest negative correlation between the amygdala and the medial prefrontal thickness.
To further examine how the correlation between amygdala volumes and mPFC thickness contributes to negative affect, the sample was split into groups with low (n = 175), medium (n = 705), and high (n = 170) negative affect. High and low groups were defined as one standard deviation above, and below, the mean negative affect score (0.00 ± 0.82) after partialing out the variance associated with age and sex. The resulting groups did not differ demographically (Table 3).
Table 3.
Summary of negative affect group demographic and self-report data
| Variable | Low negative affect group (n = 175) |
Medium negative affect group (n = 705) |
High negative affect group (n = 170) |
χ2/F | p value | |||
|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | |||
| Age | 21.54 | 3.04 | 21.25 | 2.87 | 21.67 | 3.16 | 1.78 | 0.17 |
| Percent female | 57.70 | N/A | 52.60 | N/A | 56.50 | N/A | 1.93 | 0.38 |
| Percent right handed | 90.90 | N/A | 92.50 | N/A | 92.40 | N/A | 0.52 | 0.77 |
| Years of education | 14.84 | 1.86 | 14.63 | 1.93 | 14.84 | 2.11 | 1.35 | 0.26 |
| Estimated IQ | 112.74 | 8.97 | 113.19 | 9.29 | 113.78 | 8.89 | 0.57 | 0.57 |
| Trait negative affect composite score | −1.15 | 0.27 | −0.03 | 0.45 | 1.30 | 0.41 | Selected | N/A |
| Percent family history of psychiatric illness | 18.90 | N/A | 15.90 | N/A | 25.90 | N/A | 9.38 | <0.01 |
N/A, Not applicable; selected indicates that the variable was used to define the groups and therefore, by design, differs between groups.
When considered in isolation, there was no detectable relation between amygdala volume and mPFC thickness for either the low (F(1,168) = 1.04; p = 0.31; r = 0.08; Fig. 5A) or medium groups (F(1,698) < 0.01; p = 0.97; r < −0.01; Fig. 5B). However, an inverse correlation between left amygdala volume and mPFC thickness was evident among individuals reporting the most extreme negative affect (F(1,163) = 13.55; p < 0.001; r = −0.28; Fig. 5C). The anticorrelation in the extreme negative affect participants was significantly different from the relations observed in both the low (Z = 3.34, p < 0.001) and medium (Z = 3.29, p < 0.001) groups. Of further note, participants in the high negative affect group were more likely to report a family history of psychiatric illnesses relative to the low and medium groups (Table 3). However, given the reliance on self-report, these data should be interpreted cautiously.
Figure 5.

Opposing structural differences in the amygdala and medial prefrontal cortex are present in the young adults with the most extreme negative affect. A–C, Scatter plots represent the distribution of values for the left amygdala volumes and left medial prefrontal thickness estimates for the low (A), medium (B), and high (C) negative affect groups. Reported r values reflect Pearson correlations after partialing out variance associated with collection site, scanner software, estimated IQ, age, and sex. Estimated intracranial volume was additionally partialed from the amygdala volume estimate. D–F, Surface-based analyses reflect the correlation strength between left amygdala volume and thickness estimates at each vertex for the low (D), medium (E), and high (F) negative affect groups. Color bar reflects Pearson correlations.
There is some indication that the participants reporting the highest negative affect did not solely drive the relations between negative affect, amygdala volume, and mPFC thickness. After excluding these extreme subjects from the full sample, the association between negative affect and left amygdala volume remained (F(1,872) = 9.175; p < 0.005; r = 0.10), as did the inverse relation with mPFC thickness (F(1,873) = 5.84; p < 0.05; r = −0.08). Additional analyses were run excluding participants with a family history of psychiatric illness (n = 189). The associations between negative affect with both left amygdala volume (F(1,853) = 11.27; p < 0.001; r = 0.11) and mPFC thickness (F(1,854) = 3.88; p < 0.05; r = −0.07) remained. Thus, the results show the largest effects were present in those individuals with extreme negative affect, but also that a relation in the remaining sample should not be ruled out.
Decreased medial prefrontal thickness is associated with impaired social function
Overlap exists within the neural systems believed to underlie negative affect and social functioning (Adolphs, 2001; Ochsner, 2008; Olsson and Ochsner, 2008). In line with the hypothesized role of the mPFC in social behaviors, reduced left mPFC thickness was associated with a composite score of social withdrawal and reduced social functioning within the region delineated in the negative affect analyses (F(1,200) = 12.07; p < 0.001; r = −0.24; Fig. 6A). This effect was seen in each of the three self-report measures included in the social functioning composite score (IAS aloof introverted: F(1,200) = 11.93; p < 0.001; r = −0.24; BAPQ aloof personality: F(1,200) = 10.86; p < 0.001; r = −0.23; RSRI social/school: F(1,200) = 4.31; p < 0.05; r = −0.14; Fig. 6B). While the RSRI retrospectively assesses withdrawal and cautious responding across social situations (Reznick et al., 1992), it is also a measure of childhood behavioral inhibition that shares conceptual overlap with aspects of the negative affect composite score. In line with the broad influence of amygdala–mPFC circuitry across affective and social processes, the relation with mPFC thickness remained when the RSRI was eliminated from the social functioning composite score (F(1,200) = 13.22; p < 0.001; r = −0.25). While male participants reported lower social functioning than females (−0.12 ± 0.86 vs 0.26 ± 0.78; t(204) = 3.10, p < 0.005), no sex differences emerged when considering the strength of the relation between social functioning and mPFC thickness (Z = 0.49, p = 0.62). The relation between mPFC thickness and social function was present when considering the broader FreeSurfer parcelation of the rACC (F(1,200) = 6.78; p < 0.01; r = −0.18). There was no significant association between social functioning and the volume of either the left or right amygdala (F < 2.10; p > 0.15; r < 0.08).
Figure 6.
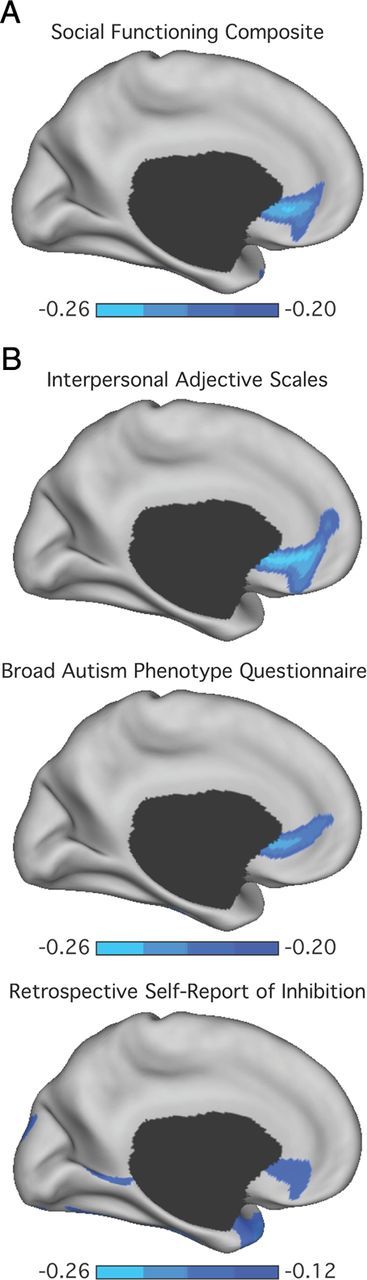
Poor social functioning is associated with reduced medial prefrontal thickness. A, The correlation strength between thickness estimates at each vertex on the left medial surface and a composite score of social functioning. B, Correlation strengths for each independent measure within the composite score. These include the aloof-introverted subscale from the Interpersonal Adjective Scales (Wiggins et al., 1988), the aloof personality component of the Broad Autism Phenotype Questionnaire (Hurley et al., 2007), and the social/school factor from the Retrospective Self-Report of Inhibition (p < 0.01) (Reznick et al., 1992). Reported r values reflect Pearson correlations after partialing out variance associated with collection site, scanner software, estimated IQ, age, and sex. Color bar reflects Pearson correlations.
Consistent with their shared anatomical associations, increased negative affect was correlated with increased social withdrawal and reduced social functioning (F(1,197) = 49.08; p < 0.001; r = 0.47). After accounting for social functioning, the relation between negative affect and amygdala volume remained for the left (F(1,170) = 4.90; p < 0.05; r = 0.13), but not the right (F(1,170) = 1.18; p = 0.28; r = 0.06), amygdala. After partialing out the variance associated with negative affect, the association between social functioning and mPFC thickness remained (F(1,171) = 5.02; p < 0.05; r = −0.16). However, the relation between negative affect and mPFC thickness became nonsignificant after partialing out variance associated with social functioning (F(1,171) = 1.63; p = 0.20; r = −0.10).
Decreased medial prefrontal thickness is associated with errors in emotion perception
We further examined the relations between social functioning, amygdala volumes, and mPFC thickness with analyses of individual differences in the detection and discrimination of the emotional states of others, a core aspect of social cognition. A benefit of this approach is the use of a performance measure of social ability, rather than relying on self-report. Analyses of amygdala volumes revealed no significant relations with errors in emotion perception (F < 0.78; p > 0.38; r < 0.03). Consistent with our prior analyses, reduced mPFC thickness (using the region defined from analyses of negative affect) was associated with increased errors in emotion perception (error rate 0.19 ± 0.06; F(1,521) = 7.33; p < 0.01; r = −0.12; Fig. 7). Follow-up analyses revealed that this effect was specific to the neutral (error rate 0.14 ± 0.13; F(1,521) = 15.68; p < 0.001; r = −0.17) and fear (error rate 0.19 ± 0.11; F(1,521) = 5.49; p < 0.05; r = −0.10) conditions with no other effects reaching significance (anger, error rate 0.39 ± 0.14; happy, error rate 0.03 ± 0.06; and sad, error rate 0.21 ± 0.14; p > 0.41, r < 0.04). Although male participants displayed a greater percentage of errors in emotion perception than females (0.19 ± 0.01 vs 0.18 ± 0.01; t(531) = 2.32, p < 0.05), there were no sex differences in the relations between task performance and mPFC thickness (Z = 1.30, p = 0.19). The association between errors in emotion perception and mPFC thickness held when considering the FreeSurfer defined parcelation of the rACC (F(1,521) = 6.63; p < 0.01; r = −0.12).
Figure 7.

Poor emotion perception is associated with reduced medial prefrontal thickness. A, Correlation strengths for each emotional condition. Reported r values reflect Pearson correlations after partialing out variance associated with collection site, scanner software, estimated IQ, age, sex, and the median response time for each condition of interest. Error bars reflect 95% confidence interval. B, The correlation strength between thickness estimates at each vertex and error rates in the emotional faces task. Color bar reflects Pearson correlations.
There was no relation between errors in emotion perception and negative affect (r = −0.05, p = 0.25). After partialing out the variance associated with negative affect, the association between errors in emotion perception and mPFC thickness remained (F(1,521) = 7.33; p < 0.01; r = −0.11). Within the participants providing data for both the emotion perception and social functioning analyses (n = 138), decreased accuracy in emotional perception was associated with reduced social functioning (r = 0.23, p < 0.01). When partialing out social functioning, the relation between emotion perception and mPFC thickness no longer reached significance (F(1,130) = 0.93; p = 0.34; r = −0.08). When accounting for errors in emotion perception, the association between mPFC thickness and reduced social functioning remained significant (F(1,130) = 8.65; p < 0.005; r = −0.24).
Decreased medial prefrontal thickness is associated with polygenic vulnerability for major depressive disorder
Consistent with evidence of dysfunction within the left mPFC in patient populations (Curran et al., 1993; Drevets et al., 1997; Hajek et al., 2008; Mayberg, 1997; Price and Drevets, 2010; Savitz and Drevets, 2009b), heightened polygenetic MDD risk (PT < 0.5) was associated with reduced mPFC thickness (F(1,430) = 8.34; p < 0.005; r = −0.14; Fig. 8A). This effect was present across five polygenic scores for depression derived at thresholds of varying association stringency from prior case control GWAS analyses (Table 4). The association between polygenic MDD risk (PT < 0.5) and mPFC thickness was reduced to a trend when considering the FreeSurfer defined parcelation of the rACC (F(1,430) = 3.73; p = 0.05; r = −0.09). Follow-up analyses confirmed the relation between cortical thickness, and polygenic burden was preferentially localized in the mPFC (Fig. 8B). No relation was observed between the polygene MDD factors (PT < 0.1 to 0.5) and negative affect (F < 0.42; p > 0.52; r < 0.03) or amygdala volumes (F < 0.85; p > 0.36; r < 0.04). The association between increasing polygenic burden (PT < 0.5) and decreasing mPFC thickness held after partialing out the variance associated with trait negative affect (F(1,392) = 7.30; p < 0.01; r = −0.13). Although brain structure is heritable, individual differences in mPFC thickness do not exclusively arise through familial factors (h2 = 0.21–0.48) (Winkler et al., 2010). The current analyses suggest that the measured polygenic MDD scores account for ∼2% of the total variance in mPFC thickness. When considering heritability estimates, this reflects that at least ∼4–9% of the phenotypic variance in mPFC thickness can be accounted for by the aggregated effect of common genetic factors, although the estimated effect may be much higher (Purcell et al., 2009).
Figure 8.

Heightened polygenic depression risk is associated with reduced mPFC thickness in clinically healthy individuals. A, Variance in cortical thickness explained on the basis of scores derived from four psychiatric and six nonpsychiatric illnesses across varying significance thresholds (PT < 0.1, 0.2, 0.3, 0.4 and 0.5, plotted left to right). BP, Bipolar Disorder; SCZ, Schizophrenia; CAD, coronary artery disease; CD, Crohn's disease; HT, hypertension; RA, rheumatoid arthritis; T1D, type I diabetes; T2D, type II diabetes. The selected mPFC region is displayed in the figure legend. Reported r values are after partialing out variance associated with collection site, scanner software, estimated IQ, multidimensional scaling components of genetic ancestry, number of nonmissing SNPs, age, and sex. Equivalent variance is explained across the MDD polygenic scores (Z < 0.12, p > 0.91). No other psychiatric or nonpsychiatric polygenic scores approached significance (p > 0.59). B, Surface-based rendering reflects the strength of the correlation between each vertex and polygenic risk for MDD (PT < 0.5) after partialing out the variance associated with the collection site, scanner software, estimated IQ, potential for population substratification, number of nonmissing SNPs, age, and sex. Display threshold is set at p < 0.01 uncorrected for multiple comparisons. Color bar reflects Pearson correlations.
Table 4.
Polygenic analyses reveal specific associations between unipolar depression risk and medial prefrontal thickness
| Discovery GWAS |
Genotype data |
Imputed data |
||||
|---|---|---|---|---|---|---|
| p value | SNP # | p | R2 | p | R2 | |
| Major depressive disorder | < 0.1 | 122,072 | 0.003 | 0.020 | 0.005 | 0.018 |
| < 0.2 | 236,773 | 0.003 | 0.020 | 0.006 | 0.017 | |
| < 0.3 | 348,603 | 0.002 | 0.021 | 0.005 | 0.018 | |
| < 0.4 | 456,525 | 0.002 | 0.021 | 0.006 | 0.017 | |
| < 0.5 | 564,290 | 0.004 | 0.018 | 0.011 | 0.011 | |
| Bipolar disorder | < 0.1 | 125,854 | 0.314 | 0.002 | 0.313 | 0.002 |
| < 0.2 | 235,719 | 0.396 | 0.002 | 0.318 | 0.002 | |
| < 0.3 | 341,228 | 0.474 | 0.001 | 0.416 | 0.001 | |
| < 0.4 | 443,095 | 0.445 | 0.001 | 0.412 | 0.002 | |
| < 0.5 | 543,664 | 0.992 | 0.001 | 0.701 | 0.001 | |
The number of risk profile SNPs that satisfy each p value threshold based on the discovery GWAS data is shown. The study datasets consist of allele information for 763,104 genotyped SNPs and dosage data for 5,597,521 imputed SNPs; R2, total variance in mPFC thickness accounted for by each polygenic factor after partialing out variance associated with collection site, scanner software, estimated IQ, multidimensional scaling components of genetic ancestry, number of nonmissing SNPs, age, and sex.
The specificity of the observed relations between polygenic MDD risk and mPFC thickness was established through a series of analyses examining genetic risk scores for ADHD, bipolar disorder, and schizophrenia (F < 0.92; p > 0.33; r < 0.05) as well as six nonpsychiatric illnesses including coronary artery disease, Crohn's disease, hypertension, rheumatoid arthritis; type I diabetes, and type II diabetes (F < 0.29; p > 0.59; r < 0.03; Fig. 8A). With the exception of ADHD (Z < 1.59; p > 0.11), the association between polygenic MDD risk and mPFC thickness was significantly greater than the relations observed for the other psychiatric polygene scores (PT < 0.1 to 0.5; Z > 2.04; p < 0.05).
Providing support for the initial genotype-data-based analyses, imputed dosage-data-based polygenic MDD risk (PT < 0.5) was associated with reduced mPFC thickness (F(1,432) = 6.53; p < 0.05; r = −0.12). This effect was present across each imputed risk score (PT < 0.1, 0.2, 0.3, 0.4; F > 7.67; p < 0.01; r > 0.13; Table 4). No relation was observed between mPFC thickness and the other imputed psychiatric (F < 1.46; p > 0.23; r < 0.06) or non-psychiatric genetic risk scores (F < 1.23; p > 0.26; r < 0.06).
Discussion
Amygdala–mPFC circuit dysfunction is hypothesized to result in negative affect, impaired social functioning, and vulnerability for the subsequent onset of MDD (Mayberg, 1997; Price and Drevets, 2010). Here, we found that negative affect associates with increased amygdala volume and reduced mPFC thickness in a cohort of clinically healthy young adults. An inverse association was found between amygdala volume and mPFC thickness among those individuals with the most extreme negative affect, representing the upper 15% of the sample. These findings are consistent with the possibility that amygdala–mPFC imbalance is linked to negative affect within the general population. A second finding of our study is that a cortical correlate of negative affect (decreased left mPFC thickness) is associated with poor social functioning and emotion identification. Thus, dysregulation of the amygdala–mPFC circuit may also be linked to impairments of social cognition. Finally, the present analyses reveal that an aggregate measure of genetic risk for MDD is associated with reduced left mPFC thickness. Suggesting the specificity of this association, no effect emerged when considering polygenic factors for bipolar disorder, schizophrenia, ADHD, or six nonpsychiatric illnesses. These results provide initial evidence that common polygenic burden is associated with anatomical variability in brain regions believed to influence vulnerability to MDD.
Amygdala–medial prefrontal imbalance
Our analyses identified opposing effects of the amygdala and mPFC on negative affect, specifically within the rACC extending into the sgACC. The sgACC participates in the dampening of fear responses as well as the experience and regulation of negative affect (Etkin, 2010; Kim et al., 2011b; Phelps and LeDoux, 2005). Amygdala–mPFC connections are concentrated within the basolateral nucleus (Price and Drevets, 2010). In rodents, mPFC inputs to the basolateral nucleus (al Maskati and Zbrozyna, 1989) and intercalated cells gate basolateral–central amygdala impulse transmission (Milad and Quirk, 2012). The central nucleus plays a role in the modulation of autonomic and endocrine responses for a host of visceral functions (LeDoux, 2000, 2012). The present findings raise the possibility that imbalance within the basolateral amygdala–mPFC network may underlie dysregulated cortical control over central nucleus output and an associated increase in visceral responsivity (Price and Drevets, 2010). What is notable about our results is that dysregulation appears to affect multiple functional domains.
The relation between psychological health and social functioning is well established (Coyne and Downey, 1991; Repetti et al., 2002; Vaughn and Leff, 1976). While these constructs are often discussed within separate literatures, they may directly or indirectly rely on overlapping neural systems (Adolphs, 2001; Ochsner, 2008; Olsson and Ochsner, 2008). Rodent and nonhuman primate research implicates the mPFC in social as well as affective, habitual, and evaluative processes (Devinsky et al., 1995; Graybiel, 2008; Price and Drevets, 2010; Rushworth et al., 2007). Reciprocal amygdala–mPFC projections suggest that adaptive social functioning may arise, in part, through visceral and affective responses to salient stimuli. Our findings indicate that shifts in social functioning and personality traits are reflected in the normal anatomical variability of a shared mPFC network. The inclusion of a behavioral index of emotion perception further supports the role of the mPFC in social function. Relations between mPFC anatomy and emotion perception were largely driven by a tendency to mistakenly ascribe emotions to neutral stimuli, rather than impaired recognition of emotional faces. Disrupted mPFC network integrity could result in the perception of emotions in nonemotional stimuli, contributing to the fear and avoidance of social situations.
While our analyses targeted the amygdala–mPFC network, a failure to modulate hippocampal hyperactivity is also hypothesized to result in affective dysregulation (Mayberg, 1997; Price and Drevets, 2010). The current data indicate that negative affect is broadly associated with the increasing volume of temporal lobe structures. Consistent with a strong cross-structure correlation within the present sample, in healthy populations, amygdala and hippocampal volumes track each other over the lifespan (Grieve et al., 2005; Mu et al., 1999; Walhovd et al., 2005). Yet prior research has suggested the presence of reduced hippocampal formation gray matter in healthy young adults with heightened neuroticism (DeYoung et al., 2010) and patients with MDD (Bremner et al., 2000), although results have been inconsistent (Price and Drevets, 2010). Similar inconsistencies have been observed when considering amygdala volumes, which are likely increased early in the course of illness, with only chronically ill patients exhibiting decreased volumes (Hamilton et al., 2008). In rodents, repeated stress results in dendritic atrophy and reduced glial cell counts in hippocampal regions (McEwen and Magarinos, 2001) as well as increased dendritic branching in the basolateral amygdala (Vyas et al., 2002). The current findings indicate that, in the absence of illness, amygdala and hippocampal volumes are largest in individuals with heightened negative affect. However, it is likely that shared relations between these structures may not be evident in chronic illness due to pathophysiological processes including elevated glucocorticoid secretion and glutamatergic transmission (Price and Drevets, 2010).
Similar effects in both women and men
There are sex differences in the prevalence of disorders of affect and social functioning (Nolen-Hoeksema and Girgus, 1994). In healthy populations, women and men exhibit differential hemodynamic responses to affective and social stimuli (Cahill, 2006; Koch et al., 2007; McRae et al., 2008; Thomas et al., 2001; But see, Wager et al., 2003). This includes sex-related hemispheric lateralization of amygdala responses for the memory of emotional material and the presentation of emotional faces (Cahill, 2006) as well as greater reductions in left amygdala response during emotion regulation in men, relative to women (McRae et al., 2008). Sex-specific responses to affective stimuli are also evident in patients with MDD where women exhibit increased left amygdala–mPFC connectivity to fearful faces (Almeida et al., 2011).
The present analyses revealed an amygdala–mPFC contribution to affective and social functioning in both sexes. Although these data suggest functionally meaningful differences in amygdala–mPFC anatomy, we are unable to make strong claims regarding the relation of structure to specific task-evoked function. While reduced mPFC thickness is independently present in men and women with increased anxiety, women as a group displayed thicker mPFC and yet heightened anxiety. It is unclear if this difference reflects adaptation to unique challenges or sex-specific differences in intrinsic architecture. Men and women react differently to environmental stressors (Hankin and Abramson, 2001) and morphometric estimates are likely insensitive to the associated subtle state dependent shifts in brain function. However, although transient amygdala–mPFC responses can be sex- (or even individual) specific, our findings suggest that the circuit's core role in affective and social processes generalizes across both sexes.
Polygenic depression risk predicts decreased medial prefrontal thickness
Depression is associated with decreased left mPFC gray matter (Hajek et al., 2008; Price and Drevets, 2010). While these reductions intensify over time in patients with psychotic mood disorders (Koo et al., 2008) and non-remitters (Frodl et al., 2008), they are immediately evident following initial diagnosis (Botteron et al., 2002) and in children with a family history of MDD (Boes et al., 2008). Suggesting an innate biological vulnerability, altered mPFC anatomy is pronounced in subgroups of patients with familial depression (Ongür et al., 1998) and individuals carrying specific genetic variants associated with increased illness risk (Canli et al., 2005; Pezawas et al., 2005; Savitz and Drevets, 2009b). The present findings highlight a relation between common polygenic burden and mPFC thickness in psychiatrically healthy young adults.
Decreased social support (Coyne and Downey, 1991; Repetti et al., 2002; Vaughn and Leff, 1976) and neuroticism (Clark et al., 1994), a facet of negative affect, predict the onset of depressive episodes. The current analyses provide evidence of a relation between an aggregate measure of genetic risk and anatomical variation in the mPFC network supporting affective and social functions. No association was observed between polygenic burden and negative affect. However, MDD is not purely genetic in origin, and stressful life events contribute to the occurrence of depressive episodes (Kendler et al., 1999). In rodents, chronic stress results in symptoms characteristic of MDD including anhedonia, changes in weight, sleep disturbances, decreased locomotion (Willner, 1997), as well as dendritic retraction and synapse loss in the mPFC (Cook and Wellman, 2004). Genetic vulnerability and life stress converge to impact the integrity of the amygdala–mPFC network. This raises the possibility that genetically mediated reductions in mPFC network integrity may contribute to an exaggerated stress response, the exacerbation of stress-induced dendritic remodeling, and heightened negative affect. Given the cross-sectional nature of the current data, longitudinal research disentangling the temporal order and convergent nature of these effects is warranted.
Abnormalities within the structure and function of the mPFC have been established in a range of psychiatric disorders. This is perhaps most evident in affective illnesses where disrupted mPFC function is theorized to underlie the occurrence of depressive and manic episodes (Mayberg, 1997; Phillips et al., 2003; Phillips and Vieta, 2007; Price and Drevets, 2010). In their landmark study, Drevets et al. (1997) established mPFC gray matter loss in unipolar and bipolar depression. Subsequent meta-analyses have replicated these mPFC volumetric reductions, predominantly in patients with familial MDD (Hajek et al., 2008). The current analyses indicate that polygenic MDD risk predicts the cortical thickness of regions within the mPFC associated with negative affect and impaired social functioning. These findings support the hypothesis that amygdala–mPFC circuit dysfunction may set the stage for MDD onset in vulnerable populations and suggest the presence of diagnostic specificity within the neuroanatomical correlates of polygenic risk.
Caveats
Several potential caveats warrant mention. First, current polygenic factors only account for ∼1–2% of the phenotypic variance in case-control analyses. It is possible that as these scores are further refined through the use of larger samples and denser SNP arrays, a shared genetic influence on mPFC thickness, amygdala volumes, and/or trait negative affect may emerge. Second, the current findings indicate relations between individual differences in affect, social functioning, polygenic MDD risk, and structural variation within the amygdala–mPFC network. However, we are unable to establish whether the observed relations solely reflect the neuroanatomical instantiation of genetic vulnerability, or arise in conjunction with factors such as increased environmental sensitivity and/or early life stress. Third, with the available anatomical data we are unable to examine individual amygdala sub-nuclei. Rodent and nonhuman primate tracing literature suggests impaired affect and reduced social functioning likely result from imbalances between the mPFC and basolateral amygdala. Subsequent work in humans, potentially leveraging diffusion-based tractographic segmentation (Saygin et al., 2011), will be necessary to explore these relations with more anatomical precision. Fourth, the participants in the present study were healthy young adults. Additional research will be necessary to establish the generalizability of the current anatomical findings.
Another limitation is the size of the effects observed. Through the use of relatively crude morphometric measures, the present findings suggest how specific neural systems differ in ways that are associated with negative affect, social functioning, and genetic risk. However, the observed associations are likely too small to be useful as predictors for individuals. For instance, the relation between negative affect and mPFC thickness accounts for ∼1% of the observed variance while the largest association accounts for ∼8% (amygdala volume and mPFC thickness in the high negative affect group). Our data establish the principles that individual differences in affect, social cognition, and polygenic risk are neurally embedded within clinically healthy individuals, that these differences are preferential to specific brain systems, and that the effects go in opposite directions for the amygdala and anatomically-linked cortical networks.
These results indicate that compromised amygdala–mPFC circuitry, which is present in a considerable percentage of healthy young adults, may simultaneously relate to emotional impairments, reduced social functioning, and polygenic vulnerability for the onset of MDD.
Footnotes
This work was supported by funding from the Simons Foundation (R.L.B.), the Howard Hughes Medical Institute (R.L.B.), and NIMH Grants R01-MH079799 (J.W.S.) and K24MH094614 (J.W.S.). The PGC analyses were supported by NIMH grant U01 MH085520 and polygene score data were obtained from http://www.broadinstitute.org/mpg/ricopili/. We are grateful to the Psychiatric Genomics Consortium, Wellcome Trust Case-Control Consortium, and DIAGRAM Consortium for sharing their genetic data analysis results. We thank Justin Baker, Eun Young Choi, Amitai Shenhav, and B. T. Thomas Yeo for their feedback on early versions of this manuscript; Timothy O'Keefe, Victor Petrov, and Gabriele Fariello for neuroinformatics; and the Harvard Center for Brain Science and the Athinoula A. Martinos Center for Biomedical Imaging for imaging support. The data were collected as part of the Brain Genomics Superstruct Project.
The authors declare declare no competing financial interests.
References
- Adolphs R. The neurobiology of social cognition. Curr Opin Neurobiol. 2001;11:231–239. doi: 10.1016/s0959-4388(00)00202-6. [DOI] [PubMed] [Google Scholar]
- al Maskati HA, Zbrozyna AW. Stimulation in prefrontal cortex area inhibits cardiovascular and motor components of the defense reaction in rats. J Auton Nerv Syst. 1989;28:117–125. doi: 10.1016/0165-1838(89)90084-2. [DOI] [PubMed] [Google Scholar]
- Almeida JR, Kronhaus DM, Sibille EL, Langenecker SA, Versace A, Labarbara EJ, Phillips ML. Abnormal left-sided orbitomedial prefrontal cortical-amygdala connectivity during happy and fear face processing: a potential neural mechanism of female MDD. Front Psychiatry. 2011 doi: 10.3389/fpsyt.2011.00069. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler DL, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Collins FS, De La Vega FM, Donnelly P, Egholm M, Flicek P, Gabriel SB, Gibbs RA, Knoppers BM, Lander ES, Lehrach H, Mardis ER, McVean GA, Nickerson DA, Peltonen L, et al. A map of human genome variation from population-scale sequencing. Nature. 2010a;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Chang K, Hawes A, Lewis LR, et al. Integrating common and rare genetic variation in diverse human population. Nature. 2010b;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodio DM, Frith CD. Meeting of minds: the medial frontal cortex and social cognition. Nat Rev Neurosci. 2006;7:268–277. doi: 10.1038/nrn1884. [DOI] [PubMed] [Google Scholar]
- Barrett LF, Bliss-Moreau E. Affect as a psychological primitive. Adv Exp Soc Psychol. 2009;41:167–218. doi: 10.1016/S0065-2601(08)00404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM. Emerging principles of altered neural circuitry in schizophrenia. Brain Res Rev. 2000;31:251–269. doi: 10.1016/s0165-0173(99)00041-7. [DOI] [PubMed] [Google Scholar]
- Boes AD, McCormick LM, Coryell WH, Nopoulos P. Rostral anterior cingulate cortex volume correlates with depressed mood in normal healthy children. Biol Psychiat. 2008;63:391–397. doi: 10.1016/j.biopsych.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botteron KN, Raichle ME, Drevets WC, Heath AC, Todd RD. Volumetric reduction in left subgenual prefrontal cortex in early onset depression. Biol Psychiat. 2002;51:342–344. doi: 10.1016/s0006-3223(01)01280-x. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. Am J Psychiat. 2000;157:115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Head D, Parker J, Fotenos AF, Marcus D, Morris JC, Snyder AZ. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: reliability and validation against manual measurement of total intracranial volume. Neuroimage. 2004;23:724–738. doi: 10.1016/j.neuroimage.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, Todd JA, Donnelly P, Barrett JC, Davison D, Easton D, Evans D, Leung HT, Marchini JL, Morris AP, Spencer CC, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill L. Why sex matters for neuroscience. Nat Rev Neurosci. 2006;7:477–484. doi: 10.1038/nrn1909. [DOI] [PubMed] [Google Scholar]
- Canli T, Omura K, Haas BW, Fallgatter A, Constable RT, Lesch KP. Beyond affect: A role for genetic variation of the serotonin transporter in neural activation during a cognitive attention task. Proc Natl Acad Sci U S A. 2005;102:12224–12229. doi: 10.1073/pnas.0503880102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS, Barch DM, Gur R, Gur R, Pinkham A, Ochsner K. CNTRICS final task selection: social cognitive and affective neuroscience-based measures. Schizophrenia Bull. 2009;35:153–162. doi: 10.1093/schbul/sbn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CS, White TL. Behavioral inhibition, behavioral activation, and affective responses to impending reward and punishment: the BIS/BAS scales. J Pers Soc Psychol. 1994;67:319–333. [Google Scholar]
- Castellanos FX, Proal E. Large-scale brain systems in ADHD: beyond the prefrontal-striatal model. Trends Cogn Sci. 2012;16:17–26. doi: 10.1016/j.tics.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LA, Watson D, Mineka S. Temperament, personality, and the mood and anxiety disorders. J Abnorm Psychol. 1994;103:103–116. [PubMed] [Google Scholar]
- Cloninger CR. A systematic method for clinical description and classification of personality variants: a proposal. Arch Gen Psychiatr. 1987;44:573–588. doi: 10.1001/archpsyc.1987.01800180093014. [DOI] [PubMed] [Google Scholar]
- Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004;60:236–248. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- Costa P, McCrae R. NEO PI-R professional manual. Odessa, FL: Psychological Assessment Resources; 1992. [Google Scholar]
- Coyne JC, Downey G. Social factors and psychopathology: stress, social support, and coping processes. Annu Rev Psychol. 1991;42:401–425. doi: 10.1146/annurev.ps.42.020191.002153. [DOI] [PubMed] [Google Scholar]
- Cremers HR, Demenescu LR, Aleman A, Renken R, van Tol MJ, van der Wee NJA, Veltman DJ, Roelofs K. Neuroticism modulates amygdala-prefrontal connectivity in response to negative emotional facial expressions. Neuroimage. 2010;49:963–970. doi: 10.1016/j.neuroimage.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Curran SM, Murray CM, Van Beck M, Dougall N, O'Carroll RE, Austin MP, Ebmeier KP, Goodwin GM. A single photon emission computerised tomography study of regional brain function in elderly patients with major depression and with Alzheimer-type dementia. Brit J Psychiat. 1993;163:155–165. doi: 10.1192/bjp.163.2.155. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatr. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Morrell MJ, Vogt BA. Contributions of anterior cingulate cortex to behaviour. Brain. 1995;118:279–306. doi: 10.1093/brain/118.1.279. [DOI] [PubMed] [Google Scholar]
- DeYoung CG, Hirsh JB, Shane MS, Papademetris X, Rajeevan N, Gray JR. Testing predictions from personality neuroscience: brain structure and the big five. Psychol Sci. 2010;21:820–828. doi: 10.1177/0956797610370159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Simpson JR, Jr, Todd RD, Reich T, Vannier M, Raichle ME. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- Etkin A. Functional neuroanatomy of anxiety: a neural circuit perspective. Curr Top Behav Neurosci. 2010;2:251–277. doi: 10.1007/7854_2009_5. [DOI] [PubMed] [Google Scholar]
- Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97:11050–11055. doi: 10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- Frodl TS, Koutsouleris N, Bottlender R, Born C, Jäger M, Scupin I, Reiser M, Möller HJ, Meisenzahl EM. Depression-related variation in brain morphology over 3 years: effects of stress? Arch Gen Psychiat. 2008;65:1156–1165. doi: 10.1001/archpsyc.65.10.1156. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. Habits, rituals, and the evaluative brain. Annu Rev Neurosci. 2008;31:359–387. doi: 10.1146/annurev.neuro.29.051605.112851. [DOI] [PubMed] [Google Scholar]
- Grieve SM, Clark CR, Williams LM, Peduto AJ, Gordon E. Preservation of limbic and paralimbic structures in aging. Hum Brain Mapp. 2005;25:391–401. doi: 10.1002/hbm.20115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek T, Kozeny J, Kopecek M, Alda M, Höschl C. Reduced subgenual cingulate volumes in mood disorders: A meta-analysis. J Psychiatry Neurosci. 2008;33:91–99. [PMC free article] [PubMed] [Google Scholar]
- Hamilton JP, Siemer M, Gotlib IH. Amygdala volume in major depressive disorder: A meta-analysis of magnetic resonance imaging studies. Mol Psychiatr. 2008;13:993–1000. doi: 10.1038/mp.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Jovicich J, Salat D, van der Kouwe A, Quinn B, Czanner S, Busa E, Pacheco J, Albert M, Killiany R, Maguire P, Rosas D, Makris N, Dale A, Dickerson B, Fischl B. Reliability of MRI-derived measurements of human cerebral cortical thickness: the effects of field strength, scanner upgrade and manufacturer. Neuroimage. 2006;32:180–194. doi: 10.1016/j.neuroimage.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Hankin BL, Abramson LY. Development of gender differences in depression: an elaborated cognitive vulnerability-transactional stress theory. Psychol Bull. 2001;127:773–796. doi: 10.1037/0033-2909.127.6.773. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, Egan MF, Weinberger DR. Serotonin transporter genetic variation and the response of the human amygdala. Science. 2002;297:400–403. doi: 10.1126/science.1071829. [DOI] [PubMed] [Google Scholar]
- Hurley RS, Losh M, Parlier M, Reznick JS, Piven J. The broad autism phenotype questionnaire. J Autism Dev Disord. 2007;37:1679–1690. doi: 10.1007/s10803-006-0299-3. [DOI] [PubMed] [Google Scholar]
- Jovicich J, Czanner S, Han X, Salat D, van der Kouwe A, Quinn B, Pacheco J, Albert M, Killiany R, Blacker D, Maguire P, Rosas D, Makris N, Gollub R, Dale A, Dickerson BC, Fischl B. MRI-derived measurements of human subcortical, ventricular and intracranial brain volumes: reliability effects of scan sessions, acquisition sequences, data analyses, scanner upgrade, scanner vendors and field strengths. Neuroimage. 2009;46:177–192. doi: 10.1016/j.neuroimage.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Whalen PJ. The structural integrity of an amygdala–prefrontal pathway predicts trait anxiety. J Neurosci. 2009;29:11614–11618. doi: 10.1523/JNEUROSCI.2335-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Gee DG, Loucks RA, Davis FC, Whalen PJ. Anxiety dissociates dorsal and ventral medial prefrontal cortex functional connectivity with the amygdala at rest. Cereb Cortex. 2011a;21:1667–1673. doi: 10.1093/cercor/bhq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Loucks RA, Palmer AL, Brown AC, Solomon KM, Marchante AN, Whalen PJ. The structural and functional connectivity of the amygdala: From normal emotion to pathological anxiety. Behav Brain Res. 2011b;223:403–410. doi: 10.1016/j.bbr.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch K, Pauly K, Kellermann T, Seiferth NY, Reske M, Backes V, Stöcker T, Shah NJ, Amunts K, Kircher T, Schneider F, Habel U. Gender differences in the cognitive control of emotion: an fMRI study. Neuropsychologia. 2007;45:2744–2754. doi: 10.1016/j.neuropsychologia.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Koo MS, Levitt JJ, Salisbury DF, Nakamura M, Shenton ME, McCarley RW. A cross-sectional and longitudinal magnetic resonance imaging study of cingulate gyrus gray matter volume abnormalities in first-episode schizophrenia and first-episode affective psychosis. Arch Gen Psychiat. 2008;65:746–760. doi: 10.1001/archpsyc.65.7.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperberg GR, Broome MR, McGuire PK, David AS, Eddy M, Ozawa F, Goff D, West WC, Williams SC, van der Kouwe AJ, Salat DH, Dale AM, Fischl B. Regionally localized thinning of the cerebral cortex in schizophrenia. Arch Gen Psychiatr. 2003;60:878–888. doi: 10.1001/archpsyc.60.9.878. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- LeDoux J. Rethinking the emotional brain. Neuron. 2012;73:653–676. doi: 10.1016/j.neuron.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus HR, Kitayama S. Culture and the self: Implications for cognition, emotion, and motivation. Psychol Rev. 1991;98:224–253. [Google Scholar]
- Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci. 1997;9:471–481. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Magarinos AM. Stress and hippocampal plasticity: implications for the pathophysiology of affective disorders. Hum Psychopharmacol. 2001;16:S7–S19. doi: 10.1002/hup.266. [DOI] [PubMed] [Google Scholar]
- McNair D, Lorr M, Droppleman L. Manual: profile of mood states. San Diego: Educational and Industrial Testing Service; 1971. [Google Scholar]
- McRae K, Ochsner KN, Mauss IB, Gabrieli JJD, Gross JJ. Gender differences in emotion regulation: an fMRI study of cognitive reappraisal. Group Process Interg. 2008;11:143–162. doi: 10.1177/1368430207088035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–151. doi: 10.1146/annurev.psych.121208.131631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morey RA, Selgrade ES, Wagner HR, 2nd, Huettel SA, Wang L, McCarthy G. Scan-rescan reliability of subcortical brain volumes derived from automated segmentation. Hum Brain Mapp. 2010;31:1751–1762. doi: 10.1002/hbm.20973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Q, Xie J, Wen Z, Weng Y, Shuyun Z. A quantitative MR study of the hippocampal formation, the amygdala, and the temporal horn of the lateral ventricle in healthy subjects 40 to 90 years of age. Am J Neuroradiol. 1999;20:207–211. [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch KP, Faraone SV, Nguyen TT, Schäfer H, Holmans P, Daly M, Steinhausen HC, Freitag C, Reif A, Renner TJ, Romanos M, Romanos J, Walitza S, Warnke A, Meyer J, et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiat. 2010;49:884–897. doi: 10.1016/j.jaac.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen-Hoeksema S, Girgus JS. The emergence of gender differences in depression during adolescence. Psychol Bull. 1994;115:424–443. doi: 10.1037/0033-2909.115.3.424. [DOI] [PubMed] [Google Scholar]
- Ochsner KN. The social-emotional processing stream: five core constructs and their translational potential for schizophrenia and beyond. Biol Psychiat. 2008;64:48–61. doi: 10.1016/j.biopsych.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Ochsner KN. The role of social cognition in emotion. Trends Cogn Sci. 2008;12:65–71. doi: 10.1016/j.tics.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Ongür D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panizzon MS, Fennema-Notestine C, Eyler LT, Jernigan TL, Prom-Wormley E, Neale M, Jacobson K, Lyons MJ, Grant MD, Franz CE, Xian H, Tsuang M, Fischl B, Seidman L, Dale A, Kremen WS. Distinct genetic influences on cortical surface area and cortical thickness. Cereb Cortex. 2009;19:2728–2735. doi: 10.1093/cercor/bhp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, Egan MF, Mattay VS, Hariri AR, Weinberger DR. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8:828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- Phelps EA. Emotion and cognition: insights from studies of the human amygdala. Annu Rev Psychol. 2006;57:27–53. doi: 10.1146/annurev.psych.56.091103.070234. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Phillips ML, Vieta E. Identifying functional neuroimaging biomarkers of bipolar disorder: Toward DSM-V. Schizophr Bull. 2007;33:893–904. doi: 10.1093/schbul/sbm060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception I: the neural basis of normal emotion perception. Biol Psychiatry. 2003;54:504–514. doi: 10.1016/s0006-3223(03)00168-9. [DOI] [PubMed] [Google Scholar]
- Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35:192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P, Ruderfer DM, McQuillin A, Morris DW, O'Dushlaine CT, Corvin A, Holmans PA, Macgregor S, Gurling H, Blackwood DH, Craddock NJ, Gill M, Hultman CM, Kirov GK, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repetti RL, Taylor SE, Seeman TE. Risky families: family social environments and the mental and physical health of offspring. Psychol Bull. 2002;128:330–366. [PubMed] [Google Scholar]
- Reznick JS, Hegeman IM, Kaufman ER, Woods SW, Jacobs M. Retrospective and concurrent self-report of behavioral-inhibition and their relation to adult mental-health. Dev Psychopathol. 1992;4:301–321. [Google Scholar]
- Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, Lin DY, Duan J, Ophoff RA, Andreassen OA, Scolnick E, Cichon S, St. Clair D, Corvin A, Gurling H, Werge T, Rujescu D, Blackwood DH, Pato CN, Malhotra AK, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, Byrne EM, Blackwood DH, Boomsma DI, Cichon S, Heath AC, Holsboer F, Lucae S, Madden PA, Martin NG, McGuffin P, Muglia P, Noethen MM, Penninx BP, Pergadia ML, et al. Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.21. Advance online publication. Retrieved April 3, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, van der Kouwe A, Jenkins BG, Dale AM, Fischl B. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 2002;58:695–701. doi: 10.1212/wnl.58.5.695. [DOI] [PubMed] [Google Scholar]
- Rushworth MFS, Behrens TEJ, Rudebeck PH, Walton ME. Contrasting roles for cingulate and orbitofrontal cortex in decisions and social behaviour. Trends Cogn Sci. 2007;11:168–176. doi: 10.1016/j.tics.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Savitz J, Drevets WC. Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev. 2009a;33:699–771. doi: 10.1016/j.neubiorev.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitz JB, Drevets WC. Imaging phenotypes of major depressive disorder: genetic correlates. Neuroscience. 2009b;164:300–330. doi: 10.1016/j.neuroscience.2009.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saygin ZM, Osher DE, Augustinack J, Fischl B, Gabrieli JD. Connectivity-based segmentation of human amygdala nuclei using probabilistic tractography. Neuroimage. 2011;56:1353–1361. doi: 10.1016/j.neuroimage.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman LJ, Valera EM, Makris N, Monuteaux MC, Boriel DL, Kelkar K, Kennedy DN, Caviness VS, Bush G, Aleardi M, Faraone SV, Biederman J. Dorsolateral prefrontal and anterior cingulate cortex volumetric abnormalities in adults with attention-deficit/hyperactivity disorder identified by magnetic resonance imaging. Biol Psychiat. 2006;60:1071–1080. doi: 10.1016/j.biopsych.2006.04.031. [DOI] [PubMed] [Google Scholar]
- Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, Edenberg HJ, Jr, Nurnberger JI, Rietschel M, Blackwood D, Corvin A, Flickinger M, Guan W, Mattingsdal M, McQuillen A, Kwan P, Wienker TF, Daly M, Dudbridge F, Holmans PA, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielberger C, Gorsuch R. Test manual for the state-trait anxiety inventory. Palo Alto, CA: Consulting Psychologists; 1970. [Google Scholar]
- Strakowski SM, DelBello MP, Sax KW, Zimmerman ME, Shear PK, Hawkins JM, Larson ER. Brain magnetic resonance imaging of structural abnormalities in bipolar disorder. Arch Gen Psychiat. 1999;56:254–260. doi: 10.1001/archpsyc.56.3.254. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiat. 2000;157:1552–1562. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- Thomas KM, Drevets WC, Whalen PJ, Eccard CH, Dahl RE, Ryan ND, Casey BJ. Amygdala response to facial expressions in children and adults. Biol Psychiat. 2001;49:309–316. doi: 10.1016/s0006-3223(00)01066-0. [DOI] [PubMed] [Google Scholar]
- Van Essen DC. A population-average, landmark-and surface-based (PALS) atlas of human cerebral cortex. Neuroimage. 2005;28:635–662. doi: 10.1016/j.neuroimage.2005.06.058. [DOI] [PubMed] [Google Scholar]
- Van Overwalle F. Social cognition and the brain: A meta-analysis. Hum Brain Mapp. 2009;30:829–858. doi: 10.1002/hbm.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn CE, Leff JP. The influence of family and social factors on the course of psychiatric illness. A comparison of schizophrenic and depressed neurotic patients. Brit J Psychiat. 1976;129:125–137. doi: 10.1192/bjp.129.2.125. [DOI] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Langenberg C, Hofmann OM, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TD, Phan KL, Liberzon I, Taylor SF. Valence, gender, and lateralization of functional brain anatomy in emotion: A meta-analysis of findings from neuroimaging. Neuroimage. 2003;19:513–531. doi: 10.1016/s1053-8119(03)00078-8. [DOI] [PubMed] [Google Scholar]
- Walhovd KB, Fjell AM, Reinvang I, Lundervold A, Dale AM, Eilertsen DE, Quinn BT, Salat D, Makris N, Fischl B. Effects of age on volumes of cortex, white matter and subcortical structures. Neurobiol Aging. 2005;26:1261–1270. doi: 10.1016/j.neurobiolaging.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Watson D, Tellegen A. Toward a consensual structure of mood. Psychol Bull. 1985;98:219–235. doi: 10.1037//0033-2909.98.2.219. [DOI] [PubMed] [Google Scholar]
- Watson D, Wiese D, Vaidya J, Tellegen A. The two general activation systems of affect: Structural findings, evolutionary considerations, and psychobiological evidence. J Pers Soc Psychol. 1999;76:820–838. [Google Scholar]
- Wiggins JS, Trapnell P, Phillips N. Psychometric and geometric characteristics of the Revised Interpersonal Adjective Scales (IAS-R) Multivar Behav Res. 1988;23:517–530. doi: 10.1207/s15327906mbr2304_8. [DOI] [PubMed] [Google Scholar]
- Williams LM, Das P, Harris AW, Liddell BB, Brammer MJ, Olivieri G, Skerrett D, Phillips ML, David AS, Peduto A, Gordon E. Dysregulation of arousal and amygdala-prefrontal systems in paranoid schizophrenia. Am J Psychiat. 2004;161:480–489. doi: 10.1176/appi.ajp.161.3.480. [DOI] [PubMed] [Google Scholar]
- Willner P. Validity, reliability and utility of the chronic mild stress model of depression: A 10-year review and evaluation. Psychopharmacology. 1997;134:319–329. doi: 10.1007/s002130050456. [DOI] [PubMed] [Google Scholar]
- Winkler AM, Kochunov P, Blangero J, Almasy L, Zilles K, Fox PT, Duggirala R, Glahn DC. Cortical thickness or grey matter volume? The importance of selecting the phenotype for imaging genetics studies. Neuroimage. 2010;53:1135–1146. doi: 10.1016/j.neuroimage.2009.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]