

Abstract
Embryonal tumors are the most common brain tumors in infants less than 36 months of age. Histologically characterized as undifferentiated small round cell tumors with divergent patterns of differentiation, these include medulloblastoma, the most common form of embryonal tumor, as well as supratentorial primitive neuroectodermal tumor, medulloepithelioma, ependymoblastoma, medullomyoblastoma, melanotic medulloblastoma, and atypical teratoid/rhabdoid tumor. All are similarly aggressive and have a tendency to disseminate throughout the central nervous system. Because of efforts to avoid craniospinal irradiation in an attempt to lessen treatment-related neurotoxicity, management of these tumors in infants is unique. Outcomes remain similarly poor among all the tumor types and, therefore, identification of specific molecular targets that have prognostic and therapeutic implications is crucial. The molecular and clinical aspects of the three most common aggressive infantile embryonal tumors, medulloblastoma, supratentorial primitive neuroectodermal tumor, and atypical teratoid/rhabdoid tumor, are the focus of this review.
Introduction
Embryonal tumors are the most common central nervous system neoplasms in infants less than 36 months of age and are described in the World Health Organization classification scheme as undifferentiated small round cell tumors with divergent patterns of differentiation.1 Tumors within this classification include medulloblastoma, the most common form of embryonal tumor, as well as supratentorial primitive neuroectodermal tumor, medulloepithelioma, ependymoblastoma, medullomyoblastoma, and melanotic medulloblastoma. 1 The atypical teratoid/rhabdoid tumor, which predominantly occurs in infants, had been viewed as a type of medulloblastoma or primitive neuroectodermal tumor, but is now recognized as a molecularly distinct embryonal tumor. 1 Despite sharing common light microscopy features, embryonal tumors appear to evolve by divergent genetic pathways.2 Whether the molecular alterations described in embryonal tumors of older children are consistent in infants remains to be confirmed. All of the infantile embryonal tumors are similarly aggressive and have a proclivity to disseminate throughout the central nervous system early in the course of illness.
The principles of management for infantile central nervous system embryonal tumors are unique because therapeutic alternatives to craniospinal irradiation, the mainstay treatment for both local and distant disease control in older children, are currently under investigation in an attempt to lessen treatment-related neurotoxicity. Outcomes remain similarly poor among all the infantile embryonal tumor types and, therefore, identification of specific molecular targets that have prognostic and therapeutic implications is crucial. The molecular and clinical aspects of the three most common aggressive infantile embryonal tumors, medulloblastoma, supratentorial primitive neuroectodermal tumor, and atypical teratoid/rhabdoid tumor, are the focus of this review.
Medulloblastoma
Medulloblastoma is the most common malignant brain tumor of infancy and comprises approximately 20% of all primary central nervous system tumors occurring in patients less than 18 years of age.3 Medulloblastoma is believed to arise from the precursor cells of the external granule layer of the developing cerebellum and is predominantly composed of densely packed cells with round to oval hyperchromatic nuclei with generally abundant mitotic activity.1
The etiologies of infant medulloblastoma are unknown for most patients. There have not been conclusive links between parental occupations or exposures in the development of medulloblastoma, although in some studies parental pesticide use, occupational contact with hydrocarbons and metals, and exposure to N-nitroso compounds have been linked with a higher likelihood of development of medulloblastoma.4,5 Several familial cancer syndromes predispose to an increase in risk of developing medulloblastoma in infancy, including TP53 germline mutation syndromes (Li-Fraumeni), PTCH mutations resulting in the nevoid basal cell carcinoma syndrome (Gorlin’s), and APC mutations characterized in Turcot’s syndrome.6
In the current World Health Organization classification, medulloblastoma has been histologically divided into two subsets, classic and desmoplastic.1 Classic medulloblastoma is more frequent, and within that grouping, an anaplastic phenotype that seems to overlap with a large-cell variant has received increasing attention (Figure 1).7 Retrospective studies have suggested that the large cell/anaplastic variant may carry a poorer prognosis.8 Desmoplastic medulloblastoma, which appears to be less frequently encountered in the infant population, may be associated with a somewhat better prognosis.9
Figure 1.
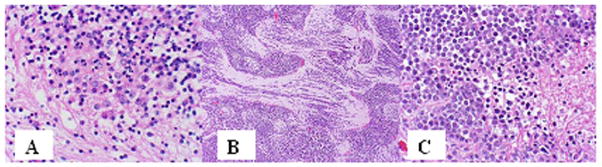
Medulloblastoma histopathologic variants include classic (A), with sheets of undifferentiated small round blue cells, desmoplastic (B), with tumor cells clustered in nodules and separated by stroma, and anaplastic (C), with relatively large undifferentiated tumor cells displaying irregular nuclear size and shape.
Molecular biology
Medulloblastoma is thought to arise from transformed granule cell precursors in the developing cerebellum.10 Under normal development, granule cell precursors undergo massive proliferation in the external granule layer upon receiving the Sonic hedgehog signal from the Purkinje cell. Granule cell precursors then exit the cell cycle and begin to differentiate and migrate downward to form the internal granule layer. Dysregulated granule cell development, including excessive signals for granule cell precursors to proliferate or an absence of signals to cease dividing, can result in the formation of medulloblastoma. Expression of the neurotrophin-3 receptor, which regulates proliferation, differentiation, and cell death of granule cell precursors, was the first molecular alteration shown to have clinical significance in embryonal tumors.11 Its expression level in medulloblastoma directly correlates with favorable outcome and may indicate a more differentiated histology.12
The nevoid basal cell carcinoma syndrome, which is caused by inherited germline mutations of the PTCH gene on chromosome 9q22, accounts for 2% of medulloblastoma. PTCH encodes the Sonic hedgehog receptor Patched1, which normally represses Sonic hedgehog signaling.13 Sonic hedgehog signaling is initiated when Sonic hedgehog binds to Patched1, which releases Smoothened from Patched1-mediated inhibition, resulting in the activation of the downstream growth-promoting transcription factors GLI1 and MYCN. Studies have shown that somatic mutations of PTCH and other members of the Sonic hedgehog pathway that similarly result in constitutive activation of Sonic hedgehog signaling are associated with 10% to 20% of sporadic medulloblastoma.14–17 Medulloblastomas arising from either nevoid basal cell carcinoma syndrome or sporadic Sonic hedgehog pathway mutations are predominantly the desmoplastic variant; however, there is overlap, with 9q22 loss also being seen in some nondesmoplastic tumors.18 These molecular alterations have potential clinical relevance, since PTCH (+/−) heterozygous mice spontaneously develop medulloblastoma, which can be induced to regress with Sonic hedgehog pathway antagonists.19,20 Furthermore, inactivation of the Sonic hedgehog target gene GLI1 has also been shown to significantly reduce medulloblastoma formation in this tumor model.21
Approximately 5% to 10% of medulloblastomas demonstrate amplification of the MYCC oncogene, which has been associated with the large cell anaplastic variant and poor prognosis.9 MYCC overexpression is more common, observed in 30% to 50% of tumors, including other histological subtypes of medulloblastoma.22 Similarly, expression of the tyrosine kinase receptor ERBB2 has been demonstrated in 40% of medulloblastoma, more frequently large cell anaplastic type, and is also an independent poor prognostic indicator.23 OTX1 and OTX2 are transcription factors essential to the developing cerebellum. Amplification of the OTX2 homeobox gene, whose product can be targeted by retinoids, has been identified in large cell anaplastic type medulloblastoma, suggesting that OTX2 is potentially another therapeutically susceptible oncogene in medulloblastoma.24 More recently, OTX1 expression was shown to correlate with desmoplastic medulloblastoma while OTX2 expression correlated with classic medulloblastoma and OTX2 protein was detected in nearly 80% of classic medulloblastoma.25 Interestingly, the OTX2-negative pediatric tumors were found predominantly in children less than 5 years of age.
Gene expression profiling has also provided some important insight into the biology of embryonal tumors. For example, expression analysis has revealed that approximately 15% of sporadic medulloblastoma have activating mutations in the Wingless (Wnt)/beta-catenin signaling pathway and that these alterations in Wnt can be used to identify a molecularly distinct subset of tumors with a more favorable prognosis.26 Upon Wnt binding to its receptor, Frizzled (FRZ), the APC complex is destabilized, liberating beta-catenin to enter the nucleus and activate downstream transcription factors.
Microarray analysis of metastatic (M+) and non-metastatic (M0) medulloblastoma at the time of diagnosis found the platelet-derived growth factor receptor-beta and members of the Ras/MAP kinase pathways to be significantly up-regulated in the M+ tumors.27 These results suggest that the platelet-derived growth factor receptor-mediated signal pathway may be critical in the control of medulloblastoma metastasis. Microarray profiling also revealed that morphologically identical tumors such as medulloblastoma and supratentorial primitive neuroectodermal tumor could be separated based solely on their specific patterns of gene expression and that desmoplastic medulloblastoma was molecularly distinct from classic medulloblastoma, predominantly due to the differential expression of the Sonic hedgehog pathway.2
Loss of genetic material from the short arm of chromosome 17p is the most common cytogenetic abnormality in medulloblastoma, occurring in 35% to 50% of tumors, most commonly as an isochrome 17q. The majority of the remaining 17p deletions are confined to distal markers near 17p13.3. Although it remains controversial, more recent studies have indicated that isochromosome 17q is an independent poor prognostic feature and that, in general, infant medulloblastoma tend to be associated with fewer genetic copy number aberrations compared with medulloblastoma in older children and hence may represent a genetically distinct form.28
Many of the molecular studies performed thus far have included a subset of medulloblastoma from infants, primarily as a means to show that specific molecules are markers of prognosis independent of young age and other high-risk clinical factors. However, no study has definitively confirmed that the incidence, distribution, and clinical impact of the molecular alterations described for medulloblastoma thus far are equally representative of the infant population in general. In fact, some evidence suggests, at least in the cases of genetic copy number aberrations and OTX2, that infant medulloblastoma may be molecularly distinct. This has important clinical implications, as the infant group is typically treated with chemotherapy alone or delayed radiation therapy and is perhaps most in need of defining valid markers that predict response to treatment as well as biologic targets for tumor-specific therapeutic intervention.
Presentation and diagnosis
Children classically present with the signs and symptoms of obstruction of cerebrospinal fluid flow and cerebellar dysfunction. Infants with medulloblastoma may present less characteristically and have intermittent vomiting, macrocephaly, and nonspecific signs of ventricular dilatation, including the “sun-setting” sign where there is an inability to elevate the eyes.29
Neuroimaging characteristically discloses a relatively well-defined mass lesion that arises in the inferior medullary velum/roof of the 4th ventricle and grows anteriorly into the 4th ventricle; it can invade the middle cerebellar peduncle or the dorsal brainstem.29–33 On magnetic resonance imaging (MRI), medulloblastoma are usually homogeneous with low T1 signal and intermediate (between gray matter and white matter) T2 signal; signal is often isointense to gray matter on fluid-attenuated inversion recovery images and hyperintense on diffusion-weighted images (Figure 2).29,30,33 In about 75% of cases, the solid portions of the tumors enhance completely and intensely.31,33 MR spectroscopy findings are similar for all the embryonal tumors. In general, these tumors have very high levels of choline and very low or absent N-acetyl-aspartate peak (decreased N-acetyl-aspartate to creatine ratio). High choline levels indicate a high degree of membrane metabolism, which usually takes place in rapidly proliferating malignant tumors. Lactate and lipid peaks can also be identified as a result of metabolic acidosis and tissue breakdown.
Figure 2.

Axial T2 (A) and contrast-enhanced T1 (B) images of a 3-year-old with a medulloblastoma. The tumor arises from the anterior midline cerebellum and fills the 4th ventricle. Tumor displays low T2 signal and near complete enhancement. Surrounding edema is evident.
Staging
Staging studies are crucial in the evaluation of most embryonal tumors, including medulloblastoma.29 Neuroimaging of the entire neuroaxis coupled with sampling of the cerebrospinal fluid for evidence of tumor dissemination are standard components of medulloblastoma management, since as many as 30% of infant patients will have disseminated disease at the time of diagnosis.29,34,35 Subarachnoid dissemination along the spinal cord is most common, although intracranial spread may be present. If possible, to avoid artifacts, patients with presumed medulloblastoma should undergo neuroaxis neuroimaging prior to surgery.36 The management of medulloblastoma is dependent on staging studies and the age of the patient. Infants less than 3 years of age are all considered to be in the high-risk treatment group.23,29 Molecular genetic parameters found to be associated with poorer prognosis include increased ERBB2, high MYCC expression, and possibly p53 accumulation.23,37–40 Recent work has questioned the utility of any single marker and has suggested that gene profile expression classification based on a multigene model may be more useful.2,23 Anaplastic/large-cell variants have been predominantly linked with more extensive and/or disseminated tumors and associated in retrospective series with poorer outcome.8,9 The identification of molecular markers that predict outcome has blurred the separations between high-risk and average-risk disease groups that have been traditionally based on clinical parameters and raises the possibility that infants may be similarly split into differing categories of disease risk.23,29,39 However, in every study to date, the single most-predictive clinical factor is extent of disease at the time of diagnosis, as independent of age, patients with disseminated disease fare less well.
Treatment and outcome
With present means of surgery, craniospinal radiotherapy, and chemotherapy, between 75% and 90% of children greater than 3 years of age with nondisseminated medulloblastoma are likely to be survivors 5 years after treatment.29,36 In contrast, overall survival for infants remains poor, although some subsets of infants, primarily those with nondisseminated localized disease and favorable molecular genetic markers, have a relatively better prognosis, with survival ranging between 50% and 60% 5 years after diagnosis.41 Of special concern with regard to the treatment of central nervous system tumors in children under the age of 3 is the immaturity of the central nervous system and its sensitivity to the long-term effects of radiation. Thus the volume and dose that can be safely given without causing extensive neurocognitive and endocrinologic sequelae is limited. It is unclear at present whether outcome is poorer in infants because of the inability to administer effective early radiotherapy or whether the biology of infant medulloblastoma is more aggressive.
Surgery
The extent of surgical resection has been associated with outcome in most series, and patients with subtotal resections, especially biopsies, fare less well than those who have undergone a total or near-total resection.42 Approximately 30% of patients will ultimately require a permanent external ventricular drainage.
Radiotherapy
Following surgery, craniospinal radiotherapy remains the single most effective means of treatment for medulloblastoma.43–45 In a recently completed study of more than 400 children with nondisseminated medulloblastoma (age, 3 to 21 years) who were treated with 2400 cGy of radiation and chemotherapy, survival was greater than 80% at 3 years.36 However, there is significant reluctance to irradiate very young children, especially those less than 3 years of age, with craniospinal treatment. After craniospinal radiotherapy, children, especially those younger than 7 years of age, will have significant intellectual compromise, related predominantly, but not completely, to whole-brain radiotherapy.46,47 Two to 3 years after radiotherapy, full-scale intelligence quotients drop 20 to 30 points and the development of significant endocrinologic sequelae are observed in most patients.48–50
With conformal radiotherapy techniques, more precise tumor delivery is possible and studies are underway to determine whether radiotherapy delivered to the primary tumor site with a 1-cm to 2-cm margin will be adequate for disease control in the infant population. Limiting the volume of local radiotherapy will often spare the cochlea and possibly result in less long-term hearing sequelae.
Chemotherapy
Because of the reluctance to use radiotherapy, especially craniospinal radiation therapy in young children, chemotherapy has been extensively explored in children with medulloblastoma who are younger than 3 years of age, and in some studies in children younger than 6 years.51–53 Various multi-agent chemotherapeutic regimens have been used and most have included combinations of an alkylator, platinum agent, and a topoisomerase inhibitor administered intravenously or orally. Outcome with such treatment has been relatively disappointing, resulting in disease control in only 20% to 30% of patients. In some studies, craniospinal and local boost radiotherapy were used after completion of chemotherapy or when the child reached 3 years of age.54 Despite this, overall disease control still remained only in the 30% to 35% range. The majority of children who had long-term benefit had nondisseminated, totally resected disease. Multiple studies are presently underway attempting to improve survival by increasing the intensification of pre-radiation chemotherapy, such as the use of higher doses of chemotherapy supported by peripheral stem cell rescue.55,56 Preliminary results using this strategy are encouraging.
In attempts to make chemotherapy even more effective and potentially avoid having to administer radiation or at least limit the radiation volume delivered, other drugs have been added to these multi-agent approaches, including intravenous and intraventricular methotrexate and intraventricular and intralumbar mafosfamide, an activated cyclophosphamide derivative.41,57–59 There does seem to be a subset of patients who can be treated with chemotherapy alone, and it is likely that the wider availability and application of molecular genetic markers will, in time, better identify this subset. Table 1 highlights the most recent treatment strategies used for infants with medulloblastoma.
Table 1.
Recent Treatment Strategies for Infant Embryonal Tumors
| Treatment | 5-y EFS (%) | (Ref) | |
|---|---|---|---|
| MB | Chemotherapy (vcr/cis/cyclo/vp16 or carbo/ifos/vp16) and delayed RT | 32 +/− 5 | (54) |
| Chemotherapy (vcr/cyclo/carbo/vp16/mtx) and IT mtx | 82 +/− 9 (CR) 50 +/− 13 (PR) 33 +/− 14 (M+) |
(41) | |
| Chemotherapy (no mtx) alone, then HDCT + RT if evidence of progression/relapse | 29 (CR) 6 (PR) 13 (M+) |
(57) | |
| sPNET | Chemotherapy (vcr/cis/cyclo/vp16 or carbo/ifos/vp16) and delayed RT | 17 +/− 6 | (54) |
| Chemotherapy (multi-agent) alone | 14 | (65) | |
| Chemotherapy (multi-agent) +/− HD-mtx followed by HDCT and autologous PBSC | 39 | (70) | |
| ATRT | Chemotherapy (vcr/cis/cyclo/vp16 or carbo/ifos/vp16) and delayed RT | 14 +/− 7 | (54) |
Abbreviations: carbo = carboplatin; CR = complete resection; cyclo = cyclophosphamide; EFS = event-free survival; ifos = ifosfamide; HDCT = high-dose chemotherapy; HD-mtx = high-dose methotrexate; IT = intrathecal; M+ = metastatic disease at diagnosis; mtx = methotrexate; PBSC = peripheral blood stem cell rescue; PR = partial resection; Ref = reference citation; RT = radiotherapy; vcr = vincristine; vp16 = etoposide.
Supratentorial Primitive Neuroectodermal Tumors
Supratentorial primitive neuroectodermal tumors, by definition occurring in the cerebrum or suprasellar region, are composed of undifferentiated or poorly differentiated neuroepithelial cells that may show differentiation along various cell lines.1 Supratentorial primitive neuroectodermal tumors are infrequent, accounting for approximately 2.5% of childhood brain tumors, with peak incidence in the first 3 years of life. Although morphologically indistinguishable from medulloblastoma, cytogenetic and molecular genetic studies have now demonstrated that supratentorial primitive neuroectodermal tumors are distinct.2,60 For example, loss of the tumor suppressor gene PTEN, observed in about 30% of medulloblastoma, has not been detected in supratentorial primitive neuroectodermal tumors.61 As of yet, no study has been reported regarding the molecular characteristics of these tumors in infants with regard to prognosis.
Presentation and diagnosis
Supratentorial primitive neuroectodermal tumors commonly present as focal cortical lesions with associated mass effect. Symptoms prior to diagnosis are usually present for less than 6 months.62 Seizures may occur, but not as frequently as focal neurologic deficits and symptoms and signs of increased intracranial pressure.62 Infants may present with macrocephaly. Neuroimaging studies usually disclose a large, well-defined mass most often located in the fronto-parietal region; supratentorial primitive neuroectodermal tumors can arise either cortically or in the deep periventricular white matter.63 Hemorrhage, necrosis, calcifications, and cyst formation are common; the tumors may be associated with large cystic areas (Figure 3).
Figure 3.
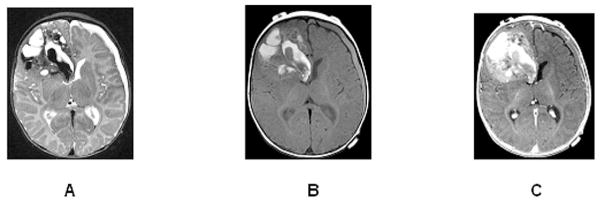
Hemorrhagic primitive neuroectodermal tumor of the right frontal lobe in a 4-month-old infant. Axial T2 (A), axial T1 pre-contrast (B), and post-contrast (C) images show a large mass with significant hemorrhagic products (bright T1 signal before contrast and mixed low and bright T2 signal). The nonhemorrhagic more peripheral components enhance mildly (C). There is no surrounding edema. A subdural hematoma is evident on the left.
Treatment and outcome
As is the case for medulloblastomas, supratentorial primitive neuroectodermal tumors require staging by cerebrospinal fluid cytology and spine MRI evaluation for the identification of subarachnoid dissemination at the time of diagnosis. In most series, 20% or less of infant patients have disseminated disease at the time of diagnosis.
The degree of surgical resection in series of patients with supratentorial primitive neuroectodermal tumors has varied, and in most series the majority of patients, whether infants or young children, have had subtotal resections.62–65 However, the extent of surgical resection has not been shown to correlate with outcome. The presence of leptomeningeal dissemination at the time of diagnosis does portend a poorer rate of survival.
Postsurgical therapy has been similar to that for children with poor-risk medulloblastoma.52,54,62–65 Treatment with chemotherapy alone, predominantly evaluated in children younger than 5 years of age at the time of diagnosis, has resulted in a poor rate of survival. Outcome may be somewhat better after the use of higher-dose chemotherapy supported by autologous bone marrow transplantation or peripheral stem cell rescue (Table 1).55,56,66
The overall outcome for children with supratentorial primitive neuroectodermal tumors is relatively poor. After treatment with chemotherapy alone, children younger than 5 years of age have a 30% or less chance of 5-year survival. As is the case for children with medulloblastoma, infants with supratentorial primitive neuroectodermal tumors are at high risk for long-term neurocognitive sequelae. This is not only due to the whole-brain radiation therapy these children are often treated with, but also due to the local effects of the tumor and the need for higher-dose boost radiotherapy to the primary tumor site, which results in the surrounding brain receiving doses of radiotherapy ranging between 4500 cGy and 5500 cGy.
Atypical Teratoid/Rhabdoid Tumors
The atypical teratoid/rhabdoid tumor is an increasingly diagnosed lesion that was first fully characterized in the 1980s.67 These lesions are composed of rhabdoid cells that are usually intermixed with variable components of primitive neuroectodermal, messenchymal, and epithelial cells. The rhabdoid cell is a medium-sized, round to oval cell with distinct borders, an eccentric nucleus, and a prominent nucleolus (Figure 4). The primitive neuroectodermal component of the atypical teratoid/rhabdoid tumor is indistinguishable from cells found in supratentorial primitive neuroectodermal tumors. Unlike medulloblastoma or supratentorial primitive neuroectodermal tumors, atypical teratoid/rhabdoid tumors display a wide range of immunoreactivity on immunohistochemical staining, with clusters of cells usually positive for epithelial membrane antigen and vimentin. There is also frequent reactivity for glial fibrillary acidic protein (GFAP), cytokeratin, and, to a lesser extent, for smooth muscle actin and neurofilament protein.
Figure 4.

Atypical teratoid/rhabdoid tumor composed of rhabdoid cells intermixed with variable components of primitive neuroectodermal, messenchymal, and epithelial cells. The rhabdoid cell (arrow) is a medium-sized, round to oval cell, with distinct borders, an eccentric nucleus, and a prominent nucleolus.
Molecular biology
Molecular genetic investigations have demonstrated that atypical teratoid/rhabdoid tumors are distinct from other embryonal tumors, in that the vast majority demonstrates monosomy 22 or deletions of chromosome band 22q11. Inactivating deletions or mutations of the tumor suppressor gene hSNF5/INI-1, located in the chromosomal region 22q11.2, are now regarded as a crucial step in the molecular pathogenesis of most atypical teratoid/rhabdoid tumors. However, at least 20% of cases do not have genomic alterations of INI-1, despite showing loss of immunostaining for the INI-1 protein.68 INI-1 encodes a subunit of the SWI/SNF family of chromatin-remodeling complexes, although its direct tumor suppressor function remains unknown.
Presentation and diagnosis
Atypical teratoid/rhabdoid tumors usually present early in life and may constitute as many as 20% of all embryonal tumors. However, the atypical teratoid/rhabdoid tumors have been reported all throughout childhood and into early adulthood. The clinical presentation of atypical teratoid/rhabdoid tumors is indistinguishable from that of medulloblastomas or supratentorial primitive neuroectodermal tumors. About 50% of atypical teratoid/rhabdoid tumors arise in the posterior fossa, 40% are supratentorial, and the rest are pineal, spinal, or multifocal.67 Their location can be intraaxial, extraaxial, or both as they often invade through the meningeal and ependymal boundaries. Leptomeningeal spread occurs in one-third of cases at presentation.
Radiological features are heterogeneous due to the frequent presence of cystic and necrotic areas, calcifications, and hemorrhage. They are usually hyperdense and enhance intensely. With MRI, T1-weighted images often feature hyperintense foci within the lesion because of the hemorrhagic components (Figure 5).
Figure 5.
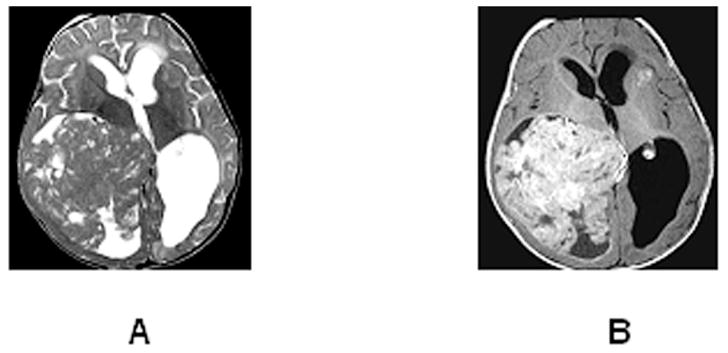
Axial T2 (A) and contrast-enhanced T1 (B) images of an 8-month-old with a large rhabdoid tumor arising in the posterior right lateral ventricle. The mass shows intense enhancement, and has low T2 signal with small islands of high T2 signal that do not enhance (small cysts or necrotic foci). A metastatic lesion is evident deep to the left anterior insula. Hydrocephalus results from posterior 3rd ventricular compression by the tumor.
Treatment and outcome
The management of atypical teratoid/rhabdoid tumors, as is the case with other embryonal tumors, begins with staging. The utility of surgery, radiotherapy, and chemotherapy for children, especially infants with atypical teratoid/rhabdoid tumors, is under active study.67 Overall survival rates for children less than 3 years of age treated with surgery and chemotherapy, independent of the type of chemotherapy used, have been poor, with survival occurring in less than 20% of patients at 12 months from diagnosis. Treatment has consisted of chemotherapy regimens used for children with medulloblastoma or supratentorial primitive neuroectodermal tumors. The use of other types of regimens, including those for patients with sarcomas and regimens including high-dose and intrathecal methotrexate, have demonstrated questionable increased efficacy. In general, studies have shown that a variety of chemotherapeutic regimens may result in tumor stabilization and in some cases objective tumor shrinkages, but have not resulted in long-term disease control; especially in patients with disseminated disease at the time of diagnosis or in those with subtotal resections. Older patients who have survived have been treated with extensive resections, radiotherapy (craniospinal and local boost), and chemotherapy.69 It is unclear whether the better survival in older patients is due to the more aggressive treatment they received or age-related biologic differences. Because of the poorer survival rates in infants, approaches are now focusing on treatment combining aggressive resection with high-dose chemotherapy using methotrexate and, if possible, peripheral stem cell support.70 Table 1 highlights the most recent treatment strategies used for atypical teratoid/rhabdoid tumors.
Acknowledgments
Presented in part at the Neurobiology of Disease in Children Symposium on Central Nervous System Tumors in conjunction with the 36th annual meeting of the Child Neurology Society, Quebec City, Quebec, October 10, 2007.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 3.Crawford JR, MacDonald TJ, Packer RJ. Medulloblastoma in childhood: new biological advances. Lancet Neurol. 2007;6:1073–1085. doi: 10.1016/S1474-4422(07)70289-2. [DOI] [PubMed] [Google Scholar]
- 4.Bunin GR, Kuijten RR, Buckley JD, et al. Relation between maternal diet and subsequent primitive neuroectodermal brain tumors in young children. N Engl J Med. 1993;329:536–541. doi: 10.1056/NEJM199308193290804. [DOI] [PubMed] [Google Scholar]
- 5.McKean-Cowdin R, Preston-Martin S, Pogoda JM, et al. Parental occupation and childhood brain tumors: astroglial and primitive neuroectodermal tumors. J Occup Environ Med. 1998;40:332–340. doi: 10.1097/00043764-199804000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Stavrou T, Bromley CM, Nicholson HS, et al. Prognostic factors and secondary malignancies in childhood medulloblastoma. J Pediatr Hematol Oncol. 2001;23:431–436. doi: 10.1097/00043426-200110000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Brown HG, Kepner JL, Perlman EJ, et al. “Large cell/anaplastic” medulloblastomas: a Pediatric Oncology Group Study. J Neuropathol Exp Neurol. 2000;59:857–865. doi: 10.1093/jnen/59.10.857. [DOI] [PubMed] [Google Scholar]
- 8.Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer. 2002;94:552–560. doi: 10.1002/cncr.10189. [DOI] [PubMed] [Google Scholar]
- 9.Eberhart CG, Kratz J, Wang Y, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol. 2004;63:441–449. doi: 10.1093/jnen/63.5.441. [DOI] [PubMed] [Google Scholar]
- 10.Oliver TG, Read TA, Kessler JD, et al. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 11.Segal RA, Goumnerova LC, Kwon YK, et al. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proc Natl Acad Sci U S A. 1994;91:12867–12871. doi: 10.1073/pnas.91.26.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grotzer MA, Janss AJ, Fung K, et al. TrkC expression predicts good clinical outcome in primitive neuroectodermal brain tumors. J Clin Oncol. 2000;18:1027–1035. doi: 10.1200/JCO.2000.18.5.1027. [DOI] [PubMed] [Google Scholar]
- 13.Rubin JB, Rowitch DH. Medulloblastoma: a problem of developmental biology. Cancer Cell. 2002;2:7–8. doi: 10.1016/s1535-6108(02)00090-9. [DOI] [PubMed] [Google Scholar]
- 14.Zurawel RH, Allen C, Chiappa S, et al. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer. 2000;27:44–51. doi: 10.1002/(sici)1098-2264(200001)27:1<44::aid-gcc6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 15.Hallahan AR, Pritchard JI, Hansen S, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 16.Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 17.Raffel C, Jenkins RB, Frederick L, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842–845. [PubMed] [Google Scholar]
- 18.Dong J, Gailani MR, Pomeroy SL, et al. Identification of PATCHED mutations in medulloblastomas by direct sequencing. Hum Mutat. 2000;16:89–90. doi: 10.1002/1098-1004(200007)16:1<89::AID-HUMU18>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 19.Berman DM, Karhadkar SS, Hallahan AR, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 20.Romer JT, Kimura H, Magdaleno S, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 21.Kimura H, Stephen D, Joyner A, et al. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene. 2005;24:4026–4036. doi: 10.1038/sj.onc.1208567. [DOI] [PubMed] [Google Scholar]
- 22.Herms J, Neidt I, Lüscher B, et al. C-MYC expression in medulloblastoma and its prognostic value. Int J Cancer. 2000;89:395–402. [PubMed] [Google Scholar]
- 23.Gajjar A, Hernan R, Kocak M, et al. Clinical, histopathologic, and molecular markers of prognosis: toward a new disease risk stratification system for medulloblastoma. J Clin Oncol. 2004;22:984–993. doi: 10.1200/JCO.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 24.Di C, Liao S, Adamson DC, et al. Identification of OTX2 as a medulloblastoma oncogene whose product can be targeted by all-trans retinoic acid. Cancer Res. 2005;65:919–924. [PubMed] [Google Scholar]
- 25.de Haas T, Oussoren E, Grajkowska W, et al. OTX1 and OTX2 expression correlates with the clinicopathologic classification of medulloblastomas. J Neuropathol Exp Neurol. 2006;65:176–186. doi: 10.1097/01.jnen.0000199576.70923.8a. [DOI] [PubMed] [Google Scholar]
- 26.Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 27.MacDonald TJ, Brown KM, LaFleur B, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29:143–152. doi: 10.1038/ng731. [DOI] [PubMed] [Google Scholar]
- 28.Pan E, Pellarin M, Holmes E, et al. Isochromosome 17q is a negative prognostic factor in poor-risk childhood medulloblastoma patients. Clin Cancer Res. 2005;11:4733–4740. doi: 10.1158/1078-0432.CCR-04-0465. [DOI] [PubMed] [Google Scholar]
- 29.Packer RJ, Cogen P, Vezina G, et al. Medulloblastoma: clinical and biologic aspects. Neurooncol. 1999;1:232–250. doi: 10.1215/15228517-1-3-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erdem E, Zimmerman RA, Haselgrove JC, et al. Diffusion-weighted imaging and fluid attenuated inversion recovery imaging in the evaluation of primitive neuroectodermal tumors. Neuroradiology. 2001;43:927–933. doi: 10.1007/s002340100603. [DOI] [PubMed] [Google Scholar]
- 31.Vezina LG, Packer RJ. Infratentorial brain tumors of childhood. Neuroimaging Clin N Am. 1994;4:423–436. [PubMed] [Google Scholar]
- 32.Zimmerman RA, Bilaniuk LT, Pahlajani H. Spectrum of medulloblastomas demonstrated by computed tomography. Radiology. 1978;126:137–141. doi: 10.1148/126.1.137. [DOI] [PubMed] [Google Scholar]
- 33.Zimmerman RA, Bilaniuk LT, Rebsamen S. Magnetic resonance imaging of pediatric posterior fossa tumors. Pediatr Neurosurg. 1992;18:58–64. doi: 10.1159/000120644. [DOI] [PubMed] [Google Scholar]
- 34.Evans AE, Jenkin RD, Sposto R, et al. The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg. 1990;72:572–582. doi: 10.3171/jns.1990.72.4.0572. [DOI] [PubMed] [Google Scholar]
- 35.Gajjar A, Fouladi M, Walter AW, et al. Comparison of lumbar and shunt cerebrospinal fluid specimens for cytologic detection of leptomeningeal disease in pediatric patients with brain tumors. J Clin Oncol. 1999;17:1825–1828. doi: 10.1200/JCO.1999.17.6.1825. [DOI] [PubMed] [Google Scholar]
- 36.Rood BR, Macdonald TJ, Packer RJ. Current treatment of medulloblastoma: recent advances and future challenges. Semin Oncol. 2004;31:666–675. doi: 10.1053/j.seminoncol.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 37.Gilbertson RJ, Pearson AD, Perry RH, et al. Prognostic significance of the c-erbB-2 oncogene product in childhood medulloblastoma. Br J Cancer. 1995;71:473–477. doi: 10.1038/bjc.1995.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min HS, Lee YJ, Park K, et al. Medulloblastoma: histopathologic and molecular markers of anaplasia and biologic behavior. Acta Neuropathol. 2006;112:13–20. doi: 10.1007/s00401-006-0073-9. [DOI] [PubMed] [Google Scholar]
- 39.Lamont JM, McManamy CS, Pearson AD, et al. Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res. 2004;10:5482–5493. doi: 10.1158/1078-0432.CCR-03-0721. [DOI] [PubMed] [Google Scholar]
- 40.Ray A, Ho M, Ma J, et al. A clinicobiological model predicting survival in medulloblastoma. Clin Cancer Res. 2004;10:7613–7620. doi: 10.1158/1078-0432.CCR-04-0499. [DOI] [PubMed] [Google Scholar]
- 41.Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352:978–986. doi: 10.1056/NEJMoa042176. [DOI] [PubMed] [Google Scholar]
- 42.Albright AL, Wisoff JH, Zeltzer PM, et al. Effects of medulloblastoma resections on outcome in children: a report from the Children’s Cancer Group. Neurosurgery. 1996;38:265–271. doi: 10.1097/00006123-199602000-00007. [DOI] [PubMed] [Google Scholar]
- 43.Polkinghorn WR, Tarbell NJ. Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol. 2007;4:295–304. doi: 10.1038/ncponc0794. [DOI] [PubMed] [Google Scholar]
- 44.Thomas PR, Deutsch M, Kepner JL, et al. Low-stage medulloblastoma: final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004–3011. doi: 10.1200/JCO.2000.18.16.3004. [DOI] [PubMed] [Google Scholar]
- 45.Taylor RE, Bailey CC, Robinson K, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma:The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21:1581–1591. doi: 10.1200/JCO.2003.05.116. [DOI] [PubMed] [Google Scholar]
- 46.Bailey CC, Gnekow A, Wellek S, et al. Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP II. Med Pediatr Oncol. 1995;25:166–178. doi: 10.1002/mpo.2950250303. [DOI] [PubMed] [Google Scholar]
- 47.Walter AW, Mulhern RK, Gajjar A, et al. Survival and neurodevelopmental outcome of young children with medulloblastoma at St Jude Children’s Research Hospital. J Clin Oncol. 1999;17:3720–3728. doi: 10.1200/JCO.1999.17.12.3720. [DOI] [PubMed] [Google Scholar]
- 48.Ris MD, Packer R, Goldwein J, et al. Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: a Children’s Cancer Group study. J Clin Oncol. 2001;19:3470–3476. doi: 10.1200/JCO.2001.19.15.3470. [DOI] [PubMed] [Google Scholar]
- 49.Radcliffe J, Packer RJ, Atkins TE, et al. Three- and four-year cognitive outcome in children with noncortical brain tumors treated with whole-brain radiotherapy. Ann Neurol. 1992;32:551–554. doi: 10.1002/ana.410320411. [DOI] [PubMed] [Google Scholar]
- 50.Packer RJ, Boyett JM, Janss AJ, et al. Growth hormone replacement therapy in children with medulloblastoma: use and effect on tumor control. J Clin Oncol. 2001;19:480–487. doi: 10.1200/JCO.2001.19.2.480. [DOI] [PubMed] [Google Scholar]
- 51.Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725–1731. doi: 10.1056/NEJM199306173282401. [DOI] [PubMed] [Google Scholar]
- 52.Geyer JR, Zeltzer PM, Boyett JM, et al. Survival of infants with primitive neuroectodermal tumors or malignant ependymomas of the CNS treated with eight drugs in 1 day: a report from the Childrens Cancer Group. J Clin Oncol. 1994;12:1607–1615. doi: 10.1200/JCO.1994.12.8.1607. [DOI] [PubMed] [Google Scholar]
- 53.Dupuis-Girod S, Hartmann O, Benhamou E, et al. Will high dose chemotherapy followed by autologous bone marrow transplantation supplant cranio-spinal irradiation in young children treated for medulloblastoma? J Neurooncol. 1996;27:87–98. doi: 10.1007/BF00146088. [DOI] [PubMed] [Google Scholar]
- 54.Geyer JR, Sposto R, Jennings M, et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. J Clin Oncol. 2005;23:7621–7631. doi: 10.1200/JCO.2005.09.095. [DOI] [PubMed] [Google Scholar]
- 55.Thorarinsdottir HK, Rood B, Kamani N, et al. Outcome for children <4 years of age with malignant central nervous system tumors treated with high-dose chemotherapy and autologous stem cell rescue. Pediatr Blood Cancer. 2007;48:278–284. doi: 10.1002/pbc.20781. [DOI] [PubMed] [Google Scholar]
- 56.Sung KW, Yoo KH, Cho EJ, et al. High-dose chemotherapy and autologous stem cell rescue in children with newly diagnosed high-risk or relapsed medulloblastoma or supratentorial primitive neuroectodermal tumor. Pediatr Blood Cancer. 2007;48:408–415. doi: 10.1002/pbc.21064. [DOI] [PubMed] [Google Scholar]
- 57.Grill J, Sainte-Rose C, Jouvet A, et al. Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol. 2005;6:573–580. doi: 10.1016/S1470-2045(05)70252-7. [DOI] [PubMed] [Google Scholar]
- 58.Kellie SJ, Wong CK, Pozza LD, et al. Activity of postoperative carboplatin, etoposide, and high-dose methotrexate in pediatric CNS embryonal tumors: results of a phase II study in newly diagnosed children. Med Pediatr Oncol. 2002;39:168–174. doi: 10.1002/mpo.10137. [DOI] [PubMed] [Google Scholar]
- 59.Blaney SM, Boyett J, Friedman H, et al. Phase I clinical trial of mafosfamide in infants and children aged 3 years or younger with newly diagnosed embryonal tumors: a pediatric brain tumor consortium study (PBTC-001) J Clin Oncol. 2005;23:525–531. doi: 10.1200/JCO.2005.06.544. [DOI] [PubMed] [Google Scholar]
- 60.Fruhwald MC, O’Dorisio MS, Dai Z, et al. Aberrant hypermethylation of the major breakpoint cluster region in 17p11.2 in medulloblastomas but not supratentorial PNETs. Genes Chromosomes Cancer. 2001;30:38–47. [PubMed] [Google Scholar]
- 61.Inda MM, Mercapide J, Munoz J, et al. PTEN and DMBT1 homozygous deletion and expression in medulloblastomas and supratentorial primitive neuroectodermal tumors. Oncol Rep. 2004;12:1341–1347. [PubMed] [Google Scholar]
- 62.Reddy AT, Janss AJ, Phillips PC, et al. Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer. 2000;88:2189–2193. doi: 10.1002/(sici)1097-0142(20000501)88:9<2189::aid-cncr27>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 63.MacDonald TJ, Rood BR, Santi MR, et al. Advances in the diagnosis, molecular genetics, and treatment of pediatric embryonal CNS tumors. Oncologist. 2003;8:174–186. doi: 10.1634/theoncologist.8-2-174. [DOI] [PubMed] [Google Scholar]
- 64.Cohen BH, Zeltzer PM, Boyett JM, et al. Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Childrens Cancer Group randomized trial. J Clin Oncol. 1995;13:1687–1696. doi: 10.1200/JCO.1995.13.7.1687. [DOI] [PubMed] [Google Scholar]
- 65.Marec-Berard P, Jouvet A, Thiesse P, et al. Supratentorial embryonal tumors in children under 5 years of age: an SFOP study of treatment with postoperative chemotherapy alone. Med Pediatr Oncol. 2002;38:83–90. doi: 10.1002/mpo.1277. [DOI] [PubMed] [Google Scholar]
- 66.Fangusaro J, Finlay Radiation therapy approaches do not currently improve overall survival in young children with sPNET as compared to the Head Start I and II experience. Pediatr Blood Cancer. 2008;50:312–318. doi: 10.1002/pbc.21307. [DOI] [PubMed] [Google Scholar]
- 67.Packer RJ, Biegel JL, Blaney S, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol. 2002;24:337–342. doi: 10.1097/00043426-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 68.Judkins AR, Burger PC, Hamilton RL, et al. INI1 protein expression distinguishes atypical teratoid/rhabdoid tumor from choroid plexus carcinoma. J Neuropathol Exp Neurol. 2005;64:391–397. doi: 10.1093/jnen/64.5.391. [DOI] [PubMed] [Google Scholar]
- 69.Tekautz TM, Fuller CE, Blaney S, et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol. 2005;23:1491–1499. doi: 10.1200/JCO.2005.05.187. [DOI] [PubMed] [Google Scholar]
- 70.Gidwani P, Levy A, Goodrich J, et al. Successful outcome with tandem myeloablative chemotherapy and autologous peripheral blood stem cell transplants in a patient with atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol. 2008 Mar 4; doi: 10.1007/s11060-008-9553-1. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]