

Abstract
T cells of both the CD4 and CD8 lineage are commonly found in affected tissues of patients with idiopathic inflammatory myopathies, but understanding the contribution of these cells to immunopathogenesis remains challenging. Given recent advances in identifying more myositis-associated autoantibodies and their putative targets, we suggest that studies on autoreactive T cells targeting those autoantigens are one way forward. Another (so far, more frequently used) approach comes from studies on effector T cells in the context of myositis. This review summarizes recent advances and current hypotheses in both of these contexts.
Background
Idiopathic inflammatory myopathies (IIMs), also known as myositis, are a group of rheumatic disorders clinically characterized by muscle weakness, leading to disability, decreased quality of life, and a reduced life expectancy. Although this is a relatively rare disease, our under-standing of risk factors and the underlying immuno-pathogenesis has increased substantially in recent years. (See reviews by Betteridge et al. [1], Chinoy et al. [2], and Rayavarapu et al. [3] published in 2011.)
Myositis shares many features with rheumatoid arthritis and systemic lupus erythematosus, namely as different examples of disabling chronic inflammatory syndromes, which can be reevaluated in the light of distinct genetic and environmental contributions [4]. Common traits between these rheumatic disorders include a major histocompatibility complex (MHC) class II association, inflammatory cell infiltrates in affected tissues, and the presence of predictive or disease activity-associated autoantibodies (or both). Taken together, these observations point to a central role for adaptive immune reactions in disease manifestation.
The spectrum of inflammatory myopathies is getting broader, and the classification criteria for the IIMs, designed by Tony Amato on behalf of the Muscle Study Group, proposed the following categories: (i) inclusion body myositis, (ii) polymyositis (PM), (iii) dermatomyositis (DM), (iv) non-specific myositis, and (v) immune-mediated necrotizing myopathy [5]. Some IIMs share common histopathological features of leukocyte infiltration, preferentially T cells and macrophages in skeletal muscle tissue, whereas others display no or spare perivascular and perimysial infiltrates. Novel studies of this latter group which are based on detailed immunopathology suggest that the predominant abnormal histological feature is instead membrane attack complex (MAC) deposition on the sarcolemma in both non-necrotic and necrotic muscle fibers [5,6]. Many patients have manifestations besides in the muscles, such as in the lungs (mostly PM), skin (DM), and sometimes in the joints. Additionally, some patients display more than one rheumatic diagnosis, and systemic sclerosis is the most common connective tissue disease associated with IIM [7].
Both CD4+ and CD8+ T cells have been described to be present and active in patients with myositis. The presence of cytotoxic CD8+ T cells has been attributed to virus or intracellular bacterial infections, which would generate potent effector cells. CD8+ T cells are often subdivided on the basis of their differentiation level, firstly into naïve and activated/memory T cells; the latter subset can be further subdivided in three groups (central memory T cells (TCM), effector memory T cells (TEM) and TEMRA) on the basis of their surface expression of different lymph node homing markers [8]. A summary of candidate infectious agents associated with myositis was published recently [9].
The presence of CD4+ T cells could also be associated with infectious agents, but in the context of myositis, it is more likely that these cells develop as a consequence of an autoimmune reaction [1]. Owing to how immune responses are orchestrated by CD4 cell-derived cytokines, CD4+ T cells are traditionally regarded as helper cells. Indeed, the most common way of subdividing CD4 T cells is based on secretion of specific cytokines, together with activity of so-called master transcription factors. In this fashion, CD4 T cells can be subdivided into different T helper subsets such as Th1, Th2, Th9, Th17, and Th22 and regulatory T (Treg) cells [10]. However, in recent years, it has become clear that CD4+ T cells can also differentiate into cytotoxic effector cells reminiscent of CD8 cells and natural killer (NK) cells [11]. Such cells have been named CD4+CD28null T cells and fall outside of the classic T helper subsets. They represent terminally differentiated cells, which in addition to being potent interferon-gamma (IFNγ) and tumor necrosis factor (TNF) producers have acquired many NK-related receptors and cytotoxic capacity by expressing both perforin and granzymes [12].
It is well established that T cells can be found at all of the different sites of disease manifestations in patients with myositis. But the importance of the presence of these cells is still a matter of debate, as is their antigen specificity. In recent years, an increasing number of myositis-associated and myositis-specific autoantibodies have been identified. The targets of those autoantibodies might represent different candidate autoantigens [1]. Clearly, such data implicate antigens that could also be studied with regard to T-cell function. Below, we will discuss T cells in the three major affected tissues in patients with myositis rather than in subgroups according to the classic disease sub-entities PM, DM, and IBM.
Muscle-infiltrating T cells
The main manifestations, shared by all three subsets of myositis, are proximal muscle weakness and muscle fatigue. Some patients have persistent cellular infiltrates, which are associated with sustained muscle weakness.
How do the first T cells migrate to muscle?
There have been ample efforts in dissecting how and which T cells (and other inflammatory cells) migrate into muscle. Here, chemokines govern the migration of leukocytes to sites of inflammation (Figure 1), and several studies have addressed this issue and demonstrated expression of the α-chemokines CXCL9 and CXCL10 and the β-chemokines CCL2, CCL3, CCL4, CCL19, and CCL21 in IIM muscle. The chemokines can be produced by infiltrating inflammatory cells but potentially also by muscle fibers themselves. The reason why muscle fibers would express chemokines could be infection, trauma, and genetic predisposition.
Figure 1.
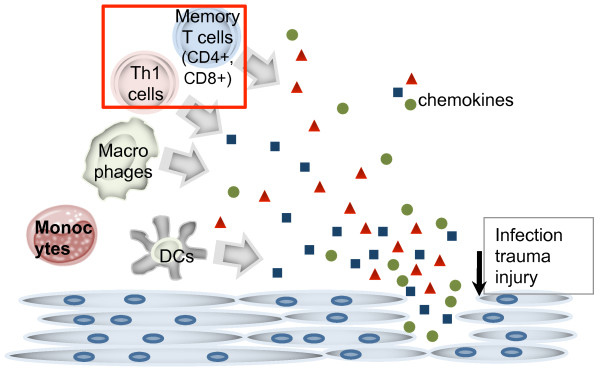
Chemokines regulate the migration of infiltrating cells. Stressed or damaged muscle fibers secrete chemokines, which will attract inflammatory cells. The infiltrating cells will also secrete chemokines that attract even more cells, creating a loop. DC, dendritic cell; Th1, T helper 1.
Although many studies have investigated mRNA from muscle biopsies, making it difficult to elucidate the cellular source of the chemokines, there are data that IIM muscle fibers themselves can produce chemokines. In this context, CCL2 expression has been demonstrated by immuno-histochemical staining of muscle biopsies [13,14] and in myoblast cell cultures in which co-stimulation with IL1β leads to elevated CCL2 mRNA levels [15]. CCL2 is a chemoattractant for CCR2- and CCR4-expressing cells, including monocytes, memory T cells, and dendritic cells. Additional chemokines are interesting in this context, such as CCL3 and CCL4, which are chemoattractants for macrophages and T cells, and CCL3 is a potent regulator of Th1-committed T cells. Indeed, a significant upregulation of CCL3 had been demonstrated in IBM myofibers. Both CCL3 and CCL4 are present in muscle-infiltrating mononuclear cells in DM, PM, and IBM [16].
Similarly to the traffic via high endothelial venules into lymph nodes, CCR7-CCL19 interaction has been suggested to contribute to amplifying/sustaining T-cell traffic to sites of inflammation. Also, muscle fibers positive for this chemokine and infiltrating lymphocytes positive for the receptor have been demonstrated [17]. Further dissection for extranodal lymphoid microstructures has been performed, and indeed such structures can be found [18]. The existence of such lymph node structures indicates that lymphocyte activation and differentiation could take place within the muscle, and there is support from studies of B cells, plasma cells, and immunoglobulin sequences that plasma cell differentiation can take place in this location [19].
A common observation in biopsies of affected muscle from patients with IIM is the focal distribution of the inflammatory infiltrates. The reasons behind this have not yet been delineated, but it is tempting to speculate that migration of the first cells into muscle is a rare event. Once a few inflammatory cells have entered the muscle, a feedback loop is started because of chemokine production by the inflammatory cells.
Which T cells are found in affected muscles?
A long-standing dogma has been that CD8 cells are more common in PM and IBM but that CD4 cells are more pronounced in DM. T-cell receptor (TCR) profiling by performing CDR3 spectratyping of the TCR Vβ chains for CD8+ T cells in PM and IBM demonstrated that CD8 T cells (potentially autoreactive) are clonally expanded and can persist for years [20-22]. A strong bias of Vβ expression in the IBM muscle as compared with the corresponding blood further suggests that the T cells are clonally expanded in situ, or are specifically recruited to the muscle, and may be driven by muscle-specific autoantigens [20]. However, within the Vβ subfamilies, there was a high variability in clonal restriction between patients for both PM [23,24] and IBM [25,26], possibly suggesting the presence of several local autoantigens and epitope spreading. For CD4+ T cells, in contrast, CDR3 spectratyping has revealed many more polyclonal patterns [21]. This, together with the observation that many muscle-infiltrating cells express perforin or granzyme B or both, could be seen as evidence for an exclusively cytotoxic CD8 T cell-mediated immunopathology. However, CD4 T cells have been less studied, but it has been demonstrated that CD4+ T cells can be a major part of the muscle infiltrate [12], so more studies are needed to fully evaluate this. Moreover, today we know that CD4+CD28null T cells predominate the infiltrate in affected muscle and those cells express cytotoxic effector substances [12,27], allowing us to reevaluate older findings in a new context.
FOXP3+ Treg cells are critical cells in maintaining immune homeostasis and preventing autoimmune and chronic inflammatory disease. Therefore, it is appealing to suspect that patients with IIM may have a numerical deficit in Treg cells. This was, however, not the case [28], and similar data exist for other rheumatic diseases. An alternative hypothesis is that the Treg cells are functionally deficient or that the inflammatory milieu does not allow Treg cell suppression as indicated in rheumatoid arthritis [29]. During investigations of biopsies before and after glucocorticoid therapy, both the overall T-cell count and the Treg cell count were reduced after therapy whereas persistent CD4+CD28null T cells could be observed [30].
How could T cells perpetuate local disease?
Cytokine staining of muscle biopsies has demonstrated only modest levels of T cell-derived cytokines. However, since many cytokines function directly on neighboring cells, this could be due in part to rapid consumption. Another interesting effector function is the perforin/granzyme B axis and its effect on muscle fibers (Figure 2). We recently started to address whether CD28null T cells could directly interact with muscle fibers by an autologous co-culture system, and we have preliminary data in support of this [31]. A granzyme attack normally would be expected to induce apoptosis in the affected cell, but one needs to remember that muscle fibers are multi-nucleated cells and, as such, may not undergo classic apoptosis [32]. Instead, one could think that the common observation of regenerating fibers could be a result of such an insult. Such regenerating fibers also express high levels of Jo-1, one of the candidate autoantigens [33]. In the context of CD28null T cells, an additional concern is their longevity, and this subset is the reason why persistent infiltrates are sometimes seen even after aggressive therapy [30].
Figure 2.
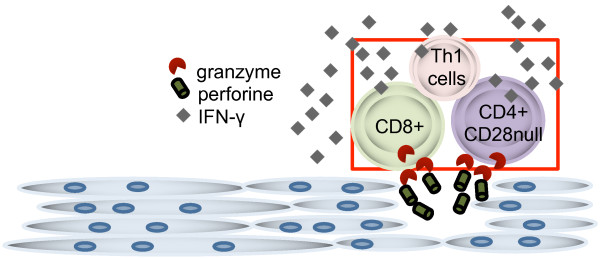
Phenotype and effector functions of muscle-infiltrating T cells. Perforin and granzyme B-positive cells are abundant in mononuclear cell infiltrates in affected muscle. Not only CD8+ but also CD4+CD28null T cells express these effector molecules. IFN-γ, interferon-gamma; Th1, T helper 1.
Skin-infiltrating T cells
DM is the IIM subtype that affects both skin and muscle. Historically, this disease has been looked upon as more CD4-driven than PM and IBM. Granzyme B-expressing T cells have been found in other rheumatic diseases with skin involvement (such as systemic lupus erythematosus) but were found to be low in DM [34]. Also, the degree of FOXP3+ cells has been reported to be low [35,36]. In contrast, type I IFN appears strong in affected skin of patients with DM [37] and could lead to accumulation of CXCR3+ lymphocytes. Indeed, enrichment of CXCR3+ cells has been reported in DM skin [38]. Another interesting T-cell population, CXCR5+ T helper cells, has been studied in peripheral blood of patients with juvenile DM and found to display Th2 and Th17 activities and to be associated with disease activity [39].
Lung involvement in myositis
Interstitial lung disease (ILD) is a heterogeneous group of non-infectious lung disorders characterized by inflammatory cell infiltration and interstitial fibrosis. It remains one of the greatest contributors to morbidity and mortality in myositis [40] and is associated with a poor quality of life for patients with myositis. Myositis-associated ILD is closely linked to the appearance of autoantibodies raised against anti-histidyl tRNA synthetase (Jo1) and the so-called anti-synthetase syndrome [41]. In anti-Jo-1+ patients, there are a few reports on T lymphocytes with specificity toward this particular autoantigen (reviewed in [42]). Additionally, T cells from bronchoalveolar lavage fluid from patients with myositis-associated ILD showed a strong bias toward Vbeta3 T-cell receptor expression as compared with healthy controls, suggesting a specific role for T cells in the development of ILD [43,44].
Since ILD often precedes myositis symptoms, it is suggested that the autoimmune reaction might start in the lungs (Figure 3). Indeed, it has been demonstrated that the Jo1 antigen is highly expressed in the lung compared with other organs [33]. Interestingly, a proteolytically sensitive conformation of Jo-1 is found in the lungs and leads to cleavage by granzyme B [45]. Granzyme B generates unique fragments of this autoantigen. It is suggested that these fragments are taken up by immature dendritic cells, which get activated, mature into professional antigen-presenting cells, and stimulate CD4+ T cells, initiating downstream immune cascades. Both CD8+ T cells and CD4+CD28null T cells may play a role in cytotoxicity in both the muscle and the lungs. They could contribute not only to destruction of target tissues in myositis but also to initiation of autoimmunity through cleavage of Jo1 mediated by granzyme B.
Figure 3.
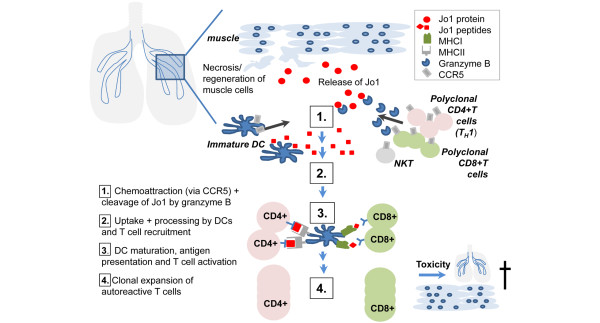
Autoimmunity could be initiated in lung and perpetuated in muscle. Epidemiologic studies suggest cigarette smoking as a key environmental risk factor. Smoking, or other irritants, could initiate Jo-1 cleavage or modification. Granzyme B-cleaved Jo-1 will attract mononuclear cells, which can process and present the Jo-1 autoantigen to the immune system. Similarly, Jo-1 is overexpressed in affected muscle and hence a similar pathway as described for the lung could also perpetuate adaptive immune reactions in the muscle. DC, dendritic cell; NK, natural killer; Th1, T helper 1.
The trigger for the initiation of the autoimmune response has not yet been clarified. On the one hand, it is hypothesized that the inflammatory cascade might be initiated by an infection with an as-yet-unidentified virus (for example, Coxsackie, influenza, HIV, hepatitis C virus, and cytomegalovirus) [42]. Those viruses may enter the respiratory tract, where they may lead not only to cell death and an associated release of Jo-1 antigen into the extracellular space but also to a modification of this enzyme. Another possibility is genetic predisposition to reduced apoptotic clearance or increased apoptosis in conjunction with environmental stimuli such as smoking [46]. The release of Jo1 and its special conformations into the extracellular milieu are believed to trigger not only activation of T cells by antigen-presenting cells but also migration of cells expressing CCR5, including dendritic cells and T cells [47], especially Th1 cells that were shown to express predominantly CCR5 [48]. This may explain an infiltration of mostly Th1 cells in the lung of patients with myositis-associated ILD [49]. In addition, it was shown that there is a disease-specific association between Jo-1, ILD, and serum levels of CXCL9 and CXCL10 [50], two IFN-γ inducible chemokines attracting CXCR3-expressing cells, including NK and Th1 cells, further enhancing the infiltration of pro-inflammatory Th1 cells and emphasizing a role for these cells in disease pathogenesis.
T cell-independent autoantibodies?
IgG autoantibodies are generally believed to be a consequence of T-cell help but could potentially arise in a T cell-independent fashion via the cytokines BAFF (B-cell activating factor) and IL-21 [51]. Moreover, high BAFF levels have been found in the circulation of anti-Jo-1+ patients and an even higher concentration of BAFF is associated with myositis-associated ILD [52]. This leads to a higher appearance of B cells and plasma cells that possibly could locally produce autoantibodies. The autoantibodies are believed to build immune complexes with Jo-1 fragments and nucleic acid released from dying cells, which may activate plasmacytoid dendritic cells (pDCs). These cells are the major source of type I IFN, and IFN-α can be detected in muscle, skin, and peripheral blood of anti-Jo-1+ myositis patients where pDCs are highly enriched [53-55]. IFN-α released by pDCs in turn upregulates expression of BAFF, creating a positive feedback loop and ultimately leading to a break of tolerance. In addition, IFN-α upregulates expression of intercellular adhesion molecule (ICAM) on lung epithelial cells, enhancing recruitment of even more inflammatory cells and further amplifying the inflammatory cascade, and furthermore may contribute toward CD28 downregulation and thereby the appearance of the CD28null phenotype [56].
Since the association between autoimmune IIM and ILD was initially described, 35 years ago, a great deal of knowledge has been added, especially regarding diagnosis and therapy of myositis-associated ILD. The initiating trigger and detailed pathogenesis of this disease remain to be elucidated. Having a closer look at how myositis-associated ILD is initiated and what major key players are involved at what time point not only will improve our understanding of disease mechanisms but also may reveal therapeutic possibilities.
How to continue dissecting T cells in myositis?
Our understanding of T-cell function and regulation is continually growing. Transferring this knowledge to clinical settings can allow the identification of new biomarkers. But to truly understand the contribution of T cells to myositis, we will need focused studies in which patient material is first stratified for autoantibodies and HLA type and in which autoreactive T cells are characterized in detail.
A third, indirect, way of increasing our understanding of T cells in this disorder involves studies before and after different treatment regimes. Assessing T-cell effector functionality at baseline and after a given time of therapy can also reveal T-cell involvement. Ultimately, we may be able to predict which patients with myositis have a disease influenced by T cells and which do not and thereby pave the way for individualized treatment strategies.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a series on Myositis, edited by Ingrid Lundberg, which can be found online at http://arthritis-research.com/series/myositis
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Abbreviations
BAFF: B-cell activating factor; DM: dermatomyositis; IBM: inclusion body myositis; IFN-γ: interferon-gamma; IIM: idiopathic inflammatory myopathy; IL: interleukin; ILD: interstitial lung disease; NK: natural killer; pDC: plasmacytoid dendritic cell; PM: polymyositis; TCR: T-cell receptor; Th: T helper; Treg: regulatory T.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Vivianne Malmström, Email: vivianne.malmstrom@ki.se.
Paulius Venalis, Email: Pauliupaulius.venalis@ki.se.
Inka Albrecht, Email: inka.albrecht@ki.se.
References
- Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther. 2011;14:209. doi: 10.1186/ar3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinoy H, Lamb JA, Ollier WE, Cooper RG. Recent advances in the immunogenetics of idiopathic inflammatory myopathy. Arthritis Res Ther. 2011;14:216. doi: 10.1186/ar3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayavarapu S, Coley W, Nagaraju K. An update on pathogenic mechanisms of inflammatory myopathies. Curr Opin Rheumatol. 2011;14:579–584. doi: 10.1097/BOR.0b013e32834b41d2. [DOI] [PubMed] [Google Scholar]
- Chinoy H, Li CK, Platt H, Fertig N, Varsani H, Gunawardena H, Betteridge Z, Oddis CV, McHugh NJ, Wedderburn LR, Ollier WE, Cooper RG. UK Adult Onset Myositis Immunogenetic Consortium and UK Juvenile Dermatomyositis Research Group. Genetic association study of NF-kappaB genes in UK Caucasian adult and juvenile onset idiopathic inflammatory myopathy. Rheumatology (Oxford) 2012;14:794–799. doi: 10.1093/rheumatology/ker379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, Vencovsky J, de Visser M, Hughes RA. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:337–345. doi: 10.1016/j.nmd.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Stenzel W, Goebel HH, Aronica E. Immune-mediated necrotizing myopathies- a heterogeneous group of diseases with specific myopathological features. Neuropathol Appl Neurobiol. 2012;14:632–646. doi: 10.1111/j.1365-2990.2012.01302.x. [DOI] [PubMed] [Google Scholar]
- Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore) 2005;14:231–249. doi: 10.1097/01.md.0000173991.74008.b0. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;14:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Gan L, Miller FW. State of the art: what we know about infectious agents and myositis. Curr Opin Rheumatol. 2011;14:585–594. doi: 10.1097/BOR.0b013e32834b5457. [DOI] [PubMed] [Google Scholar]
- Noelle RJ, Nowak EC. Cellular sources and immune functions of interleukin-9. Nat Rev Immunol. 2010;14:683–687. doi: 10.1038/nri2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa T, Wagner UG, Goronzy JJ, Weyand CM. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998;14:2108–2116. doi: 10.1002/1529-0131(199812)41:12<2108::AID-ART5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Fasth AE, Dastmalchi M, Rahbar A, Salomonsson S, Pandya JM, Lindroos E, Nennesmo I, Malmberg KJ, Söderberg-Nauclér C, Trollmo C, Lundberg IE, Malmström V. T cell infiltrates in the muscles of patients with dermatomyositis and polymyositis are dominated by CD28null T cells. J Immunol. 2009;14:4792–4799. doi: 10.4049/jimmunol.0803688. [DOI] [PubMed] [Google Scholar]
- Baird GS, Montine TJ. Multiplex immunoassay analysis of cytokines in idiopathic inflammatory myopathy. Arch Pathol Lab Med. 2008;14:232–238. doi: 10.5858/2008-132-232-MIAOCI. [DOI] [PubMed] [Google Scholar]
- De Paepe B, Creus KK, De Bleecker JL. Chemokine profile of different inflammatory myopathies reflects humoral versus cytotoxic immune responses. Ann N Y Acad Sci. 2007;14:441–453. doi: 10.1196/annals.1398.050. [DOI] [PubMed] [Google Scholar]
- Marino M, Scuderi F, Provenzano C, Scheller J, Rose-John S, Bartoccioni E. IL-6 regulates MCP-1, ICAM-1 and IL-6 expression in human myoblasts. J Neuroimmunol. 2008;14:41–48. doi: 10.1016/j.jneuroim.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Barthel K, Wrede A, Salajegheh M, Bahr M, Dalakas MC. Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain. 2008;14(Pt 5):1228–1240. doi: 10.1093/brain/awn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateyama M, Fujihara K, Misu T, Itoyama Y. CCR7+ myeloid dendritic cells together with CCR7+ T cells and CCR7+ macrophages invade CCL19+ nonnecrotic muscle fibers in inclusion body myositis. J Neurol Sci. 2009;14:47–52. doi: 10.1016/j.jns.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Lopez De Padilla CM, Vallejo AN, Lacomis D, McNallan K, Reed AM. Extranodal lymphoid microstructures in inflamed muscle and disease severity of newonset juvenile dermatomyositis. Arthritis Rheum. 2009;14:1160–1172. doi: 10.1002/art.24411. [DOI] [PubMed] [Google Scholar]
- Salajegheh M, Pinkus JL, Amato AA, Morehouse C, Jallal B, Yao Y, Greenberg SA. Permissive environment for B-cell maturation in myositis muscle in the absence of B-cell follicles. Muscle Nerve. 2010;14:576–583. doi: 10.1002/mus.21739. [DOI] [PubMed] [Google Scholar]
- Salajegheh M, Rakocevic G, Raju R, Shatunov A, Goldfarb LG, Dalakas MC. T cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology. 2007;14:1672–1679. doi: 10.1212/01.wnl.0000265398.77681.09. [DOI] [PubMed] [Google Scholar]
- Hofbauer M, Wiesener S, Babbe H, Roers A, Wekerle H, Dornmair K, Hohlfeld R, Goebels N. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc Natl Acad Sci USA. 2003;14:4090–4095. doi: 10.1073/pnas.0236183100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntzing K, Lindberg C, Moslemi AR, Oldfors A. Inclusion body myositis: clonal expansions of muscle-infiltrating T cells persist over time. Scand J Immunol. 2003;14:195–200. doi: 10.1046/j.1365-3083.2003.01251.x. [DOI] [PubMed] [Google Scholar]
- Benveniste O, Chérin P, Maisonobe T, Merat R, Chosidow O, Mouthon L, Guillevin L, Flahault A, Burland MC, Klatzmann D, Herson S, Boyer O. Severe perturbations of the blood T cell repertoire in polymyositis, but not dermatomyositis patients. J Immunol. 2001;14:3521–3529. doi: 10.4049/jimmunol.167.6.3521. [DOI] [PubMed] [Google Scholar]
- O'Hanlon TP, Dalakas MC, Plotz PH, Miller FW. Predominant TCR-alpha beta variable and joining gene expression by muscle-infiltrating lymphocytes in the idiopathic inflammatory myopathies. J Immunol. 1994;14:2569–2576. [PubMed] [Google Scholar]
- Fyhr IM, Moslemi AR, Lindberg C, Oldfors A. T cell receptor beta-chain repertoire in inclusion body myositis. J Neuroimmunol. 1998;14:129–134. doi: 10.1016/S0165-5728(98)00163-5. [DOI] [PubMed] [Google Scholar]
- Dimitri D, Benveniste O, Dubourg O, Maisonobe T, Eymard B, Amoura Z, Jean L, Tiev K, Piette JC, Klatzmann D, Herson S, Boyer O. Shared blood and muscle CD8+ T-cell expansions in inclusion body myositis. Brain. 2006;14(Pt 4):986–995. doi: 10.1093/brain/awl020. [DOI] [PubMed] [Google Scholar]
- Pandya JM, Fasth AE, Zong M, Arnardottir S, Dani L, Lindroos E, Malmstrom V, Lundberg IE. Expanded T cell receptor Vbeta-restricted T cells from patients with sporadic inclusion body myositis are proinflammatory and cytotoxic CD28null T cells. Arthritis Rheum. 2010;14:3457–3466. doi: 10.1002/art.27665. [DOI] [PubMed] [Google Scholar]
- Waschbisch A, Schwab N, Ruck T, Stenner MP, Wiendl H. FOXP3+ T regulatory cells in idiopathic inflammatory myopathies. J Neuroimmunol. 2010;14:137–142. doi: 10.1016/j.jneuroim.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Herrath J, Muller M, Amoudruz P, Janson P, Michaelsson J, Larsson PT, Trollmo C, Raghavan S, Malmstrom V. The inflammatory milieu in the rheumatic joint reduces regulatory T-cell function. Eur J Immunol. 2011;14:2279–2290. doi: 10.1002/eji.201041004. [DOI] [PubMed] [Google Scholar]
- Loell IM, Pandya J, Raghavan S, Zong M, Malmstrom V, Lundberg IE. Persisting CD28(null) T cells, but not regulatory T cells, in muscle tissue of myositis patients after immunosuppressive therapy. Arthritis Rheum. 2011;14:S86–S86. doi: 10.1002/acr.20531. [DOI] [Google Scholar]
- Venalis P, Pandya J, Stache V, Nader G, Malmstrom V, Lundberg IE, Fasth A. CD28null T cells from myositis patients are cytotoxic to autologous muscle cells in vitro. Arthritis Rheum. 2011;14:S323–S324. [Google Scholar]
- Nagaraju K, Casciola-Rosen L, Rosen A, Thompson C, Loeffler L, Parker T, Danning C, Rochon PJ, Gillespie J, Plotz P. The inhibition of apoptosis in myositis and in normal muscle cells. J Immunol. 2000;14:5459–5465. doi: 10.4049/jimmunol.164.10.5459. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, Corse A, Rosen A. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005;14:591–601. doi: 10.1084/jem.20041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi M, Capello F, Bertolino L, Seia Z, Pippione M. Identification of granzyme B-expressing CD-8-positive T cells in lymphocytic inflammatory infiltrate in cutaneous lupus erythematosus and in dermatomyositis. Clin Exp Dermatol. 2009;14:910–914. doi: 10.1111/j.1365-2230.2009.03297.x. [DOI] [PubMed] [Google Scholar]
- Solomon GJ, Magro CM. Foxp3 expression in cutaneous T-cell lymphocytic infiltrates. J Cutan Pathol. 2008;14:1032–1039. doi: 10.1111/j.1600-0560.2007.00969.x. [DOI] [PubMed] [Google Scholar]
- Antiga E, Kretz CC, Klembt R, Massi D, Ruland V, Stumpf C, Baroni G, Hartmann M, Hartschuh W, Volpi W, Del Bianco E, Enk A, Fabbri P, Krammer PH, Caproni M, Kuhn A. Characterization of regulatory T cells in patients with dermatomyositis. J Autoimmun. 2010;14:342–350. doi: 10.1016/j.jaut.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Wenzel J, Schmidt R, Proelss J, Zahn S, Bieber T, Tuting T. Type I interferon-associated skin recruitment of CXCR3+ lymphocytes in dermatomyositis. Clin Exp Dermatol. 2006;14:576–582. doi: 10.1111/j.1365-2230.2006.02150.x. [DOI] [PubMed] [Google Scholar]
- Caproni M, Torchia D, Cardinali C, Volpi W, Del Bianco E, D'Agata A, Fabbri P. Infiltrating cells, related cytokines and chemokine receptors in lesional skin of patients with dermatomyositis. Br J Dermatol. 2004;14:784–791. doi: 10.1111/j.1365-2133.2004.06144.x. [DOI] [PubMed] [Google Scholar]
- Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, Foucat E, Dullaers M, Oh S, Sabzghabaei N, Lavecchio EM, Punaro M, Pascual V, Banchereau J, Ueno H. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;14:108–121. doi: 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottin V, Thivolet-Bejui F, Reynaud-Gaubert M, Cadranel J, Delaval P, Ternamian PJ, Cordier JF. Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur Respir J. 2003;14:245–250. doi: 10.1183/09031936.03.00026703. [DOI] [PubMed] [Google Scholar]
- Mammen AL. Autoimmune myopathies: autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol. 2011;14:343–354. doi: 10.1038/nrneurol.2011.63. [DOI] [PubMed] [Google Scholar]
- Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest. 2010;14:1464–1474. doi: 10.1378/chest.10-0180. [DOI] [PubMed] [Google Scholar]
- Chino Y, Murata H, Goto D, Matsumoto I, Tsutsumi A, Sakamoto T, Ohtsuka M, Sekisawa K, Ito S, Sumida T. T cell receptor BV gene repertoire of lymphocytes in bronchoalveolar lavage fluid of polymyositis/dermatomyositis patients with interstitial pneumonitis. Int J Mol Med. 2006;14:101–109. [PubMed] [Google Scholar]
- Englund P, Wahlstrom J, Fathi M, Rasmussen E, Grunewald J, Tornling G, Lundberg IE. Restricted T cell receptor BV gene usage in the lungs and muscles of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 2007;14:372–383. doi: 10.1002/art.22293. [DOI] [PubMed] [Google Scholar]
- Levine SM, Raben N, Xie D, Askin FB, Tuder R, Mullins M, Rosen A, Casciola-Rosen LA. Novel conformation of histidyl-transfer RNA synthetase in the lung: the target tissue in Jo-1 autoantibody-associated myositis. Arthritis Rheum. 2007;14:2729–2739. doi: 10.1002/art.22790. [DOI] [PubMed] [Google Scholar]
- Chinoy H, Adimulam S, Marriage F, New P, Vincze M, Zilahi E, Kapitány A, Gyetvai A, Ekholm L, Novota P, Remakova M, Charles P, McHugh NJ, Padyukov L, Alfredsson L, Vencovsky J, Lundberg IE, Danko K, Ollier WE, Cooper RG. Interaction of HLA-DRB1*03 and smoking for the development of anti-Jo-1 antibodies in adult idiopathic inflammatory myopathies: a European-wide case study. Ann Rheum Dis. 2012;14:961–965. doi: 10.1136/annrheumdis-2011-200182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard OM, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, Casciola-Rosen L, Härtlein M, Kron M, Yang D, Yiadom K, Dwivedi S, Plotz PH, Oppenheim JJ. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002;14:781–791. doi: 10.1084/jem.20020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C, Dayer JM. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;14:344–345. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- Kurasawa K, Nawata Y, Takabayashi K, Kumano K, Kita Y, Takiguchi Y, Kuriyama T, Sueishi M, Saito Y, Iwamoto I. Activation of pulmonary T cells in corticosteroid-resistant and-sensitive interstitial pneumonitis in dermatomyositis/polymyositis. Clin Exp Immunol. 2002;14:541–548. doi: 10.1046/j.1365-2249.2002.01933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards TJ, Eggebeen A, Gibson K, Yousem S, Fuhrman C, Gochuico BR, Fertig N, Oddis CV, Kaminski N, Rosas IO, Ascherman DP. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum. 2009;14:2183–2192. doi: 10.1002/art.24631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;14:491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- Krystufkova O, Vallerskog T, Helmers SB, Mann H, Putova I, Belacek J, Malmstrom V, Trollmo C, Vencovsky J, Lundberg IE. Increased serum levels of B cell activating factor (BAFF) in subsets of patients with idiopathic inflammatory myopathies. Ann Rheum Dis. 2009;14:836–843. doi: 10.1136/ard.2008.091405. [DOI] [PubMed] [Google Scholar]
- Lopez de Padilla CM, Vallejo AN, McNallan KT, Vehe R, Smith SA, Dietz AB, Vuk-Pavlovic S, Reed AM. Plasmacytoid dendritic cells in inflamed muscle of patients with juvenile dermatomyositis. Arthritis Rheum. 2007;14:1658–1668. doi: 10.1002/art.22558. [DOI] [PubMed] [Google Scholar]
- Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, Pachman LM. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of masT cells and mature plasmacytoid dendritic cells. Arthritis Rheum. 2010;14:2813–2822. doi: 10.1002/art.27529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg IE, Helmers SB. The type I interferon system in idiopathic inflammatory myopathies. Autoimmunity. 2010;14:239–243. doi: 10.3109/08916930903510955. [DOI] [PubMed] [Google Scholar]
- Borthwick NJ, Lowdell M, Salmon M, Akbar AN. Loss of CD28 expression on CD8(+) T cells is induced by IL-2 receptor gamma chain signalling cytokines and type I IFN, and increases susceptibility to activation-induced apoptosis. Int Immunol. 2000;14:1005–1013. doi: 10.1093/intimm/12.7.1005. [DOI] [PubMed] [Google Scholar]