

Abstract
Recent advancements in T cell immunotherapy suggest that T cells engineered with high affinity T cell receptors (TCR) can offer better tumor regression. However, whether a high affinity TCR alone is sufficient to control tumor growth, or the T cell subset bearing the TCR is also important remains unclear. Using the human tyrosinase epitope reactive, CD8 independent, high affinity TCR isolated from MHC class-I restricted CD4+ T cells obtained from tumor infiltrating lymphocytes of a metastatic melanoma patient, we developed a novel TCR transgenic mouse with a C57BL/6 background. This HLA-A2 restricted TCR was positively selected on both CD4+ and CD8+ single-positive (SP) cells. However, when the TCR transgenic mouse was developed with an HLA-A2 background, the transgenic TCR was primarily expressed by CD3+CD4-CD8- double-negative (DN) T cells. TIL 1383I TCR transgenic CD4+, CD8+ and CD4-CD8- T cells were functional and retained the ability to control tumor growth without the need for vaccination or cytokine support in vivo. Furthermore, the HLA-A2+/human tyrosinase TCR double transgenic mice developed spontaneous hair depigmentation and had visual defects that progressed with age. Our data show that the expression of the high affinity TIL 1383I TCR alone in CD3+ T cells is sufficient to control the growth of murine and human melanoma and the presence or absence of CD4 and CD8 co-receptors had little effect on its functional capacity.
Keywords: TCR, transgenic, human, mouse, melanoma, melanocytes, vitiligo
Introduction
Adoptive transfer of tumor infiltrating lymphocytes (TIL) is a promising approach for providing patients with anti-tumor immunity (1). Alternatively, the use of recombinant viral vectors encoding T cell receptor (TCR) genes to engineer normal PBL-derived human T cells to provide patients with a source of autologous tumor reactive T cells is another promising approach that was pioneered by our group (2). We also described the first use of TCR gene-modified T cells in cancer patients (3). This trial, and subsequent trials, suggests that the clinical use of TCR gene-modified T cells for patient treatment is feasible and generally safe (4). However, the therapeutic efficacy of TCR gene-modified T cells has not approached the success observed with TIL (5). Therefore, additional improvements are being actively explored.
Much of what we know about the behavior of T cells in vivo has come from clinical trials and adoptive T cells transfer studies in tumor-bearing mice. In mouse models, the primary source of tumor reactive T cells has come from TCR transgenic mice that target non-self (6) or mouse self-antigens (7, 8) expressed by tumor cells. A recent study has generated transgenic mice with the entire human TCR αβ gene loci, whose T cells express a diverse TCR repertoire similar to humans (9). In the present study, we describe a novel TCR transgenic mouse (designated h3T for human TIL1383I Tyrosinase TCR) developed using the HLA-A2 restricted, tyrosinase368-376 reactive human TIL 1383I TCR that is planned to treat patients with advanced melanoma by utilizing a TCR transduction approach. Since TIL1383I TCR was isolated from an MHC class I restricted CD4+ T cell, the development of transgenic mice bearing this receptor is also important in understanding T cell selection (10). In C57BL/6 mice, we observed that this HLA-A2 restricted TCR was functionally expressed on both CD4+ and CD8+ T cells. When crossed to HLA-A2 transgenic mice, the resulting h3T-A2 mice had their peripheral lymphoid compartments dominated by a novel CD3+CD4-CD8- T cell effector population expressing the TIL 1383I TCR, and the mice developed spontaneous vitiligo. Our data presented here show that h3T and h3TA2 mice are resistant to challenge with HLA-A2+ B16 melanoma cells and that their splenocytes mediated rejection of established tumors in vivo irrespective of the presence of the CD4 and CD8 co-receptors. Because the CD4 and CD8 co-receptors are thought to play an important role in stabilizing the peptide-MHC complex and activation of T cells, the transgenic TCR bearing CD4+, CD8+ and CD4-CD8- T cells obtained from h3T and h3TA2 mouse model provided us a unique opportunity to compare the role of co-receptors in anti-tumor T cell responses.
Material and Methods
Generation of h3T TCR transgenic mice
Transgenic mice bearing TIL1383I TCR reactive to human tyrosinase epitope were developed on a C57BL/6 background. Genomic DNA was isolated from TIL1383I, an HLA-A2+ human tyrosinase368-376 (YMDGTMSQV) specific CD4+ T cell line. Fragments containing the genomic V-J and V-D-J regions of the TCR α- and β-chains were cloned, sequenced, and sub cloned into the TCR cassette vectors described earlier (11) and co-injected into fertilized C57BL/6 embryos yielding transgenic founder lines using the transgenic core facility at the Medical University of South Carolina (MUSC). Animals were maintained in pathogen-free facilities and under the approved procedures of the Institutional Animal Care and Use Committee.
Mice and tumor cells
HLA-A2+ human melanoma 624-MEL and its HLA-A2- variant 624-28-MEL were established earlier at Surgery Branch, NCI (12). T2 cells are transporter-associated protein-deficient and its empty surface HLA-A2 molecules were used for direct presentation of epitopes to the antigen-reactive CTL. B16 (H2b) is a tyrosinase-positive murine melanoma. HLA-A2+ murine B16 melanoma cells (referred as B16-A2 in text) were obtained from Dr. Rolf Kiessling at Karolinska Institute, Stockholm, Sweden. C57BL/6, HLA-A2, Rag-1-/-, SCID/beige mice were obtained from The Jackson Laboratories, MI.
Culture medium and reagents
Human tyrosinase peptide (YMDGTMSQV), murine tyrosinase (FMDGTMSQV), melanoma-associated antigen recognized by T cells (MART-1)27-35 peptide (ELAGIGILTV), and influenza matrix protein (MP)58-66 peptide (GILGFVFTL) were purchased from Genzyme Corporation (Cambridge, MA). Culture medium was Iscove’s Modified Dulbecco’s Medium (GIBCO BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (Gemini Bioproducts, Calabasas, CA). Fluorochrome conjugated antibodies for CD3 (clone 145-2C11), CD4 (clone GK1.5), CD8 (53-6.7), CD25 (clone 3C7), CD69 (H1.2F3), CD44 (clone IM7), CD62L (MEL-14), CD107a (clone 1D4B) were obtained from BioLegend, San Diego, CA. Human Vβ12 antibody (clone S511) was purchased from Thermo Scientific, Rockford, IL. HLA-A2 antibody (clone BB7.2) was obtained from BD Biosciences, San Jose, CA. Major Histocompatibility Complex (MHC) class I tetramers for human tyrosinase368-376 and human gp100209-217 were purchased from Beckman Coulter (Fullerton, CA).
Cytokine release assay
Cytokine release by effector cells was measured by co-culturing 1×104 to 1×105 effector cells in a 1:1 ratio with melanoma tumor cells or peptide-pulsed T2 cells as described previously (13). After 16 to 24 h, culture supernatants were harvested and cytokine concentrations measured by sandwich ELISA per the manufacturer’s protocol (R & D Systems, MN) using a spectrophotometer (BioTek, Winooski, VT).
Flow cytometry
Staining for cell surface markers was performed by incubation of Abs at a 1:100 dilution in FACS buffer (2% FCS, PBS) for 30 min at 4°C. Samples were acquired on a FACScan or FACSCalibur (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (Tree Star, OR). FACSAria cell sorter (BD Biosciences, San Jose, CA) was used to obtain greater than 99% Vβ12+CD4+ or Vβ12+CD8+ T cells from h3T mice splenocytes and Vβ12+CD4-CD8- T cells from h3T-A2 mice splenocytes.
Adoptive transfer and tumor treatment
HLA-A2 mice were subcutaneously injected with 2.5 × 105 B16-A2 cells and palpable tumors were treated with cyclophosphamide (4 mg/mouse) one day before adoptively transferring 1.0 × 106 Vβ12+ freshly isolated splenocytes from h3T or h3T-A2 mice. Tumors were measured with calipers twice a week. For lung metastases experiments, HLA-A2 mice intravenously injected with 2.5 × 105 B16-A2 melanoma cells were administered cyclophosphamide (4 mg/mouse) 4 days after tumor inoculation. One day later, mice were left untreated or received an intravenous adoptive transfer of freshly isolated 1 × 106 Vβ12+ fresh splenocytes from h3T or h3T-A2 mice. Animals were sacrificed 3 weeks after adoptive transfer, and tumor foci were counted. To test the ability of h3T and h3T-A2 derived T cells in treating human tumors SCID/beige mice subcutaneously injected with 5 × 106 624 MEL cells and palpable tumors were treated with intravenous adoptive transfer of 1 × 105 freshly sorted h3T CD4+Vβ12+, h3T CD8+Vβ12+, or h3T-A2 CD4-CD8-Vβ12+ T cells. Tumors were measured with calipers twice a week. All experiments were performed at least twice.
In Situ Reverse Transcriptase Polymerase Chain Reaction
Human Vβ12 TCR mRNA expression was examined in formalin fixed, paraffin embedded eye sections from HLA-A2 and H3T-A2 mice using the in situ RT-PCR methods as previously described (14). After removing the anterior segment of eye and the lens, eyes were further fixed in 4% PFA for another 2 hours. Eyes were transferred to a 30% sucrose solution for 12-14 hours and cut into 10 μm thick cryosections. Further, the tissue was de-waxed in xylene, gradually de-hydrated with alcohol and washed in phosphate buffered saline (PBS) before Proteinase K treatment for 27 minutes at room temperature. Proteinase K treatment was inactivated by incubating slides at 105 °C for 3 min. This was then followed by DNase treatment at 37 °C overnight. The In situ RT-PCR procedure was then performed using the forward primer (5’-ATGCTGAAATCACCCAGAGCCCAA-3’) and reverse primer (5’-TATACAGATGTCTGGGAGGAGGCA-3’). The iScript one-step RT-PCR kit (BioRad, Hercules, CA) was used to form the cDNA copy of the template and to amplify. After amplification, slides were washed in 2X SSC buffer for a total of 3 times and then amplicons were detected by in situ hybridization methods as previously described (14). Hybridizations were performed by using a green-fluorescent probe (ACATGGGCTGAGGCTGATCCATTACT), specific for human Vβ12 chain. Hybridizations were performed using EKONO buffer (G-Biosciences, Maryland Heights, MO) at 95°C for 5 min, then 42°C overnight. RT-PCR and hybridization reactions were performed in a MJR-BioRad-Twin Tower, PTC-200 thermocycler (Waltham, MA). The cycling reactions started with 30 min at 50°C, then 95°C for 5 min (cDNA step) followed by 42 cycles of cDNA amplification at 95°C denaturing, 60 °C annealing, and 72°C extension. Subsequently, images were acquired using Olympus BX61 motorized microscope equipped with Olympus DP72 digital camera and CellSens digital imaging software.
Histology
Eight μm cryosections were cut from Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetek USA, Torrance, CA) embedded tissue of pigmented, non-transgenic control mice and depigmented, h3T-A2 double transgenic mice, fixed in cold acetone. After blocking with a specific antibody binding using Superblock and Biotin block (ScyTek, Logan, UT), sections were stained with mouse monoclonal antibody Ta99 to TRP-1 (Covance, Princeton, NJ) and biotinylated Armenian hamster monoclonal antibody to CD3ε (BD Pharmingen, San Jose, CA), followed by a combination of alkaline phosphatase-labeled secondary goat antiserum to mouse IgG2a (Southern Biotech, Birmingham, AL) and horseradish peroxidase-labeled streptavidin (Southern Biotech, Birmingham, AL). Immunoenzymatic staining was completed using Fast blue BB as a substrate for alkaline phosphatase (Sigma, St. Louis, MO), followed by detection of peroxidase using aminoethylcarbazole (Sigma, St. Louis, MO) with hydrogen peroxide in sodium acetate buffer essentially as previously described (15). Following the similar procedure as described above immunohistochemical staining of mouse eyes, the sections were performed using CD3ε-Biotinylated (BD Pharmingen, 145-2C11), CD25-Biotinylated (PC61, BioLegend), Trp-1 (Covance, Ta99), CD68 (FA-11, abcam), and Vβ12-FITC (S511, Thermo Fisher Scientific) and detected using secondary antibodies Strepavidin-HRP (Southern Biotech), anti-Mouse IgG2a-HRP (Southern Biotech), Anti-Rat-HRP (Southern Biotech), and anti-FITC-HRP (MyBioSource), respectively. Slides were developed in 3-Amino-9-ethylcarbazole (Sigma-Aldrich, A5754) substrate solution to detect antigens and counterstained with haematoxylin for nuclear localization.
Mouse melanocyte culture
Strips of HLA-A2 transgenic or C57BL/6 mouse skin adding up to 3 × 2 cm were incubated in a cocktail of 30 U/ml DNAse-I (Roche Diagnostics Corporation, Indianapolis, IN), 50 μg/ml thermolysin, 0.1 mg/ml trypsin and 0.5 mg/ml collagenase IV (all from Sigma, St. Louis, MO), in RPMI (Mediatech Inc., Manassas, VA) shaking overnight at room temperature. Tissue was passed through a 70 μm cell strainer (BD Biosciences, Bedford, MA) and resulting cells were plated in Ham’sF12 (Hyclone Laboratories Inc., Logan, UT) medium with 10% FBS (Mediatech Inc., Manassas, VA), 0.1% bovine pituitary extract (Invitrogen, Carlsbad, CA), 16 nM phorbol 12 myristate 13 acetate (Sigma), 0.1 mM isobutyl methyl xanthine (Sigma), 100 IU/ml penicillin/ 100 μg/ml streptomycin/ 100 μg/ml amphotericin (Invitrogen, Carlsbad, CA). Established cultures were treated with 50 μg/ml G418 sulphate (Invitrogen, Carlsbad, CA) for 4 days to remove cells other than melanocytes that are insensitive to this agent (16). Cells were split 1:4 at confluency and used in experiments.
Electroretinography (ERG)
To quantitate retinal function, we performed ERGs as previously described (17). Briefly, mice were dark-adapted overnight, and the following day they were anesthetized with ketamine and xylazine. Pupils were dilated with a phenylephrine HCl (2.5%) and tropicamide (1%) (Akorn, Inc., Buffalo Grove, IL). A needle ground-electrode was placed subcutaneously in the back of the animal and a reference electrode on the tongue. A stimulus intensity-series of ERGs was recorded in response to single-flash intensities using 40 dB attenuation (low-intensity flash) through no attenuation (high-intensity flash). The single-flash responses were an average of 3 flashes with an inter-stimulus interval of 2 minutes to ensure that ERG amplitudes at a given intensity were identical between the first and the last flash. Each single-flash ERG response was measured using a contact lens containing a gold-ring electrode held in place by a drop of methylcellulose. Electroretinograms were recorded using UTAS-2000 system (LKC Technologies, Gaithersburg, MD). Corneal electrical responses to a single, 10 μs white-light flash were delivered by a Ganzfeld stimulator.
Results
Development and characterization of h3T mice
The TIL 1383I TCR has high affinity for HLA-A2/tyrosinase368-376 complexes and T cells transduced to express this TCR could make potent anti-tumor effectors for treating melanoma patients. However, what remains unclear is how this unique MHC class I restricted TCR developed and how effective T cells expressing the TIL 1383I TCR would be in treating patients with advanced melanoma.
To address these and other questions, we generated a TCR transgenic mouse encoding the TIL 1383I TCR and named it h3T (short for hTTT or human TIL 1383I Tyrosinase TCR). The h3T mouse was developed with the C57BL/6 background so the T cells would be syngeneic with the mouse B16 melanoma and to take advantage of the numerous transgenic and knockout strains available with the H-2b haplotype. As shown in Fig. 1A the human Vβ12 transgene was detected on double-positive (DP) thymocytes in transgenic (h3T) but not in non-transgenic (C57BL/6) littermates, indicating the human TCR transgenes could be expressed on mouse T cells. Analysis of the thymocyte subsets revealed that, compared to C57BL/6 littermates, h3T mice have an increased frequency of CD4-CD8- DN (23.3% vs. 2.4%) thymocytes, a decreased frequency of CD4+CD8+ DP (69.8% vs. 87.3%) and CD4+CD8- SP (4.2% vs. 8.0%) thymocytes, and a similar frequency of CD4-CD8+ SP (2.7% vs. 2.4%) thymocytes (Fig. 1B). These results suggested that the expression of the TIL 1383I TCR affected T cell development in the h3T mouse.
FIGURE 1. Expression of human tyrosinase TCR on h3T mouse T cells and its function.
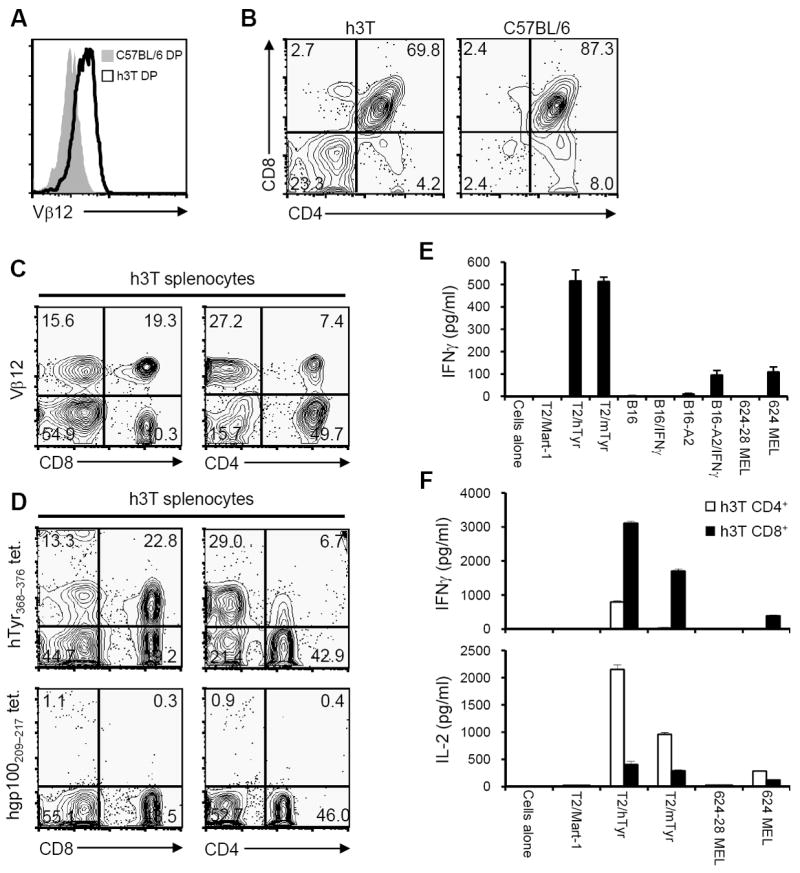
A). Thymocytes from h3T and C57BL/6 control mice were stained using a TCR subunit specific human Vβ12 antibody. B). Thymocytes from h3T and C57BL/6 mice were stained with fluorochrome conjugated anti-CD4 and anti-CD8 antibody to determine single positive, double positive and double negative populations. C). h3T splenocytes were stained with fluorochrome conjugated anti-CD4, anti-CD8 followed by Vβ12 antibody. D). h3T splenocytes were stained with fluorochrome conjugated anti-CD4, anti-CD8 followed by human tyrosinase368-376 epitope reactive cognate tetramer (upper panel) or human glycoprotein100209-217 epitope reactive control tetramer reagent (lower panel). E). IFN-γ was measured by ELISA using supernatant obtained after overnight stimulation of h3T splenocytes as effectors and human or mouse tyrosinase peptide pulsed T2 cells, HLA-A2 transduced mouse melanoma B16 used untreated or after treatment with IFN-γ overnight, HLA-A2+ human melanoma 624 MEL as cognate stimulators. MART-127-35 peptide pulsed T2 cells, mouse melanoma B16 untreated or after treatment with IFN-γ overnight and HLA-A2- human melanoma 624-28 MEL served as controls. F). Supernatants obtained after overnight stimulation of sorted Vβ12+CD4+ and Vβ12+CD8+ effector T cells from h3T mouse were co-cultured with human or mouse tyrosinase peptide pulsed T2 cells and HLA-A2+ human melanoma 624 MEL as stimulators. Flu-MP58-66 peptide pulsed T2 cells and HLA-A2- human melanoma 624-28 MEL served as controls. Data shown in A-E are from one representative of five experiments performed and in F from one of two experiments.
Given the altered thymocyte distribution in the h3T mice, we examined human Vβ12 expression on their mature T cells. Despite lacking HLA-A2 expression, Vβ12+CD8+ and Vβ12+CD4+ T cells were present in the spleens of h3T mice (Fig. 1C). These mouse CD4 and CD8 SP T cells expressed the properly assembled human TIL 1383I TCR as indicated by staining with HLA-A2/tyrosinase368-376 tetramers but not control HLA-A2/gp100209-217 tetramers (Fig. 1D). Therefore, the TIL1383I TCR was expressed on the surface of mature T cells in h3T mice even in the absence of HLA-A2 expression in their thymus.
To determine if h3T T cells were antigen reactive, unfractionated splenocytes from h3T and C57BL/6 littermates were co-cultured overnight with a panel of stimulators, and the amount of interferon-γ (IFN-γ) released was measured using ELISA. T2 cells loaded with the mouse or human tyrosinase peptide stimulated h3T splenocytes to secrete IFN-γ (Fig. 1E). More importantly, h3T splenocytes recognized HLA-A2+ B16-A2 (mouse) and 624 MEL (human) cells but not HLA-A2- B16 and 624-28 MEL cells. In addition to IFN-γ, h3T T cells were poly-functional and secreted cytokines IL-2, TNF-α, GM-CSF upon antigen stimulation (Supplementary Fig. S1A). Importantly, the cytokine secretion observed was from h3T derived naïve T cells that had not underwent any prior activation with cytokines or TCR engagement as required in the other widely used transgenic mouse model Pmel that expresses a mouse MHC class I restricted TCR specific for gp10025-33 (7). Of particular importance is the fact that these fresh naïve splenic T cells recognized naturally processed antigen on the surface of tumor cells. Also, despite developing in the thymus of H-2b mice, they do not recognize B16 melanoma cells (Fig. 1E) or other H-2b tumor cell lines (not shown). These results indicate that h3T mice possess multi-functional transgenic T cells with the HLA-A2 restricted tyrosinase reactivity of the parent TIL 1383I T cells.
The TIL 1383I TCR is CD8-independent meaning that both CD4+ and CD8+ T cells transduced to express the TIL 1383I TCR can recognize HLA-A2+ melanoma cells. Therefore, it was important to determine if both the CD4+ and CD8+ T cell subsets from h3T mice were functional. Spleens from h3T mice were FACS sorted to purify the individual Vβ12+CD8+ and Vβ12+CD4+ T cell populations for use in functional assays. When co-cultured with mouse or human tyrosinase peptide loaded T2 cells or 624-MEL tumor cells, the purified CD4+ T cells principally secreted IL-2 and the CD8+ T cells principally secreted IFN-γ (Fig. 1F). And finally, despite being able to recognize processed antigen presented on tumor cells, h3T T cells are similar to the parent TIL1383I T cells and TIL 1383I TCR transduced CD4+ and CD8+ T cells having relatively low functional avidity by requiring T2 cells to be loaded with ≥10 nM peptide to stimulate IFN-γ or IL-2 secretion (Supplementary Fig. S1B). Therefore, despite lacking HLA-A2 expression, h3T mice contain multi-functional CD4+ and CD8+ T cells that have properties similar to TIL 1383I TCR transduced CD4+ and CD8+ human T cells.
Influence of HLA-A2 expression on transgenic T cells in h3T mice
Given that h3T T cells were positively selected in the absence of HLA-A2 expression in C57BL/6 mice, the next question was to assess the impact of HLA-A2 on T cell development and function. We crossed h3T mice to HLA-A2 transgenic mice (expressing the complete HLA-A2 molecule) and analyzed the resulting progeny (h3T-A2). In contrast to h3T mice, the human Vβ12 transgene was expressed on the DN thymocytes in h3T-A2 mice but not on DP thymocytes (Fig. 2A). These DN cells represented a higher proportion of the h3T-A2 thymus compared to their HLA-A2 non-transgenic littermates (~60% vs. 6%, Fig. 2B). The increased frequency of DN cells in the thymus of h3T-A2 mice appeared to be at the expense of the DP cells (~26% vs. 68%) with only a slight reduction in the CD4 SP (~7% vs. 18%) and CD8 SP (~3% vs. 6%) cells. These results indicate that HLA-A2 expression had a profound effect on T cell development in the thymus of h3T-A2 mice.
FIGURE 2. Expression of human tyrosinase TCR on h3T-A2 mouse T cells and its function.
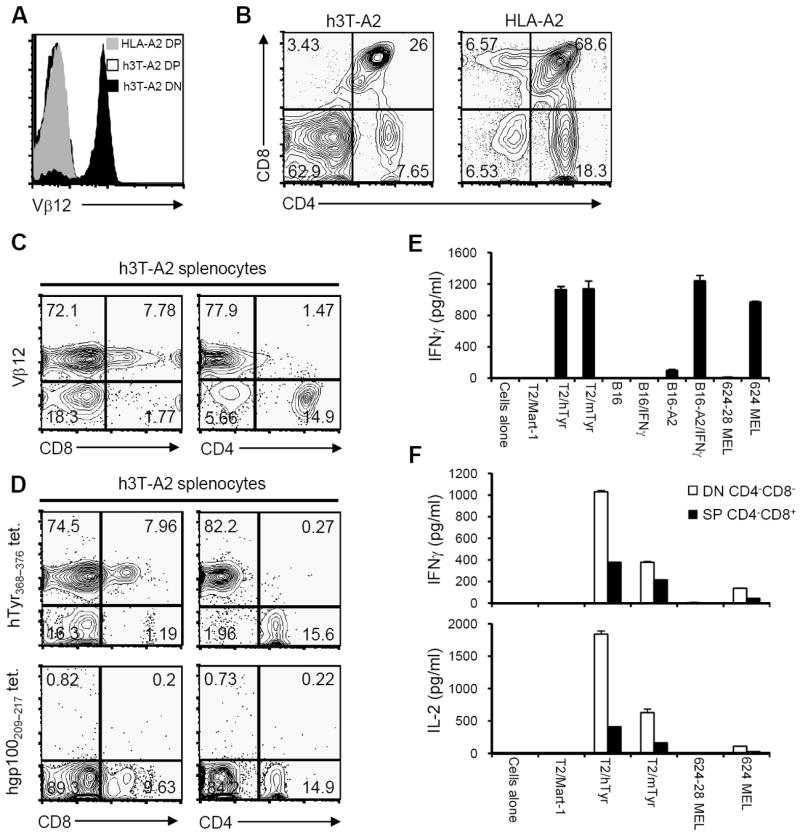
A). Thymocytes from h3T-A2 and HLA-A2 control mouse were stained using TCR specific human Vβ12 antibody. Vβ12 expression on CD4+CD8+ positive cells was determined on HLA-A2 mice (grey histogram) and h3T-A2 mice (open histogram). Black histogram represents Vβ12 expression on CD4- CD8- cells from h3T-A2 mice. B). Thymocytes from h3T-A2 and HLA-A2 mice were stained with fluorochrome conjugated anti-CD4 and anti-CD8 antibody to determine single positive, double positive and double negative populations. C). h3T-A2 splenocytes were stained with fluorochrome conjugated anti-CD4, anti-CD8 followed by Vβ12 antibody. D). h3T-A2 splenocytes were stained with fluorochrome conjugated anti-CD4, anti-CD8 followed by human tyrosinase368-376 epitope reactive cognate tetramer (upper panel) or human glycoprotein100209-217 epitope reactive control tetramer reagent (lower panel). E). IFN-γ was measured by ELISA using supernatant obtained after overnight stimulation of h3T-A2 splenocytes as effectors and human or mouse tyrosinase peptide pulsed T2 cells, HLA-A2 transduced mouse melanoma B16 used untreated or after treatment with IFN-γ overnight or HLA-A2+ human melanoma 624 MEL as cognate stimulators. MART-127-35 peptide pulsed T2 cells, mouse melanoma B16 used untreated or after treatment with IFN-γ overnight and HLA-A2- human melanoma 624-28 MEL served as controls. F). Supernatant using FACS sorted Vβ12+CD4-CD8- double negative T cells from h3T-A2 mouse was obtained as mentioned in E) and different cytokine were evaluated using the BD Cytometric Bead Array System. Data shown in A-E are from one representative of five experiments performed and in F from one of two experiments.
Because of the altered thymocyte distribution in h3T-A2 mice, we examined the distribution of Vβ12+ T cells in their spleens and lymph nodes. The vast majority of the T cells (~80%) in the spleen of h3T-A2 mice expressed the human Vβ12 transgene (Fig. 2C). However, only 8-10% of the splenic T cells were CD8+ SP T cells with very few (generally <1%) being CD4+ SP T cells. The remaining Vβ12+ T cells in the spleen of h3T-A2 mice (generally >90%) expressed neither the CD4 nor CD8 co-receptor. Four color staining with anti-Vβ12, anti-CD3, anti-CD4, and anti-CD8 determined the majority of the Vβ12+ T cells were indeed CD4/CD8 DN T cells with a minor population of CD8+ SP T cells whereas the Vβ12- T cells were mostly SP T cells with the majority being CD4+ T cells (Supplementary Fig. S2A). As seen with the h3T mice, expression of the Vβ12 transgene represented properly assembled TIL 1383I TCR’s since the same proportion of Vβ12+ T cells (approximately 80%) stained with HLA-A2/tyrosinase368-376 tetramers but not control HLA-A2/gp100209-217 tetramers (Fig. 2D). This tetramer staining was CD8 co-receptor independent: both the CD8+ and CD8- T cells, obtained from h3T and h3T-A2 mice respectively, stained with the HLA-A2/tyrosinase368-376 tetramers. Despite their expression of HLA-A2 and tyrosinase, h3T-A2 mice had a minor population of Vβ12+ CD8+ SP T cells and a dominant population of Vβ12+CD4-CD8- DN T cells in their periphery. These results indicate that while HLA-A2 expression does affect development of T cells bearing the high affinity TIL 1383I TCR that somehow evade negative selection in the thymus and are positively selected to the periphery.
Functionally, h3T-A2 T cells are similar to h3T T cells in that fresh splenocytes secrete IFN-γ (Fig. 2E) and other cytokines (Supplementary Fig. S2B) when stimulated with tyrosinase peptide loaded T2 cells or HLA-A2+ tyrosinase+ tumor cells. Despite the high affinity TIL 1383I TCR and the expression of HLA-A2 during T cell development, h3T-A2 mice possess multi-functional tumor reactive T cells in their periphery. Given the altered T cell subset distribution in h3T-A2 mice, we wanted to determine if the Vβ12+ CD8+ T cells and in particular the Vβ12+ DN T cells were functional. h3T-A2 splenocytes were FACS sorted to purify the Vβ12+ CD8+ T cells and the Vβ12+ DN T cells for use in functional assays. The Vβ12+ DN T cells secreted IFN-γ, IL-2, TNF-α and GM-CSF when stimulated with tyrosinase368-376 peptide loaded T2 cells and HLA-A2+ human melanoma cells (Fig. 2F). The Vβ12+ CD8+ T cells secreted low levels of IFN-γ and TNF-α and no IL-2 when stimulated with peptide pulsed targets and melanoma cells. These results indicate that despite the expression of a CD8-independent TCR with high affinity for antigen and the HLA-A2 restriction element, h3T-A2 mice have functional transgenic CD8+ T cells. Also, these mice have a unique population of DN T cells that are HLA-A2 restricted and tyrosinase reactive.
Phenotypic characterization of h3T and h3T-A2 splenocytes
To investigate any phenotypic differences between the transgenic TCR-bearing splenic T cells from both h3T and h3T-A2 mice, we evaluated baseline levels of the activation/effector markers CD25, CD44 and CD69. Transgenic h3T mouse-derived Vβ12+CD4+ T cells showed a slightly higher level of CD25 expression and a reduced CD62L expression as compared to h3T Vβ12+CD8+ and h3T-A2 Vβ12+CD4-CD8- (Fig. 3A). Thus, h3T Vβ12+CD8+ and h3T-A2 Vβ12+CD4-CD8- T cells exhibited a naïve phenotype while h3T Vβ12+CD4+ T cells showed and activated phenotype. Further, the expression level of human Vβ12 on various subsets of T cells from h3T and h3T-A2 mice were found to be almost similar (Fig. 3B). An evaluation of the cytolytic ability as measured by up-regulation of CD107a expression upon antigen stimulation revealed that TIL1383I TCR bearing Vβ12+CD4+ T cells, h3T Vβ12+CD8+ T cells and h3T-A2 Vβ12+CD4-CD8- are cytolytic-although not to the same extent (Fig. 3C). However, despite having large numbers of functional cytolytic tyrosinase reactive CD4+ and CD8+ T cells in their periphery, h3T mice remain pigmented throughout their life. In contrast, h3T-A2 mice are born pigmented and starting about the time of weaning (3-4 weeks of age) they begin to exhibit vitiligo that progresses with age, leading to complete depigmentation (Fig. 3D).
FIGURE 3. Phenotypic characterization of h3T and h3T-A2 splenocytes.
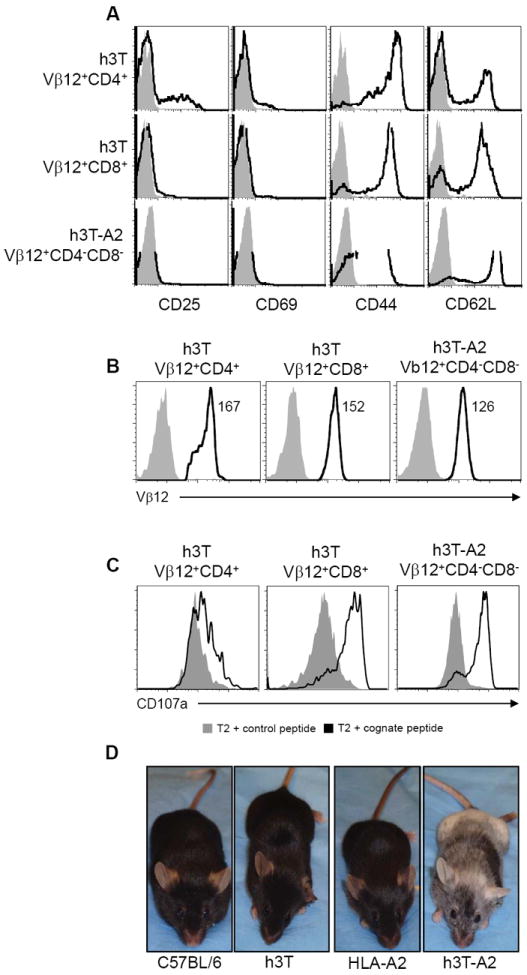
A). Splenocytes from h3T and h3T-A2 mouse were stained using fluorochrome conjugated TCR specific human anti-Vβ12, anti-CD4, anti-CD8 antibody along with the indicated cell surface antibody. Grey histogram indicates staining with isotype control and open histogram indicate the cell surface expression level on the gated T cells as indicated on left. B). Splenocytes from h3T and h3T-A2 mouse were stained using fluorochrome conjugated TCR specific human anti-Vβ12, anti-CD4, anti-CD8 antibody. Grey histogram indicates staining with isotype control and open histogram indicate the Vβ12 expression level on the gated T cells as indicated on top. C). h3T and h3T-A2 splenocytes were stimulated overnight with human tyrosinase cognate peptide-pulsed T2 cells. CD107a expression was determined by flow cytometry on Vβ12+CD4+, Vβ12+CD8+ and Vβ12+CD4-CD8- T cells. MART-127-35 peptide-pulsed T2 cells were used as control. Grey histogram indicates staining with isotype control and open histogram indicate the CD107a expression level on the gated T cells as indicated on top. D). Spontaneous depigmentation at 10 weeks of age in h3T-A2 mouse as compared to age matched C57BL/6, h3T and HLA-A2 controls.
Melanocyte destruction in h3T-A2 mice
The presence of tyrosinase reactive T cells in h3T and h3T-A2 mice led us to further investigate their ability to mediate autoimmunity by the destruction of melanocytes in vitro and in vivo. The progressive vitiligo observed in h3T-A2 from two weeks onwards is due to melanocyte destruction because there is no evidence of pigmented cells remaining in the skin of h3T-A2 mice after active depigmentation (Fig. 4A). By comparison to non-TCR transgenic mice h3T-A2 mice show an increase in depigmentation with age (Fig. 4B). While depigmentation was noticeable at the time of weaning, it continually progressed to be around 60 – 70 % by 30 weeks of age in h3T-A2 mice. Splenocytes from h3T-A2 mouse also secreted IFN-γ when stimulated with HLA-A2+ but not HLA-A2- mouse (Fig. 4C) and human (Fig. 4D) melanocytes in vitro. Cytotoxicity of these tyrosinase-reactive and HLA-A2 restricted T cells was also confirmed by the loss of melanocytes from the hair follicles of depigmenting mice as demonstrated by loss of reactivity with antibodies to tyrosinase-related protein 1 (TRP-1) (Fig. 4E). Further, infiltration of the skin by antigen-specific, Vβ12 expressing T cells was also observed (Fig. 4F, lower panel). These results indicate that the expression of the TIL 1383I TCR on h3T and h3T-A2 T cells can lead to melanocyte destruction resulting in autoimmunity. However, in experiments performed to evaluate if vitiligo could be transferred by adoptively transferring 1 × 106 transgenic T cells in HLA-A2 or Rag-A2 mice we did not notice any development of vitiligo despite the persistence of cells after 90 days of injection (Supplementary Fig. S2C).
FIGURE 4. Effect of TCR transgenic T cells on skin pigmentation.
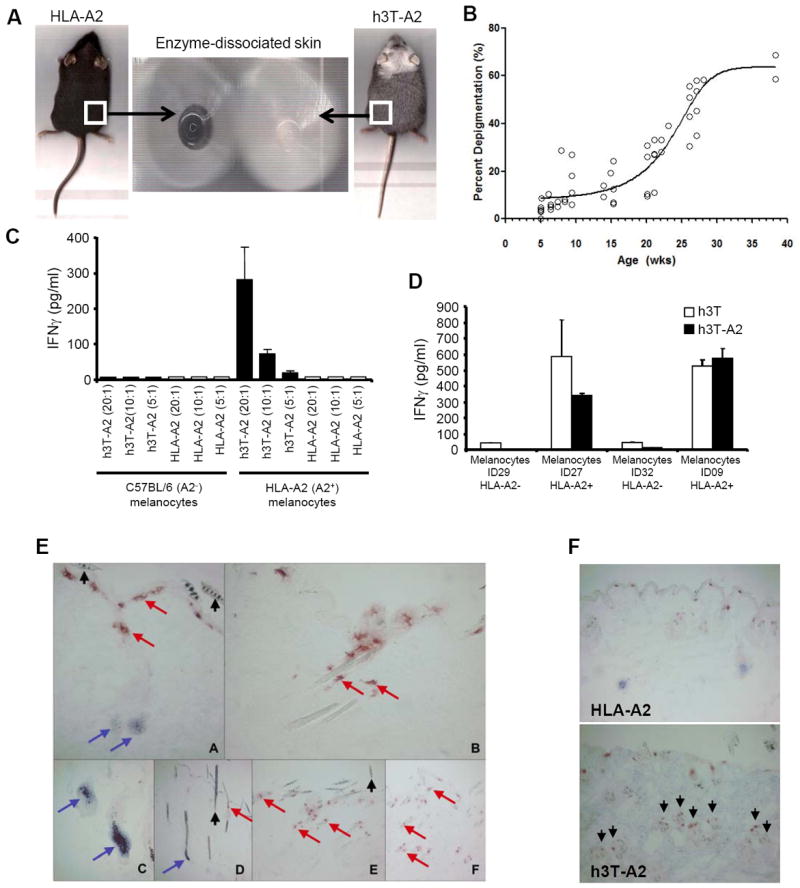
A). Enzyme dissociated skin section was obtained from h3T-A2 mice as described under material and methods with depigmentation and age matched HLA-A2 as controls, and imaged. B). h3T-A2 depigmentation over time was assessed after isofluorane sedation of mice and using a flatbed scanner. Mice were scanned, and images were corrected for ambient light from scan to scan using Adobe Photoshop. Mouse ventral images were inverted and a marquee was drawn to sample the scanned luminosities compared to a non-depigmented C57BL/6 mouse to determine percent depigmentation. A consistent pixel number was sampled for every time point. Percent depigmentation was determined by comparison to non-TCR transgenic mice. C). h3T-A2 splenocytes were co-cultured with mouse melanocytes generated from either C57BL/6 or HLA-A2 mice at various effector to target ratios and supernatants were evaluated for IFN-γ content by ELISA. (D). h3T and h3T-A2 splenocytes were co-cultured with human melanocytes generated from HLA-A2+ (ID27, ID09) or HLA-A2- (ID29, ID32) normal healthy donors and supernatant obtained was evaluated for IFN-γ secretion by ELISA. (E). Immunohistology of depigmenting skin in h3TA2. Melanocytes and T cells detected in skin obtained from HLA-A2 Tg (A, C, D) and h3T-A2 (B, E, F) mice using primary antibodies to TRP-1 and CD3, respectively. B, E at 5weeks; F at 20 weeks. Note complete loss of melanocytes (but not melanin) and T cell infiltration of hair follicles by 5 weeks of age. Arrows: Blue, TRP-1; Red, CD3, black, melanin. (F). Melanocytes and T cells were detected in skin obtained from HLA-A2 wild type mice (upper panel) and h3T-A2 mice (lower panel) using primary antibodies to TRP-1 and CD3, respectively. Note increased T cell infiltration by 5 wks. of age. Arrows mark CD3 staining.
Clinical trials using TCR transduced T cells have also found that T cells bearing high affinity TCR’s can induce reversible ocular and auditory changes in some patients (18-21). Despite their coat color changes, h3T-A2 mice initially have pigmented eyes suggesting the melanocytes in the eye may be immune to destruction by h3T-A2 T cells. To determine if vision changes occur in h3T-A2 mice, a stimulus intensity-series of electroretinograms (ERG) were recorded in twelve month old age-matched wild type and h3T-A2 mice in response to single-flash intensities using 44.3 dB attenuation (low-intensity flash) to no attenuation (high-intensity flash; 0 dB). Although, h3T-A2 mice had mild changes in a-wave amplitudes, their b-waves and OPs were attenuated by more than 58% compared to the wild-type mice (Fig. 5A). In h3T-A2 mice b-wave amplitude were significantly reduced at -30 dB to 0 dB when compared to wild-type mice (wild-type 121±11 versus h3T-A2 80±21, n=6-12, p=0.073; -40dB), (wild-type 278±25 versus h3T-A2 141±29, n=6-15, p=. 006; -30dB), (wild-type 370±31 versus h3T-A2 129±39, n=5-15, p=0.0007; -20dB), (wild-type 526±37 versus h3T-A2 217±44, n=5-15, p=0.0003; -10dB), and (wild-type 667±41 versus h3T-A2 278±38, n=8-13, p=0.0001; 0dB). At lower flash intensities, b-wave amplitudes were attenuated more drastically in h3T-A2 mice. Despite the presence of pigment in their eyes, we have seen 34–65% loss in b-wave amplitudes in year old h3T-A2 mice when compared to the wild-type mice indicating some visual defects in h3T-A2 mice. No significant differences were noticed in mice younger than one-year old (data not shown). To further confirm the infiltration of transgenic T cells in the retina of h3T-A2 animals, we performed in situ hybridization using specific probes for human Vβ12 chain. Expression of RNA transcript for Vβ12 was distinctly observed in the retina of h3TA2 mice as compared to retina of an age matched HLA-A2 non-transgenic mice. Most of the Vβ12 mRNA staining was detected in the inner nuclear layer (Fig. 5B, white arrows), which was migrating to the outer plexiform layer. Additionally, bright field pictures demonstrate that retinal layers were intact in HLA-A2 and greatly disturbed only in the h3T-A2 mice. In addition, images from h3T-A2 mice of a retracting eye showing infiltration by transgenic T cells and by macrophages with interruption of the RPE in comparison to the intact, contra lateral eye (Fig. 5C) convincingly implicate transgenic T cell infiltrates for the pathology of eye.
FIGURE 5. Effect of TCR transgenic T cells on eye.
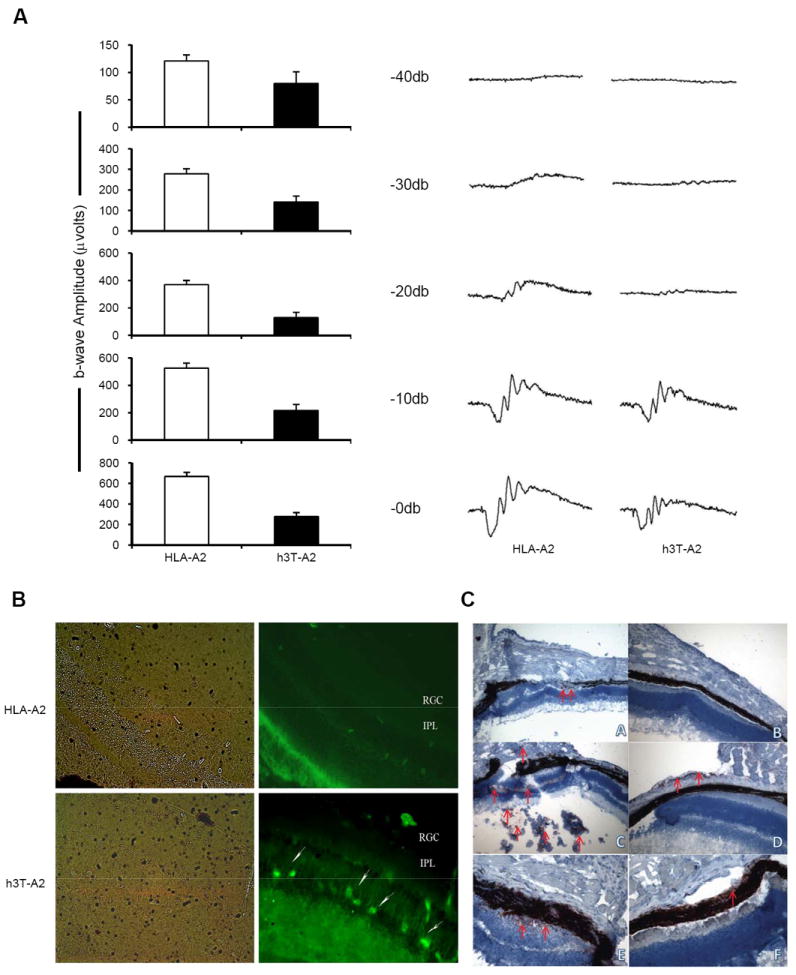
(A). Retinal function in h3T-A2 mice. Representative electroretinograms (ERGs) from wild type and h3T-A2 mice eyes is shown on left panel. Bar diagrams on right panel demonstrate combined data for individual light intensity. Waveforms were recorded from over night dark-adapted wild type and h3T-A2 mice in response to a single 10 μs flash using UTAS-2000 system. Data are expressed as a mean ± SE (-40 dB, n=6-12, p=0.073; -30dB, n=6-15, **p=0.006; -20 dB, n=5-15, ***p=0.0007; -10 dB, n=5-15, ***p=0.0003; 0 dB, n=8-13, ***p=0.0001). (B). Localization of Vβ12 mRNA transcript in the retina was detected by in situ hybridization using human TCR specific fluorescein-labeled probes. Micrographs are representative of results obtained from eyes of four different animals. Left panel shows pictures taken in bright field and right panel shows fluorescent-labeled mRNA detection for Vβ12 in HLA-A2 and h3T-A2 mice. RGC, retinal ganglion cells; IPL, inner plexiform layer. White arrows indicate positive staining for Vβ12+ transgenic T-cells, 40X. (C). Affected (L) and unaffected (R) eye, 8 μm frozen section were stained indirectly with anti-Vβ12 1:200 (A, B); anti-CD68 Ab 1:50 (C, D); and anti-TRP-1 1:50 (E, F) (original magnification 200x), detecting antigen-specific T cells infiltrating through the retina, macrophage infiltrates and leaky retinal pigment epithelial cells in a representative affected eye, respectively. AEC staining using indirect immunoperoxidase labeling and hematoxilin counter staining.
h3T and h3T-A2 T cells are therapeutic in vivo
The presence of highly antigen reactive T cells in h3T and h3T-A2 mice capable of mediating vitiligo suggested that these T cells might also have strong anti-tumor activity in vivo. To this end, we tested the ability of transgenic T cells to protect against tumor challenge. Both h3T and h3T-A2 mice were resistant to subcutaneous challenges of 2.5 × 105 B16-A2 tumor cells but solid tumors formed and grew progressively when challenged with the same number of B16 tumor cells (Fig. 6A). Similarly, h3T and h3T-A2 mice were resistant to intravenous challenges of 2.5 × 105 B16-A2 tumor cells but lung metastases formed when challenged with the same number of B16 tumor cells (Fig. 6B). The HLA-A2 transgenic mice without the TIL1383I TCR showed progressive growth of B16-A2 tumor cells (Supplementary Fig. S3A). These results indicate that transgenic T cells expressing the TIL 1383I TCR can protect mice against tumor challenge.
FIGURE 6. In vivo function in h3T and h3T-A2 mouse.
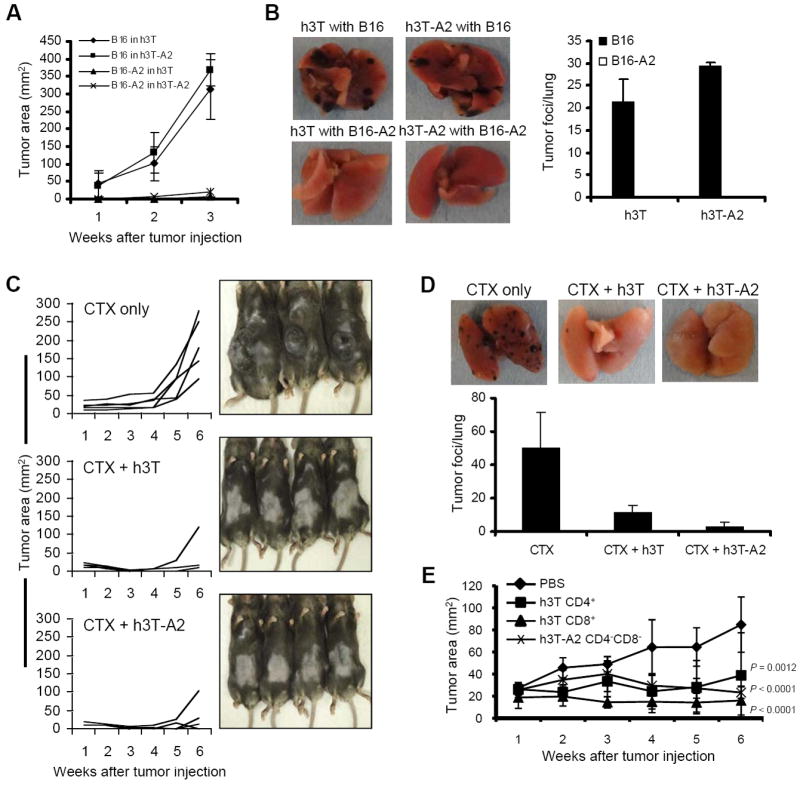
A). Murine melanoma B16 and B16-A2 (2.5 × 105) were injected sub-cutaneously in h3T and h3T-A2 mice. Tumor growth was measured using digital calipers every fourth day. B). Murine melanoma B16 and B16-A2 (2.5 × 105) were injected intravenously through tail vein in h3T and h3T-A2 mice. Mice were sacrificed after 21 days and tumor foci were evaluated in the lungs. Left panel shows picture of a representative lung from each group. Right panel shows the histogram representing the mean number of tumor foci observed per lung obtained from all five mice in each group. C). HLA-A2 mice (n = 5/group) were inoculated (sc) with 2.5 × 105 murine B16-A2 melanoma cells and palpable tumors were treated with or without cyclophosphamide (CTX, 4 mg/mouse). CTX-injected mice were either left without further manipulation or adoptively transferred one day later with 1.0 ×106 Vβ12+ cells harvested from h3T or h3T-A2 mice spleens. Tumor size was then recorded at the indicated time points. Experiment was repeated three times with similar results. D). HLA-A2 mice (n = 3/group) were inoculated (iv) with 2.5 × 105 murine B16-A2 melanoma cells and treated with CTX (4 mg/mouse) after four days. CTX-injected mice were either left without further manipulation or adoptively transferred one day later with 1.0×106 Vβ12+ cells harvested from h3T or h3T-A2 mice spleens. Four weeks after tumor injection, lungs were harvested from mice, and the number of metastatic foci was counted. The experiment was repeated three times with similar results. E). Human HLA-A2+ (624 MEL) and HLA-A2- (624-28 MEL) human melanomas were established in SCID/beige mice before palpable tumors were treated by adoptively transferring purified populations of 105 Vβ12+CD4+, Vβ12+CD8+ and Vβ12+CD4-CD8- h3T T cells. Mice that received PBS were used as controls. Tumor growth was measured using digital calipers every fourth day. Data in figure demonstrate mean tumor size at each time point per group. To compare growth trajectories across groups, random effects linear regression models were fit with log of tumor size as the outcome to adhere to linearity assumptions. Log of tumor size was regressed on time and statistical significance of differences in slopes (compared to the reference group) was assessed by p-values. P-values less than 0.05 were considered to be statistically significant. Data represent two independent experiments. N = 5 mice/group.
Tumor protection is just one measure of anti-tumor effectiveness in vivo. The real test of an effector T cell is in the therapeutic setting. HLA-A2 transgenic mice bearing established subcutaneous B16 or B16-A2 tumors were treated with 1 × 106 h3T or h3T-A2 T cells intravenously after pretreatment with 4 mg/mouse Cyclophosphamide (CTX). The adoptive transfer of h3T or h3T-A2 T cells had no impact on the growth of B16 tumors but led to rejection or delayed growth of established B16-A2 tumors (Fig. 6C) until seven weeks. When 1 × 106 h3T splenocytes were transferred alone in absence of CTX the tumors grew at the same rate as in untreated control, whereas in the CTX administered animals h3T transgenic T cells controlled the tumor growth (Supplementary Fig. S3B). Mice challenged intravenously with B16 or B16-A2 tumor cells were also treated with h3T or h3T-A2 T cells to measure the impact on established lung metastases. Splenic T cells from h3T or h3T-A2 transgenic strains effectively treated mice bearing established B16-A2 lung metastases but were ineffective in mice bearing established B16 lung metastases (Fig. 6D). These results indicate that h3T and h3T-A2 T cells are potent anti-tumor effectors in vivo.
We have shown that the TIL 1383I TCR can be expressed on mature CD4+ (h3T), CD8+ (h3T and h3T-A2), and CD4-CD8- (h3T-A2) T cells and each of these populations function in vitro. We wanted to determine if each of the subsets had anti-tumor activity in vivo and the transgenic T cells can control growth of human melanoma. Groups of five SCID/beige mice were challenged subcutaneously with the HLA-A2+ human melanoma 624 MEL. Once palpable tumors were established, mice were treated intravenously with FACS sorted (≥ 98 % pure) subset of TIL1383I TCR bearing T cells. As shown in Fig. 6E, mice treated with purified Vβ12+CD4+, Vβ12+CD8+, or Vβ12+CD4-CD8- T cells had a statistically significant reduction in tumor growth compared to untreated mice. In addition, these TIL1383I TCR bearing T cell subsets can be traced even three months after adoptive transfer in the peripheral blood of both immunocompetent HLA-A2 (Supplementary Fig. S4A) and immunodeficient SCID/beige recipient mice (Supplementary Fig. S4B) with CD62L+CD44+ phenotype. These results indicate that each of the mature T cell subsets in h3T mice that express the TIL 1383I TCR have anti-tumor activity in vivo.
Discussion
The introduction of cloned genes into the germ line of mice has proven to be a powerful tool to investigate the role of the respective gene products on the immune system. Specificity of the TCR may control T cell fate by specific receptor-ligand interactions either at early or late stages of T cell development (22). Earlier studies for analyzing the role of TCR were carried out by developing transgenic mice with the TCRs of interest (23). Given the belief that T cells bearing high affinity TCR would be more effective at targeting self antigens expressed by tumors, we developed a novel TCR transgenic mouse using HLA-A2 restricted high affinity TCR reactive to human tyrosinase-derived peptide YMDGTMSQV isolated from a class-I restricted CD4+ T cells of tumor infiltrating lymphocytes (TILs) of a patient with metastatic melanoma (24). Since the TCR used to develop our TCR transgenic mice was HLA-A2 restricted, TIL 1383I TCR expressing T cells in our C57BL/6J founder were not expected to be positively selected in the thymus and to be found in the periphery. However, we have found the expression of the transgene in the CD4+ and CD8+ T cells in peripheral blood and lymphoid organs of C57BL/6 mice. Twenty five percent of CD4+ and seventy five percent of CD8+ T cells in the h3T mouse express the transgenic TCR. Surprisingly, analysis of peripheral blood and splenocytes from h3T-A2 mouse indicated that the majority of TCR transgenic CD3+ T cells mouse do not express CD4 or CD8 co-receptors. Interestingly, we notice that these h3T-A2 mice gradually develop spontaneous autoimmune vitiligo. Further, T cells from both h3T and h3T-A2 mice were functional and secreted cytokine after stimulation with cognate peptide pulsed T2 cells, human melanoma cells, or melanocytes and both murine B16-A2 and human melanoma.
The development of transgenic mice often results in an unusual phenotype due to forced expression of the TCR αβ chain (25). In Fas deficient mice, a significant number of T cells exhibit the DN phenotype (26), however in the h3T strain the DN phenotype was more prevalent in the double transgenic h3T-A2 mice that were on correct HLA matched genetic background. Whether this phenotype is a result of co-receptor down-regulation prior to positive selection or that TCR expressing DN thymocytes bypassed the DP stage and were positively selected needs to be investigated. Notably, another founder animal obtained on C57BL/6 and HLA-A2 background showed the same phenotype of transgenic T cells (data not shown). This consistent set of data confirms that expression of this CD8 independent TCR on both CD4+ and CD8+ T cells and CD3+CD4-CD8- T cells is less likely a random phenomenon related to the forced expression of the human transgene in the mouse genome. While the CD3+CD4-CD8- phenotype may seem to be physiologically less relevant for an effector T cell, peripheral blood (PB) mononuclear cells comprise 1–3% of such cells in a normal healthy individual. A recent study has used a novel protocol by which DNT cells can be expanded ex vivo to therapeutic levels in 2 weeks from acute myeloid leukemia (AML) patients during chemotherapy and induced complete remission. The expanded DNT cells expressed similar or higher levels of IFN-γ, TNF-α and Granzyme B as that seen in bulk activated CD8 T cells from the same patient (27). Another study reported isolation of a human DN T cell clone recognizing an HLA-A2-restricted melanoma-associated antigenic gp100-peptide from the peripheral blood of a melanoma patient (28). Like h3T-A2 derived CD3+CD4-CD8- T cells this study showed that gp100-specific DN T cell clone was able to confer antigen-specific cytotoxicity against gp100-pulsed target cells as well as HLA-A2+ gp100 expressing melanoma cells. In addition, it has also been shown recently that the CD8 co-receptor enhances susceptibility to TGF-β-mediated immune suppression (29). Thus, in the absence of the co-receptor the CD4-CD8- T cells from h3T-A2 mice would be less susceptible to immunosuppression and would be able to control tumor growth more efficiently. Together, these data indicate that functionally active antigen-specific DN T cells recognizing MHC class I-restricted tumor-associated antigen (TAA) contribute to anti-tumor immunity in vivo.
The immunogenic phenotype of CD4-CD8- T cells is contradictory to other reports that have attributed a regulatory phenotype to this unusual T cell population (30). In contrast, transgenic T cells from h3T-A2 mice with a CD4-CD8- phenotype did not secrete IL-4, IL-5 or IL-10 on antigen stimulation (data not shown). The development of spontaneous depigmentation has been attributed to either self-reactive CD4+ T cells (31) or CD8+ T cells (32). However, the h3T-A2 model carries predominantly αβTCR+CD3+CD4-CD8- T cells. Thus, the mere presence of a high affinity human tyrosinase specific functional TCR seems to be sufficient to mediate killing of melanocytes despite differences in anchor residues between human and mouse tyrosinase epitope. An earlier study that established a model using murine tyrosinase-derived homologue (FMDGTMSQV) of human tyrosinase-derived antigen (YMDGTMSQV) in transgenic mice expressing the HLA-A*0201, showed that murine peptide was naturally processed and presented in vivo by the HLA-A*0201 similarly to its human counterpart, and that this presentation leads to incomplete self-tolerance (33). The development of spontaneous depigmentation in our h3T-A2 mice suggests that T cells bearing the receptor reactive with YMDGTMSQV are non-tolerant for the FMDGTMSQV, mouse tyrosinase peptide. Whether it is the high affinity TCR or the loss of co-receptor in the positively selected immunogenic T cells in h3T-A2 mice that is responsible for the non-tolerant phenotype is under investigation. It must be noted that despite the positive selection of the tyrosinase TCR in h3T strain, we did not observe any vitiligo in those animals as in case of the h3T-A2 mouse strain. However, these h3T splenocytes recognized HLA-A2+ mouse melanocytes in an in vitro assay. This implies that peripheral T cells recognized the murine tyrosinase antigen when presented in the right context (by HLA-A2+ APC’s). Additionally, the loss of melanocytes from the hair follicles of depigmenting h3T-A2 mice was similar to loss of melanocytes from the skin as a hallmark of vitiligo (34). While h3T-A2 mice develop spontaneous vitiligo, adoptive transfer of the splenocytes either in HLA-A2 or Rag-A2 recipient did not result in transfer of vitiligo despite the presence if transgenic T cells even after 90 days. This is in contrast to the FH transgenic mouse model that bears MHC class I restricted murine tyrosinase TCR or MHC class II-restricted TCR transgenic mouse model in which CD4+ T cells recognize a novel epitope in TRP-1 (8). It is notable that despite the difference in anchor residues between murine and human tyrosinase epitopes, the TIL1383I transgenic TCR bearing T cells were able to recognize murine epitope but vitiligo was still not adoptively transferable. This raises the possibility that other innate mechanisms could also be responsible for vitiligo development in h3T-A2 mice, or besides adoptively transferring freshly isolated h3T-A2 splenocytes alone additional stimuli might be required for development of vitiligo (8, 32).
Subsequently the data obtained for electroretinogram (ERG) also demonstrate that h3T-A2 mice develop abnormalities in the retina function as compared to age-matched controls. The ERG data suggested that there is a marginal decrease in a-wave amplitudes whereas major changes were seen in b-wave amplitudes of h3T-A2 mice. These results support the idea that the inner retina of h3T-A2 mice is more severely damaged than photoreceptors in the outer segment of the eye. The causative factors and cellular mechanisms that could be involved in the retina damage in h3T-A2 mice need further investigation. Various studies undertaken to understand the biological role of melanin in the retinal function have utilized genetically manipulated and spontaneous mutant animal models, e.g. oculo-cutaneous albinism type 1 (OCA1) model that is characterized by congenital hypo-pigmentation due to a mutation in the tyrosinase gene. These studies in OCA1 (Tyrc-2j) model have shown that several retinal functional abnormalities are associated with photoreceptor loss. Moreover, AAV-mediated retinal gene delivery of the human tyrosinase gene was able to activate melanosome biogenesis and preserve retinal function (35). Tyrosinase-deficient mice lack pigmentation in melanocytes and retinal pigment epithelium (RPE) cells which ultimately affects the retina function (36). Despite expressing a functional tyrosinase gene in h3T-A2 mice, spontaneous depigmentation due to melanocyte killing can be observed. Our ocular data show 34-65% loss in b-wave amplitudes in h3T-A2 mice when compared to the year old age-matched wild-type mice. The loss of retina function in h3T-A2 mice could not be only due to hypo-pigmentation of RPE, because such condition affects mainly outer segment photoreceptor activity (36). In our studies, photoreceptor activity (e.g., a-wave) was marginally affected in h3T-A2 mice, suggesting that RPE pigmentation does not play a key role to maintain retina function in h3T-A2 mice. It is reasonable to speculate that infiltrated T-cells could have caused an increased accumulation of cytokines (e.g.; TNF-α, IF-γ, IL-1β) in the eye, which subsequently led to the retinal degeneration. Further studies are required to comprehensively determine the mechanisms responsible for retinal degeneration and address if the noted defect are specific to tyrosinase TCR transgenic mice, since we also observe complete occlusion of one eye in up to 5% of h3T-A2 mice by 14 weeks of age.
Traditionally, CD8+ T cytotoxic lymphocytes have been considered necessary for controlling tumor growth. Despite earlier reports that CD4+ T cells alone were capable of protecting experimental animals against solid tumors without providing any CD8+ T cells (37, 38), the role of CD4+ T cells has remained unappreciated. However, recent studies have shown that CD4+ T cells possess an intrinsic ability to orchestrate broad anti tumor responses lacking in CD8+ T cells (39). That CD4+ TCR transgenic cells were capable of controlling tumor growth did not raise much discussion due to difference in TCR on CD8+ and CD4+ T cells used in another study (40), and issues about difference in class I and class II epitopes and TCR avidities were raised (41). Lately a few studies using class II restricted gp75/TRP-1 transgenic CD4+ T cells have shown acquisition of cytotoxic activity in vivo and the ability to eradicate large established melanoma in lymphopenic host (8, 42). Our data show that the mere presence of adoptively transferred class I restricted high affinity TCR on either CD4+, CD8+ or CD4-CD8- T cells is sufficient to control tumor growth both in immunodeficient SCID/beige recipients that lack T, B and NK cells and in immunocompetent HLA-A2+ recipients. The persistence of human Vβ12+ transgenic T cells in mice even months after adoptive transfer is intriguing. Importantly, it should be noted that in our adoptive transfer experiments, TCR transgenic cells from h3T or h3T-A2 mice were neither stimulated ex vivo nor were the tumor bearing animals given cytokines after T cell transfer for their sustained maintenance. This is in accordance with the study by Hunder et al. (2008), who reported successful treatment of a patient with ex vivo generated CD4+ T cell clones recognizing TAA NY-ESO (43) without IL-2 administration. Similarly, a study in mice revealed the capacity of CD4+ T cells to control tumor in the absence of any exogenous cytokine administration (20). Recently, TCR-αβ genes derived after amino acid modifications to increase cell surface expression and reactivity after gene transfer were also used to genetically engineer CD8+ and CD4+ T cells with class I restricted TCR and had equal tumor regression in a murine melanoma model (44). However, our data suggest that irrespective of the presence or complete absence of CD4 and CD8 co-receptors, the mere presence of a high affinity TIL 1383I TCR directs its ability to mount an effector immune response. We believe that the novel mouse models reported in this study will not only be useful in designing future tumor immunotherapy protocols but will also help in understanding the unique thymic selection of the tyrosinase-specific high affinity TIL1383I TCR and TCR signaling issues of CD4 and CD8 T cells upon activation with the same antigen.
Supplementary Material
Acknowledgments
We would like to thank Dr. Bijay Mukherji at University of Connecticut Health Center (Farmington, CT) for his critiques while preparing this manuscript. We also thank Dr. Alexander Awgulewitsch and Ms. Donna Jacobs at MUSC transgenic core facility for their valuable help in developing the h3T TCR transgenic mice. Helpful suggestions regarding the optimization of the In situ RT-PCR technique from Dr. Omar Bagasra and Ms. Leslie Johnson at Claflin University, Orangeburg, SC, and Dr. James Norris at MUSC are duly acknowledged. Authors also acknowledge help from Dr. Jennifer G. Schnellmann in the Office of Scientific Editing & Publications, and Ms. Lauro Colombo at the Oral Histology Core at MUSC.
The work was supported by NIH R21 CA137625 to SM, NIH RO1 AR057643 to CLP, NIH R01 CA104947, NIH CA104947-S1 (ARRA Supplement) and PO1 CA154778 to MIN.
References
- 1.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nature reviews. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–513. [PubMed] [Google Scholar]
- 3.Duval L, Schmidt H, Kaltoft K, Fode K, Jensen JJ, Sorensen SM, Nishimura MI, von der Maase H. Adoptive transfer of allogeneic cytotoxic T lymphocytes equipped with a HLA-A2 restricted MART-1 T-cell receptor: a phase I trial in metastatic melanoma. Clin Cancer Res. 2006;12:1229–1236. doi: 10.1158/1078-0432.CCR-05-1485. [DOI] [PubMed] [Google Scholar]
- 4.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (New York N Y. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature medicine. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becerra JC, Arthur JF, Landucci GR, Forthal DN, Theuer CP. CD8+ T-cell mediated tumor protection by Pseudomonas exotoxin fused to ovalbumin in C57BL/6 mice. Surgery. 2003;133:404–410. doi: 10.1067/msy.2003.112. [DOI] [PubMed] [Google Scholar]
- 7.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, Antony PA. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207:651–667. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li LP, Lampert JC, Chen X, Leitao C, Popovic J, Muller W, Blankenstein T. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nature medicine. 2010;16:1029–1034. doi: 10.1038/nm.2197. [DOI] [PubMed] [Google Scholar]
- 10.von Boehmer H. Developmental biology of T cells in T cell-receptor transgenic mice. Annual review of immunology. 1990;8:531–556. doi: 10.1146/annurev.iy.08.040190.002531. [DOI] [PubMed] [Google Scholar]
- 11.Kouskoff V, Signorelli K, Benoist C, Mathis D. Cassette vectors directing expression of T cell receptor genes in transgenic mice. Journal of immunological methods. 1995;180:273–280. doi: 10.1016/0022-1759(95)00002-r. [DOI] [PubMed] [Google Scholar]
- 12.Rivoltini L, Barracchini KC, Viggiano V, Kawakami Y, Smith A, Mixon A, Restifo NP, Topalian SL, Simonis TB, Rosenberg SA, et al. Quantitative correlation between HLA class I allele expression and recognition of melanoma cells by antigen-specific cytotoxic T lymphocytes. Cancer research. 1995;55:3149–3157. [PMC free article] [PubMed] [Google Scholar]
- 13.Mehrotra S, Stevens R, Zengou R, Chakraborty NG, Butterfield LH, Economou JS, Dorsky DI, Mukherji B. Regulation of melanoma epitope-specific cytolytic T lymphocyte response by immature and activated dendritic cells, in vitro. Cancer Res. 2003;63:5607–5614. [PubMed] [Google Scholar]
- 14.Bagasra O. Protocols for the in situ PCR-amplification and detection of mRNA and DNA sequences. Nature protocols. 2007;2:2782–2795. doi: 10.1038/nprot.2007.395. [DOI] [PubMed] [Google Scholar]
- 15.Le Poole IC, Riker AI, Quevedo ME, Stennett LS, Wang E, Marincola FM, Kast WM, Robinson JK, Nickoloff BJ. Interferon-gamma reduces melanosomal antigen expression and recognition of melanoma cells by cytotoxic T cells. The American journal of pathology. 2002;160:521–528. doi: 10.1016/s0002-9440(10)64871-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halaban R, Alfano FD. Selective elimination of fibroblasts from cultures of normal human melanocytes. In vitro. 1984;20:447–450. doi: 10.1007/BF02619590. [DOI] [PubMed] [Google Scholar]
- 17.Husain S, Potter DE, Crosson CE. Opioid receptor-activation: retina protected from ischemic injury. Investigative ophthalmology & visual science. 2009;50:3853–3859. doi: 10.1167/iovs.08-2907. [DOI] [PubMed] [Google Scholar]
- 18.Yang AS, Bird P. Intracranial metastatic melanoma presenting with bilateral hearing loss. Otol Neurotol. 2009;30:243–244. doi: 10.1097/MAO.0b013e31816c7c88. [DOI] [PubMed] [Google Scholar]
- 19.Yeh S, Karne NK, Kerkar SP, Heller CK, Palmer DC, Johnson LA, Li Z, Bishop RJ, Wong WT, Sherry RM, Yang JC, Dudley ME, Restifo NP, Rosenberg SA, Nussenblatt RB. Ocular and systemic autoimmunity after successful tumor-infiltrating lymphocyte immunotherapy for recurrent, metastatic melanoma. Ophthalmology. 2009;116:981–989. e981. doi: 10.1016/j.ophtha.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer DC, Chan CC, Gattinoni L, Wrzesinski C, Paulos CM, Hinrichs CS, Powell DJ, Jr, Klebanoff CA, Finkelstein SE, Fariss RN, Yu Z, Nussenblatt RB, Rosenberg SA, Restifo NP. Effective tumor treatment targeting a melanoma/melanocyte-associated antigen triggers severe ocular autoimmunity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8061–8066. doi: 10.1073/pnas.0710929105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Currie L, Tomma A. Malignant melanoma presenting as sudden onset of complete hearing loss. Annals of plastic surgery. 2001;47:336–337. doi: 10.1097/00000637-200109000-00020. [DOI] [PubMed] [Google Scholar]
- 22.von Boehmer H, Kishi H, Borgulya P, Scott B, van Ewijk W, Teh HS, Kisielow P. Control of T-cell development by the TCR alpha beta for antigen. Cold Spring Harbor symposia on quantitative biology. 1989;54(Pt 1):111–118. [PubMed] [Google Scholar]
- 23.von Boehmer H. Learning by the immune system studied in T-cell receptor transgenic mice. Thymus. 1989;13:19–26. [PubMed] [Google Scholar]
- 24.Nishimura MI, Avichezer D, Custer MC, Lee CS, Chen C, Parkhurst MR, Diamond RA, Robbins PF, Schwartzentruber DJ, Rosenberg SA. MHC class I-restricted recognition of a melanoma antigen by a human CD4+ tumor infiltrating lymphocyte. Cancer research. 1999;59:6230–6238. [PubMed] [Google Scholar]
- 25.Huesmann M, Scott B, Kisielow P, von Boehmer H. Kinetics and efficacy of positive selection in the thymus of normal and T cell receptor transgenic mice. Cell. 1991;66:533–540. doi: 10.1016/0092-8674(81)90016-7. [DOI] [PubMed] [Google Scholar]
- 26.Haas JP, Grunke M, Frank C, Kolowos W, Dirnecker D, Leipold G, Hieronymus T, Lorenz HM, Herrmann M. Increased spontaneous in vitro apoptosis in double negative T cells of humans with a fas/apo-1 mutation. Cell death and differentiation. 1998;5:751–757. doi: 10.1038/sj.cdd.4400426. [DOI] [PubMed] [Google Scholar]
- 27.Merims S, Li X, Joe B, Dokouhaki P, Han M, Childs RW, Wang ZY, Gupta V, Minden MD, Zhang L. Anti-leukemia effect of ex vivo expanded DNT cells from AML patients: a potential novel autologous T-cell adoptive immunotherapy. Leukemia. 2011 doi: 10.1038/leu.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voelkl S, Moore TV, Rehli M, Nishimura MI, Mackensen A, Fischer K. Characterization of MHC class-I restricted TCRalphabeta+ CD4- CD8- double negative T cells recognizing the gp100 antigen from a melanoma patient after gp100 vaccination. Cancer Immunol Immunother. 2009;58:709–718. doi: 10.1007/s00262-008-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zloza A, Jagoda MC, Lyons GE, Graves MC, Kohlhapp FJ, O’Sullivan JA, Lacek AT, Nishimura MI, Guevara-Patino JA. CD8 co-receptor promotes susceptibility of CD8+ T cells to transforming growth factor-beta (TGF-beta)-mediated suppression. Cancer Immunol Immunother. 2011;60:291–297. doi: 10.1007/s00262-010-0962-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomson CW, Lee BP, Zhang L. Double-negative regulatory T cells: non-conventional regulators. Immunologic research. 2006;35:163–178. doi: 10.1385/IR:35:1:163. [DOI] [PubMed] [Google Scholar]
- 31.Lambe T, Leung JC, Bouriez-Jones T, Silver K, Makinen K, Crockford TL, Ferry H, Forrester JV, Cornall RJ. CD4 T cell-dependent autoimmunity against a melanocyte neoantigen induces spontaneous vitiligo and depends upon Fas-Fas ligand interactions. J Immunol. 2006;177:3055–3062. doi: 10.4049/jimmunol.177.5.3055. [DOI] [PubMed] [Google Scholar]
- 32.Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J Immunol. 2010;184:1909–1917. doi: 10.4049/jimmunol.0902778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colella TA, Bullock TN, Russell LB, Mullins DW, Overwijk WW, Luckey CJ, Pierce RA, Restifo NP, Engelhard VH. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J Exp Med. 2000;191:1221–1232. doi: 10.1084/jem.191.7.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Poole IC, van den Wijngaard RM, Westerhof W, Dutrieux RP, Das PK. Presence or absence of melanocytes in vitiligo lesions: an immunohistochemical investigation. The Journal of investigative dermatology. 1993;100:816–822. doi: 10.1111/1523-1747.ep12476645. [DOI] [PubMed] [Google Scholar]
- 35.Gargiulo A, Bonetti C, Montefusco S, Neglia S, Di Vicino U, Marrocco E, Corte MD, Domenici L, Auricchio A, Surace EM. AAV-mediated tyrosinase gene transfer restores melanogenesis and retinal function in a model of oculo-cutaneous albinism type I (OCA1) Mol Ther. 2009;17:1347–1354. doi: 10.1038/mt.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raposo G, Marks MS. Melanosomes--dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 2007;8:786–797. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenberg PD, Cheever MA, Fefer A. Eradication of disseminated murine leukemia by chemoimmunotherapy with cyclophosphamide and adoptively transferred immune syngeneic Lyt-1+2- lymphocytes. J Exp Med. 1981;154:952–963. doi: 10.1084/jem.154.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mumberg D, Monach PA, Wanderling S, Philip M, Toledano AY, Schreiber RD, Schreiber H. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8633–8638. doi: 10.1073/pnas.96.15.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z, Noh HS, Chen J, Kim JH, Falo LD, Jr, You Z. Potent tumor-specific protection ignited by adoptively transferred CD4+ T cells. J Immunol. 2008;181:4363–4370. doi: 10.4049/jimmunol.181.6.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, Matzinger P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109:5346–5354. doi: 10.1182/blood-2006-10-051318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muranski P, Restifo NP. Adoptive immunotherapy of cancer using CD4(+) T cells. Current opinion in immunology. 2009;21:200–208. doi: 10.1016/j.coi.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, Restifo NP, Allison JP. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. The New England journal of medicine. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frankel TL, Burns WR, Peng PD, Yu Z, Chinnasamy D, Wargo JA, Zheng Z, Restifo NP, Rosenberg SA, Morgan RA. Both CD4 and CD8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid TCR targeting tyrosinase. J Immunol. 2010;184:5988–5998. doi: 10.4049/jimmunol.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.