

Abstract
During the last two decades there has been considerable growth in the development of catalytic reactions capable of activating unreactive C–H bonds. These methods allow for the synthesis of complex molecules from easily available and cheaper precursors in a fewer number of steps. Naturally, the development of C–H activation methods for direct functionalization of heterocyclic molecules, invaluable building blocks for pharmaceutical and synthetic chemistry and material science, has received substantial attention as well.
Introduction
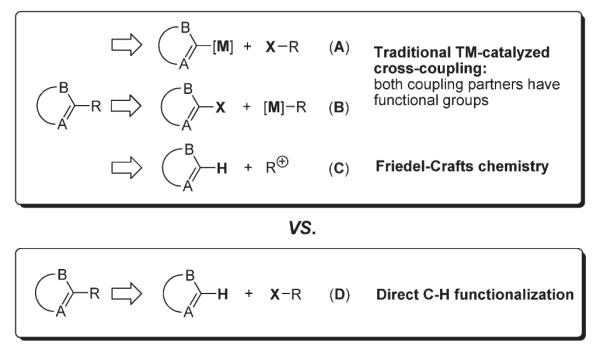
The traditional methods for functionalization of heterocyclic compounds usually employ different types of cross-coupling reactions, where the heterocyclic partner bears either a metal-containing functionality (nucleophilic component) or a halogen atom (electrophilic component) (A and B, Scheme 1). Friedel–Crafts-type alkylation (C) is also well-established, however it is limited to electron-rich substrates only and often is not regioselective. Alternatively, direct functionalization of heterocycles via the activation of unreactive C–H bonds (D) constitutes a much more attractive approach.1
Scheme 1.
Pd-catalyzed arylation of heteroaromatics with aryl halides is the most developed type of C–H functionalization of heterocyclic compounds. The earliest report containing an example of a very low yielding intramolecular arylation of pyridine appeared in 1984.2 Since then, this chemistry has been rapidly growing and new types of direct intra- and intermolecular reactions, as well as cascade transformations of heterocycles, such as C–H arylation, heteroarylation, vinylation, and formal alkylation, have been developed. Palladium complexes are among the most frequently used catalysts; however, there are some reports on efficient rhodium-catalyzed transformations.
This review provides mechanistic discussions on transition metal-catalyzed direct C–H functionalization of electron-rich and electron-deficient heterocycles, discusses different sets of conditions used for functionalization of both types of heterocycles, and covers the most essential synthetic applications.
1. C–H arylation of electron-rich heterocyclic systems
1.1. Reactions involving Pd0/PdII manifold: mechanistic studies
One of the most abundant protocols for transition metal-catalyzed functionalization of heteroaromatic compounds involves a Pd0/PdII catalytic cycle. For C–H arylation of heterocyclic substrates, four distinct mechanisms have been proposed to date: electrophilic aromatic substitution (A),3 C–H activation (B),4 cross-coupling (C),3 and Heck-type arylation (D)5 (Scheme 2).
Scheme 2.
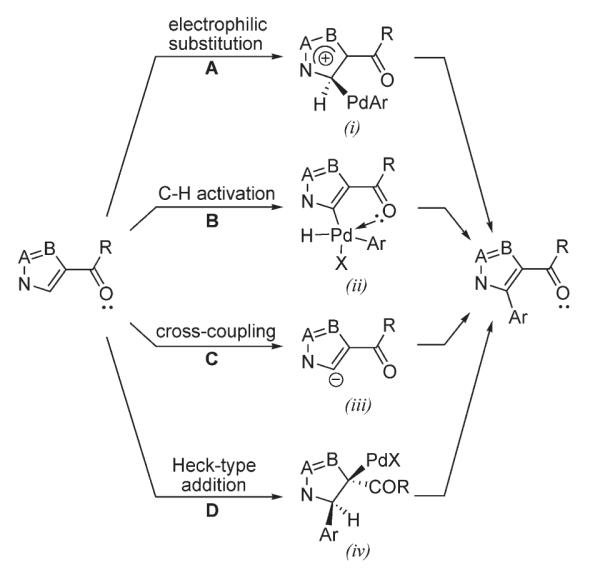
Proposed mechanisms for Pd-catalyzed arylation of electron-rich hetroaromatic compounds.
The first systematic mechanistic studies on the palladium-catalyzed arylation of heterocycles were reported by Gevorgyan.6 Indolizine, one of the most electron-rich heterocycles known,7 was chosen for these studies. Experimental and theoretical studies were performed to evaluate possible involvement of any of the four mechanisms depicted in Scheme 3.
Scheme 3.
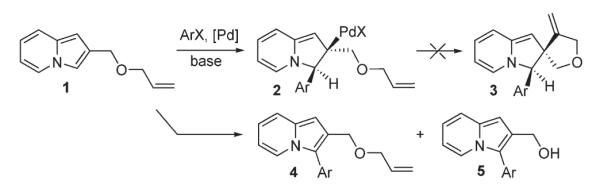
Attempts to perform cascade Heck reaction on indolizine.
First, to evaluate the possible involvement of a Heck-type process D, a cascade Heck reaction8 of 1 (Scheme 3) was tested, aiming at spirocyclic compound 3. The latter would form via the second carbopalladation of the first Heck reaction product 2. However, trials on this cascade transformation with allyl ether 1 were unsuccessful, producing a mixture of C-3 arylated indolizines 4 and 5 instead. In another set of experiments, indolizine 6 was subjected to reductive Heck reaction conditions,9 aiming at dihydroindolizine derivative 7 (Scheme 4). However, numerous efforts to perform this transformation under a variety of reduction conditions failed. It was rationalized that if this reaction does operate via a Heck-type mechanism,5 the arylation of 6-carboethoxyindolizine 8 should lead to 7-arylindolizine 10 (Scheme 5). However, 8 was selectively arylated at C-3 instead, producing 9 as a sole regioisomer.
Scheme 4.
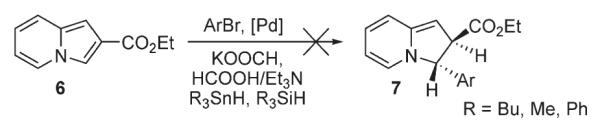
Attempts to perform reductive Heck reaction.
Scheme 5.
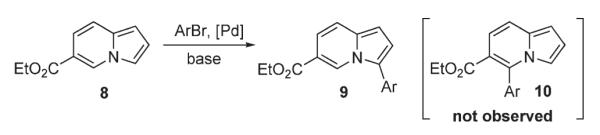
Arylation of 6-carboethoxyindolizine.
Next, the possibility of C–H activation pathway (B), proposed by Miura for the “coordination-assisted” C-3 arylation of 2-carbamoyl-substituted thiophenes,10 was tested. Normally reactions proceeding via C–H activation motif experience a substantial isotope effect.11 However, studies performed by the Gevorgyan group on indolizine 11 (Scheme 6) indicated no isotope effect (kH/D = 1.0), thus disfavoring involvement of C–H activation (Path B, Scheme 3) in this transformation.6
Scheme 6.
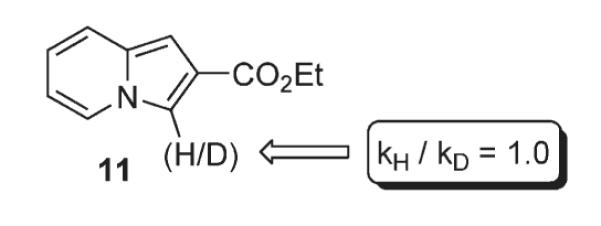
Possible C–H activation mechanism: isotope effect study.
Furthermore, the absence of KIE at position C-3 of indolizine 11 contradicts another possible mechanism: a cross-coupling type process (Path C, Scheme 2), as it is known that reactions proceeding via proton–metal exchange should also exhibit a substantial KIE.12 The possibility of involvement of this mechanism was originally proposed by Miura for the copper-assisted C-2 arylation of azoles, where arylation of “acidic” sites of the heterocycles was promoted by the addition of Cu salts.3 However, addition of copper salts during the arylation of indolizine 6 resulted in greatly prolonged reaction times and reduced yields, thus, not supporting Path C.6
Next, experimental and theoretical studies were performed to elucidate the possible involvement of the electrophilic aromatic substitution process (A). This mechanism has often been considered as the most probable mechanism for arylation of heterocycles.3,5,13,14 Naturally, the absence of an isotope effect in the arylation of 11 (Scheme 6) does not contradict pathway A, as the deprotonation event in the electrophilic mechanisms is not a rate-limiting step.15 Additionally, it is well known that the pyrrole ring of an indolizine is electron-rich and easily undergoes electrophilic substitution reactions.7 DFT calculations perfectly confirmed this point, revealing that the pyrrole ring has an extended HOMO density, whereas the LUMO mostly resides at the pyridine ring.6 To gather further support for electrophilic mechanism A, kinetic studies were performed (Table 1).6 As expected for an electrophilic path, the EWG-substituted indolizine underwent the slowest arylation among tested substrates (Method I, Table 1). A similar trend of reactivity was observed in the Lewis acid-mediated Friedel–Crafts acylation of the same indolizines (Method II, Table 1), thus strongly supporting electrophilic nature of the arylation (Path A, Scheme 2).6
Table 1.
Relative rates of Pd-catalyzed arylation (Method I) and Lewis acid-mediated acylation (Method II) of selected indolizines
| Relative rates |
||
|---|---|---|
| R1 | Method I | Method II |
| H | 1.00 | 1.00 |
| Me | 0.97 | 0.67 |
| CO2Et | 0.66 | 0.33 |
Shortly thereafter, Sames reported detailed mechanistic studies on C-2 and C-3 arylation of indoles.16 Three mechanisms that rationalize strong preference for the palladium-catalyzed C-2 arylation of indole were considered (Scheme 7): Heck-type reaction (A), non-electrophilic metalation of the C-2 position (B), and the electrophilic metalation at C-3 with or without subsequent migration to C-2 (C or D, respectively). While pathways A and B were briefly commented on, electrophilic mechanisms C and D received the most attention in this work.
Scheme 7.
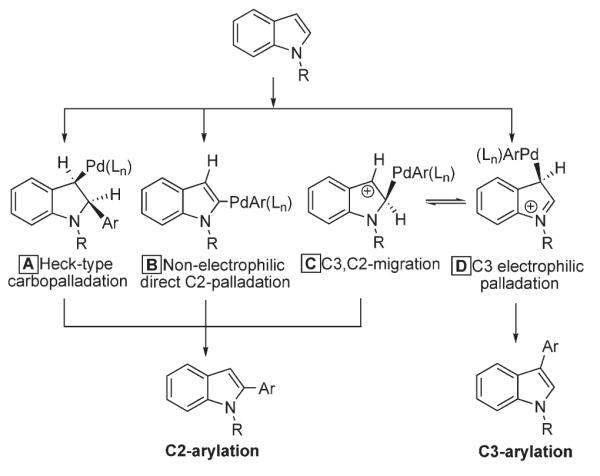
Possible pathways for C-2 and C-3 arylation of indoles.
First, it was mentioned that a Heck-type mechanism (Path A) might be operational in this process.5,17,18 In this case, the process would involve a carbopalladation step, followed by anti-dehydropalladation or by isomerization and syn-elimination.19 However, the isomerization step requires a reversible α-hydride elimination to form a carbene intermediate, a process unknown for palladium (unlike for Pt or Ru).20
The next possible pathway is a non-electrophilic C-2- palladation of indole (Path B), which has been proposed by Tollari21 and Nonoyama.22 However, it was concluded that involvement of this scenario for non-functionalized indoles is unlikely, as the presence of a strong directing group is a strict requirement.21,22
Sames further evaluated the feasibility of pathways C and D, which involve an electrophilic palladation of indole.16 It is well known that electrophilic substitution at indole has a strong preference for the C-3 position.23 On the other hand, multiple examples of palladium-catalyzed arylation demonstrate predominant or exclusive C-2 selectivity. Thus, an interesting suggestion was made: the initial metalation does occur at the more nucleophilic C-3 carbon, followed by the C-3 to C-2 migration of an aryl palladium moiety, resulting in the apparent C-2 arylation (Path C). Better stabilization of the C-2–Pd bond by the adjacent nitrogen was proposed to be the driving force for this migration.24 Kinetic isotope effect studies performed by Sames revealed an unexpectedly large kinetic isotope effect of 1.6 at C-3, a position in 15 where no substitution occurs (Scheme 8), which was rationalized in terms of secondary KIE. However, it was pointed out, an unusual value of 1.6 may be the sum of several rate-contributing steps. At the same time, the KIE at C-2 was ~1.2, evidently too small for the cleavage of C-2–H bond at the rate-limiting step. Sames concluded, that these results, if not clearly in support migratory pathway C, seem to be somewhat contradictory with the mechanism wherein a direct palladation at position C-2 takes place.
Scheme 8.
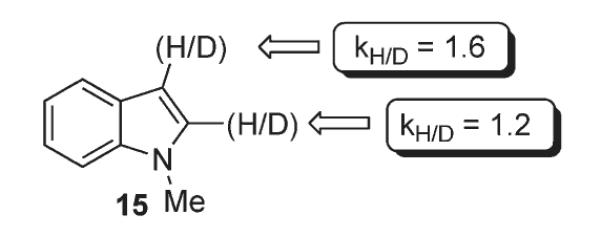
Kinetic isotope effect in the phenylation of C-2 and C-3 positions of indole.
In conclusion, the mechanisic pathway for Pd0/PdII-catalyzed arylation of electron-rich heterocycles, proposed by Miura3 (Path A, Scheme 2), has received the most unambiguous experimental support so far. The key step of the process involves an electrophilic attack of arylpalladium species at the heterocycle. The regioselectivity of this step is governed by the distribution of electron density in the substrate. In indoles, however, the altering regiochemical outcome may be a result of palladium migration (Path C, Scheme 7). Nevertheless, the potential involvement of other aforementioned pathways or mixed mechanisms can not, at this point, be completely ruled out.
1.2. Reactions involving Pd0/PdII manifold: synthetic applications
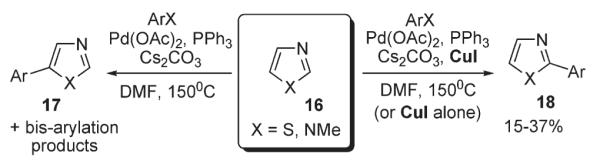
Miura investigated reactions of bromo- and iodobenzene with imidazoles, oxazoles, and thiazoles 16 (Scheme 9).3 It was found that in the presence of catalytic amounts of Pd(OAc)2 and PPh3 in DMF, the coupling products, 5-arylated azoles 17, can be synthesized selectively in good yields.3 Interestingly, it was shown that the addition of stoichiometric Cu(I) salt significantly increases reactivity of the C-2 position of azoles leading to 2-arylated products 18. Although the trend of reactivity in the electrophilic reactions of the azole ring is known to be C-5>C-4>C-2, in the presence of CuI alone (no Pd catalysis), a substantial amount of C-2 substituted product formed, which was attributed to an alternative operating mechanism. It was proposed that exchange of the most acidic proton at C-2 with Cu results in an organocopper intermediate, which is enabled to undergo cross-coupling with aryl halide to form C-2 arylated product 18 (Scheme 9).3
Scheme 9.
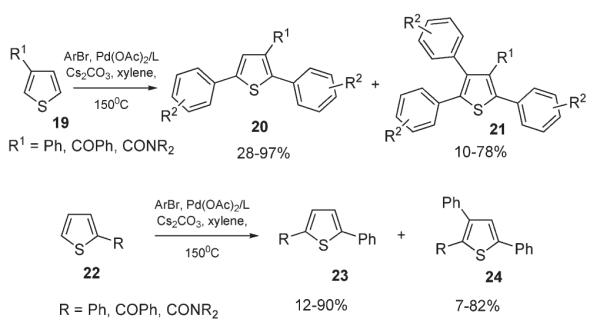
Furthermore, Miura reported a method for palladium-catalyzed multiple arylation of thiophenes 19 and 22 (Scheme 10).25 Thiophene carboxamides were shown to undergo triarylation, accompanied by formal decarbamoylation in the presence of Pd(OAc)2, Buchwald’s ligand (P(o-biphenyl)(tBu)2) or P(tBu)3, and Cs2CO3 in refluxing xylene.25 3-Substituted thiophenes 19, bearing EWGs, can also be converted to triarylated products 21 in high yields. At the same time, selective diarylation can also be achieved in the presence of an excess of ArBr (Scheme 10).
Scheme 10.
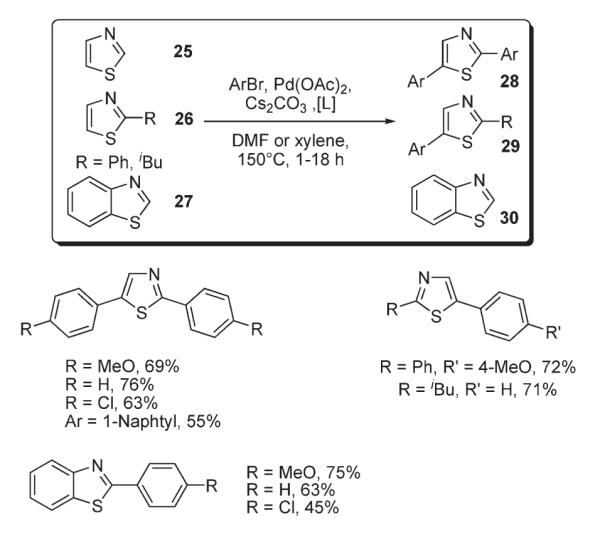
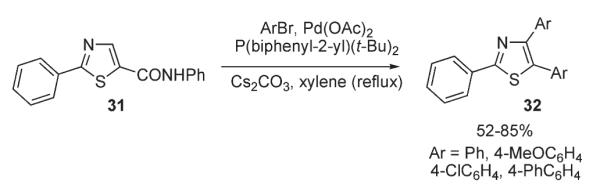
Later, Miura disclosed a similar protocol for palladium-catalyzed C-2 and C-5 arylation of thiazoles 25, 26 and benzothiazoles 27 with aryl bromides (Scheme 11).26 The best yields of diarylated products were obtained by employment of bulky phosphine ligands (P(o-biphenyl)(tBu)2) or P(tBu)3 in DMF at 150 °C. At the same time, arylation of thiazole 31 bearing a carboxanilide function as sacrificial group in less polar o-xylene afforded 4,5-diarylated thiazole 32 exclusively, (Scheme 12).26
Scheme 11.
Scheme 12.
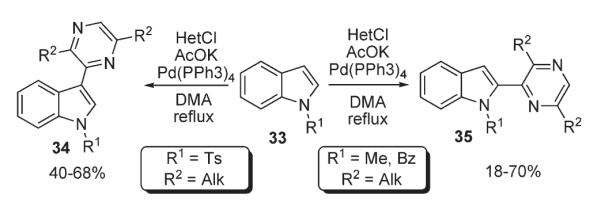
One of the first direct heteroarylations of heterocycles was reported in 1989 by Ohta who investigated palladium-catalyzed coupling of 2-chloro-3,6-dialkylpyrazines with protected indoles 33 (Scheme 13).27,28 Reactions of 1-tosylindole with chloropyrazine in the presence of catalytic Pd(PPh3)4 led predominately to 3-heteroaryl indoles 34 in moderate to good yields. Alternatively, under the same conditions, 1-alkyl- and benzylindoles were shown to undergo substitution at C-2, affording the corresponding 2-heteroarylated products 35 in good yields (Scheme 13).27,28
Scheme 13.
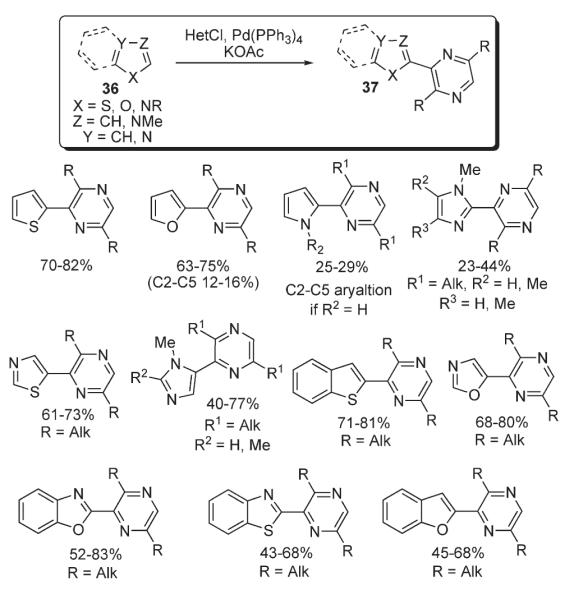
Later, Ohta demonstrated palladium-catalyzed heteroarylation of other electron-rich heterocycles 36 (Scheme 14).28 Coupling of chloropyrazines with pyrroles, furans, thiophenes, and a number of azoles in the presence of Pd(PPh3)4 gave the corresponding pyrazine-substituted products 37 in moderate to very good yields.28
Scheme 14.
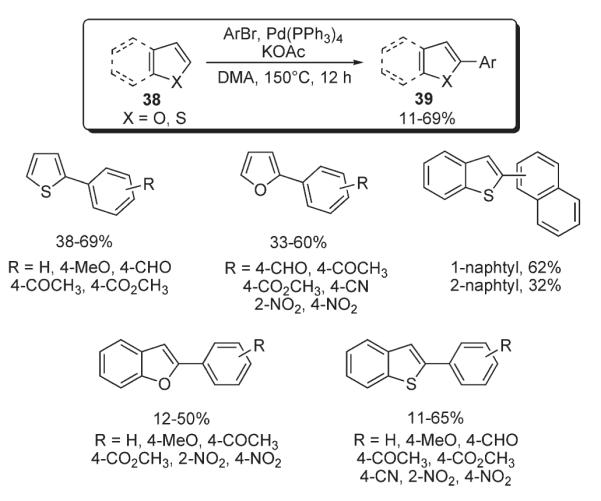
Furthermore, this methodology was extended by Ohta for the arylation of furan, thiophene, and their benzo-analogues 38 (Scheme 15).29 The developed protocol allowed for the synthesis of a variety of compounds arylated selectively at the C-2 position (39). It was noted that electron-deficient aryl components generally result in lower yields, while aryl bromides bearing EWGs react very smoothly.29
Scheme 15.
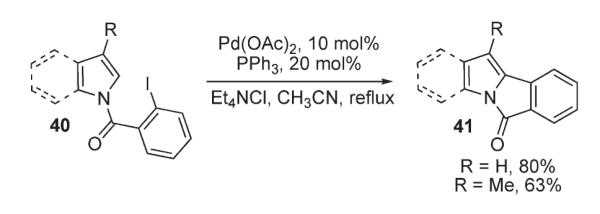
In 1990, Grigg reported an intramolecular arylation of iodo-1-aroylpyrroles and indoles 40 (Scheme 16).30,31 It was demonstrated that in the presence of Pd(OAc)2, PhPh3 and tetraethylammonium chloride the reaction affords good yields of tri- and tetracyclic products 41.30,31
Scheme 16.
Later, Kozikowski disclosed a methodology towards polycyclic indoles 43 featuring intramolecular cyclization of bromoaryl-bearing indoles 42 (Scheme 17).32 It was demonstrated that intramolecular C–H arylation proceeded readily in the presence of Pd(PPh3)4 and KOAc in DMA, affording polycyclic products 43 in very good yields.
Scheme 17.
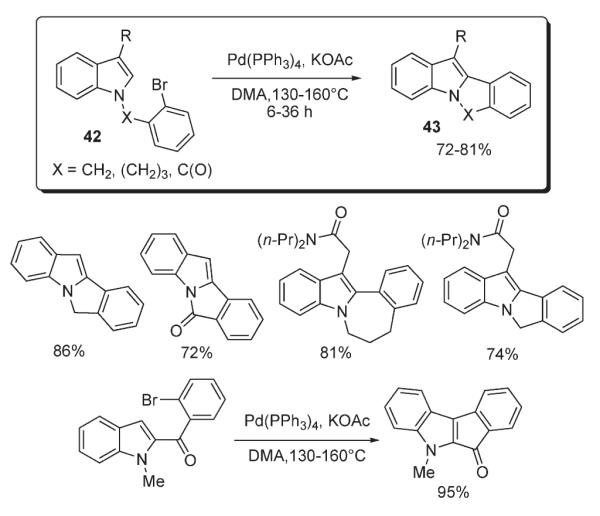
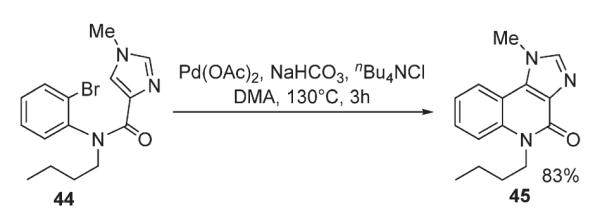
Suzuki reported synthesis of imidazo[4,5-c]quinolin-4(5H)-one ring systems 45 via an intramolecular palladium-catalyzed cyclization of 44 (Scheme 18).33 The effect of solvent and type of base used in the C–H arylation of imidazole ring were thoroughly investigated.33
Scheme 18.
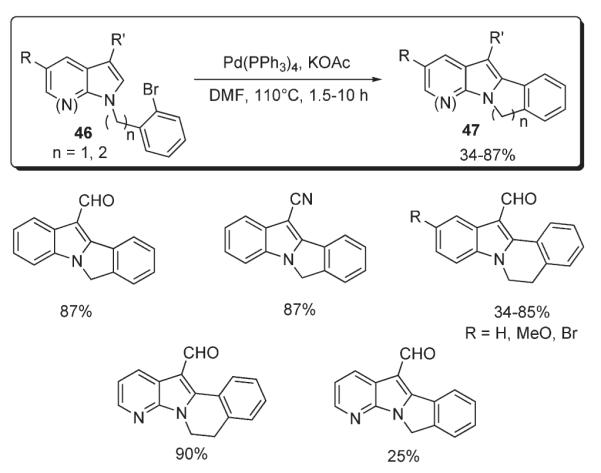
In 1995, Merour reported a protocol for the intramolecular annulation of 46, leading to polycyclic indole- and pyrrolo[2,3-b]pyridine-containing structures 47 (Scheme 19).34 The employment of Pd(PPh3)4–KOAc catalytic system allowed for obtaining annulated products 47 in moderate to very high yields. Notably, it was demonstrated that this method perfectly tolerates sensitive functional groups, such as aldehyde and halogen (Scheme 19).34
Scheme 19.
In 1997, Lemaire reported a method for the arylation of thiophenes 48 under Jeffery’s conditions35 (Scheme 20).36,37 It was found that in thiophenes bearing an EWG at C-2, arylation occurred at C-5 regiospecifically. In the case of 3-substituted substrates 48, the reaction was not regiospecific, leading to the formation of C-2–C-5 disubstituted products. The arylation of 2-carboxaldehyde thiophene proceeded in rather moderate yields; however, the reaction of 2-cyanothiophene gave arylated products in excellent yields (Scheme 20).36,37
Scheme 20.
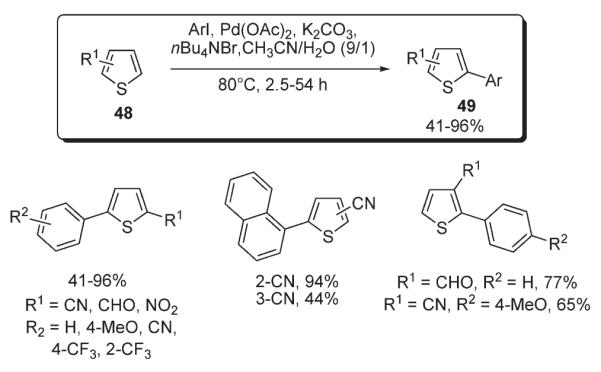
Later, Lemaire disclosed an approach towards 2-aryl- and 2-heteroaryl-benzothiophenes 51 (Scheme 21).38 It utilized a phosphine-free system: Pd(OAc)2, K2CO3, and crown ether. In particular, good results were obtained in the presence of dicyclohexyl-18-crown-6. In the case of sensitive functional groups, such as aldehyde, a quaternary ammonium salt was used as an additive instead of a crown ether. Importantly, this protocol allowed for obtaining high yields of arylated products 51 from benzothiazoles 50 bearing both electron-donating and electron-withdrawing groups (Scheme 21).38
Scheme 21.
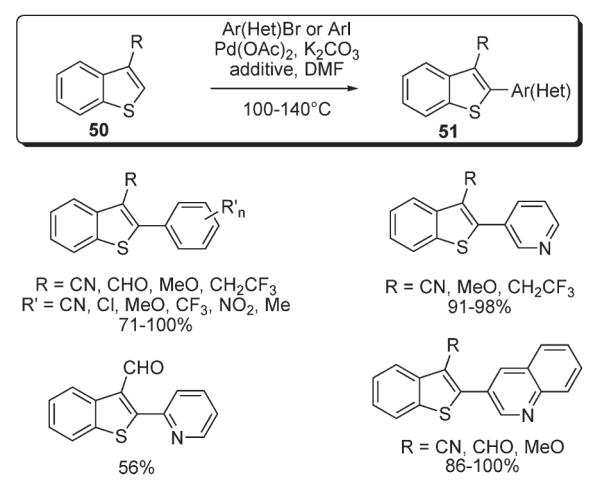
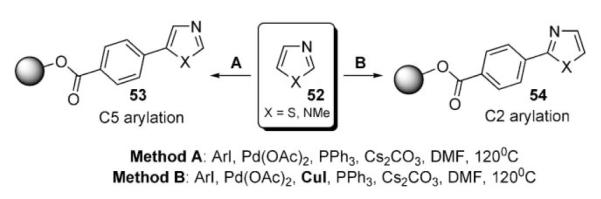
A method for regiodivergent palladium-catalyzed monoarylation of azoles 52 with aryl iodides, immobilized on a solid polymer support, was developed by Kondo (Scheme 22).39 The switch of regioselectivity (C-2 vs C-5) was primarily achieved by the employment of CuI, which led to the exclusive formation of C-2-functionalized product 54, while only C-5 arylation occurred in the absence of copper additive. Remarkably, unsymmetrical diarylation was demonstrated in the sequential derivatization mode (Scheme 22).39
Scheme 22.
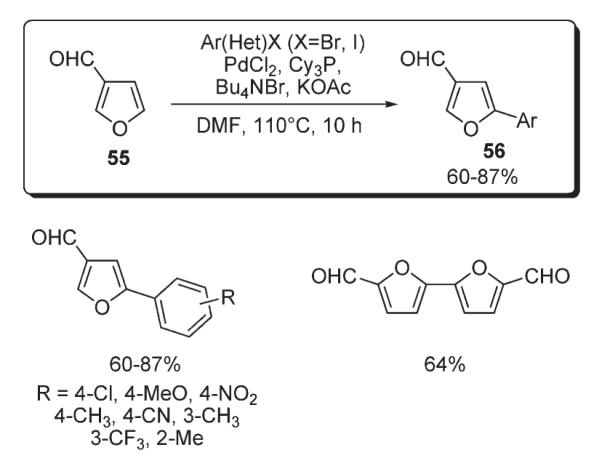
McClure reported a regioselective method for the C-5 arylation of furaldehyde 55 (Scheme 23).40 In the presence of PdCl2, PCy3, Bu4NBr, and KOAc, a variety of 5-arylfurfurals 56 were obtained in high yields. The mechanistic rationale proposed included both Heck-type carbopalladation and electrophilic metalation pathways. However, no experimental support for these mechanisms was provided (Scheme 23).40
Scheme 23.
Sharp developed a method for the regioselective Heck-type arylation of 3-ester-substituted furans and thiophenes (Schemes 24 and 25).5 Notably, it was suggested that a change in the reaction mechanism can occur depending on the reaction conditions (Scheme 24). Particularly, a nonpolar solvent and phosphine ligands used in Method A stabilize the σ-bonded Pd(II) species, favoring the Heck-type carbopalladation and affording intermediate i (see above, Scheme 2), which leads to the C-2-arylated product 58. Alternatively, in Method B, a polar solvent and an absence of stabilizing phosphine ligands are likely to promote the ionization of the Pd–X to form an electrophilic Pd(II) species. These species would be expected to react preferentially at the more electron-rich C-5 position of the heterocycle, resulting in the regioisomer 59. Application of these methods (A, B, C) for arylation of furans and thiophenes 60 led to selectively functionalized heterocycles 61 in moderate to good yields (Scheme 25).5
Scheme 24.
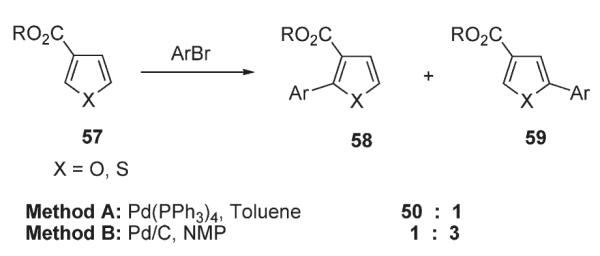
Condition-dependent Heck vs electrophilic arylation.
Scheme 25.
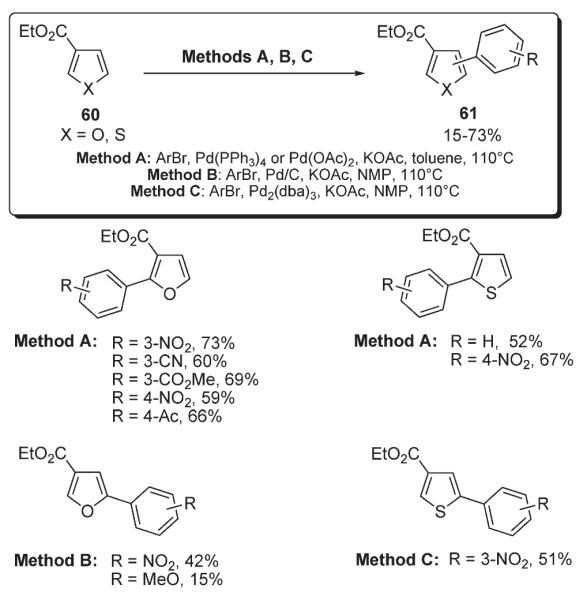
Mori reported a mild and regioselective C-2 arylation of thiazole 25 employing a palladium/copper(I) catalytic system in the presence of tetrabutylammonium fluoride (Scheme 26).41 A variety of 2,5-diarylthiazoles 63 bearing two different aryl groups were synthesized in a subsequent second-fold arylation of 62 under standard Cu-free conditions (Scheme 26).41
Scheme 26.
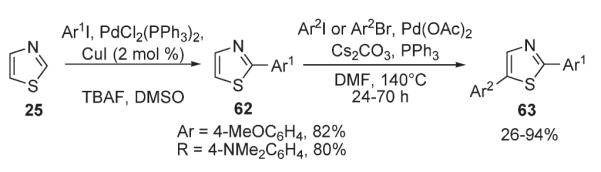
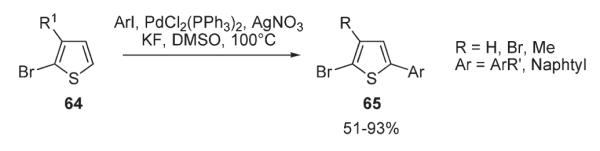
Later, Mori investigated Pd-catalyzed arylation reactions of thiophenes 64 possessing Br-substituent at C-2 with aryl iodides, while preserving the carbon–bromine bond (Scheme 27).42 The arylation with ArI was shown to proceed selectively at C-5 in the presence of PdCl2(PPh3)2, AgNO3, and KF in DMSO, affording high to excellent yields of arylated bromothiophenes 65, building blocks for further derivatization via standard cross-coupling techniques. Remarkably, no homocoupling reaction of bromothiophenes 64 occurred under these conditions.
Scheme 27.
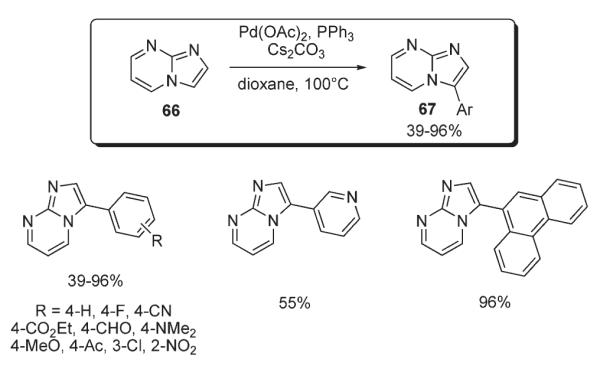
A method for direct arylation and heteroarylation of imidazopyrimidine 66 was reported by Li (Scheme 28).43 The protocol involved employment of the Pd(OAc)2/PPh3 catalytic system in the presence of Cs2CO3 in dioxane, affording the corresponding aryl derivatives 67 with good to excellent yields (Scheme 28).43 It was suggested that this arylation proceeds via Miura’s electrophilic mechanism.3
Scheme 28.
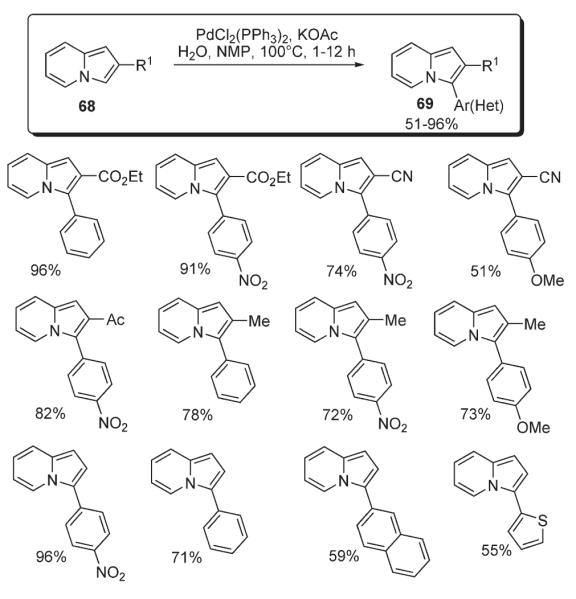
Gevorgyan reported an efficient and regioselective method for C-3 arylation and heteroarylation of indolizines 68 (Scheme 29).6 It was demonstrated that a variety of substituents on both the indolizine and aryl bromide are tolerated, providing easy access to substituted indolizines 69 in yields ranging from good to very high. Detailed mechanistic studies (Schemes 3–6, Table 1) strongly supported an electrophilic mechanism for this transformation.6
Scheme 29.
Sames reported a method for the selective C-2-arylation of N-substituted indoles 70 (Scheme 30).44 Optimization of the reaction conditions and catalyst loads has identified an optimal protocol which allowed for up to good yields of arylated derivatives 71. This method exhibited good functional group tolerance. Interestingly, it was demonstrated that employment of haloarenes possessing bulky ortho-substituents afforded mixtures of C-2 and C-3 substituted indoles 72 and 73 (Scheme 30).16,44 A selective C-3 arylation of N-unprotected indole (74) was achieved by the employment of magnesium bases (Table 2).16 Interestingly, arylation of 75, in which nitrogen is protected via treatment of 74 with a combination of MeMgCl with TMEDA (tetramethylethylenediamine) or Mg(HMDS)2, selectively led to 3-phenylindole 76. This result was explained in terms of the formation of a sterically demanding magnesium which, in combination with bulky phosphine ligand at the arylpalladium (see above: Path C, Scheme 7), governed the regioselectivity of arylation. Systematic mechanistic studies on this transformation were discussed above (Schemes 7 and 8).16
Scheme 30.
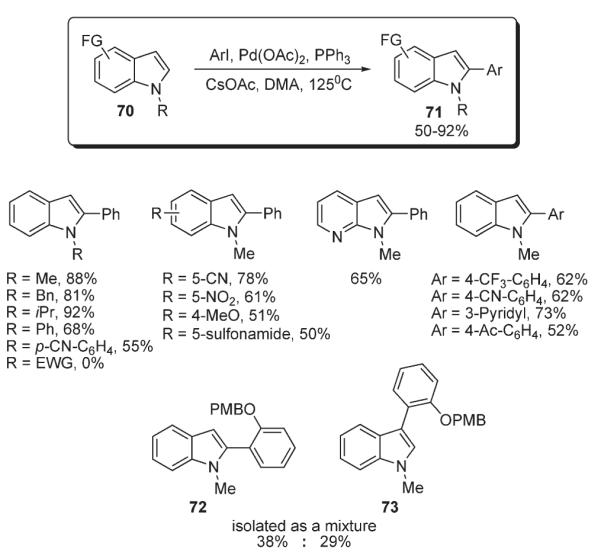
Table 2.
Control of regioselectivity by the choice of magnesium salt
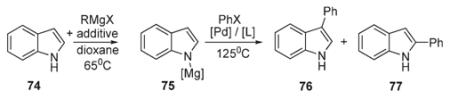
| X | [Mg] | Yield | 75:76 |
|---|---|---|---|
| I | MgCl | 24% | 7 : 1 |
| I | MgCl–TMEDA | 61% | 14 : 1 |
| Br | MgCl–TMEDA | 96% | 67 : 1 |
| I | MgN(TMS)2 | 77% | 26 : 1 |
Conditions: Pd(OAc)2 (2.5 mol%), Ph3P (10 mol%), IMes (2.5 mol%)
Sames also disclosed an efficient method of C–H arylation of 2-(trimethylsilyl)ethoxymethyl (SEM) protected azoles 78, including pyrroles, indoles, imidazoles, and imidazopyridines (Scheme 31).45 The reaction was catalyzed by employment of palladium imidazolyl carbene complexes 80. Remarkably, simple deprotection of the SEM group allowed for convenient access to aryl-substituted azoles, not available by direct arylation (Scheme 31).45
Scheme 31.
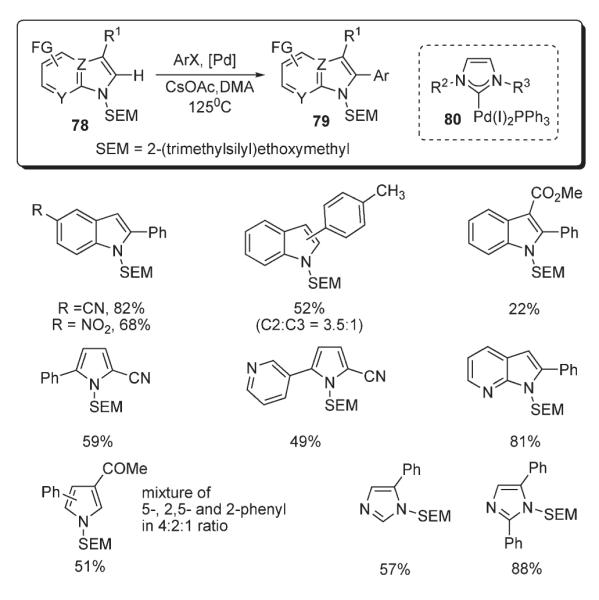
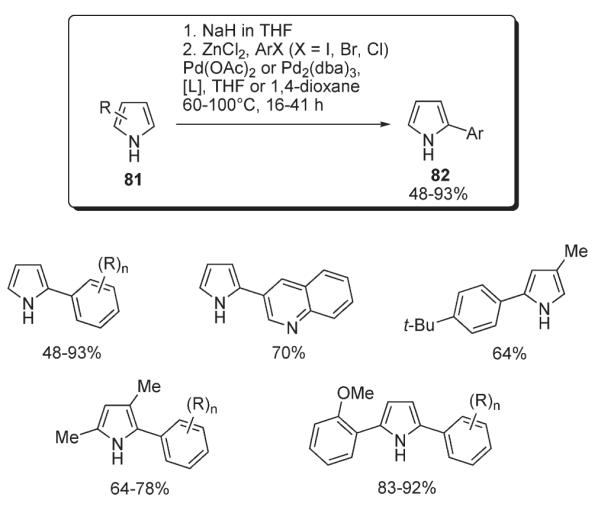
A general method for the conversion of pyrrole N-anions to 2-aryl derivatives 82 was proposed by Sadighi (Scheme 32).46 It was shown that in the presence of Pd(OAc)2 or Pd2(dba)3, along with sterically-demanding 2-(dialkylphosphino)biphenyl ligands, the Zn salt of pyrrole 81 smoothly undergoes cross-coupling with chloro- and bromoarenes and heteroarenes to afford arylated products 82 in moderate to very high yields (Scheme 32).46
Scheme 32.
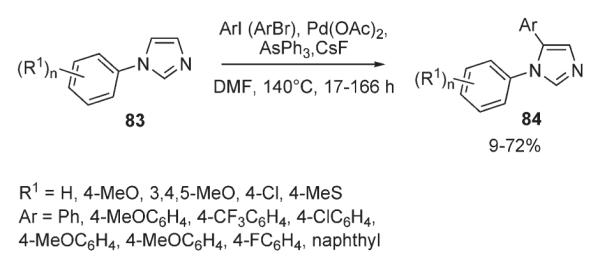
Rossi demonstrated that a variety of 1,5-diarylimidazoles 84 can be synthesized via a direct palladium-catalyzed C–H arylation of 1-arylimidazoles 83 (Scheme 33).47 The protocol involves a Pd(OAc)2/AsPh3 catalyst system. Although the yields are often moderate, very high degrees of regioselectivity were achieved. It was demonstrated that experimental data on the reaction selectivity supports the electrophilic mechanism proposed by Miura3 for arylation of azoles and are in accordance with the trend of nucleophilicity in azole ring C-5>C-4>C-2.
Scheme 33.
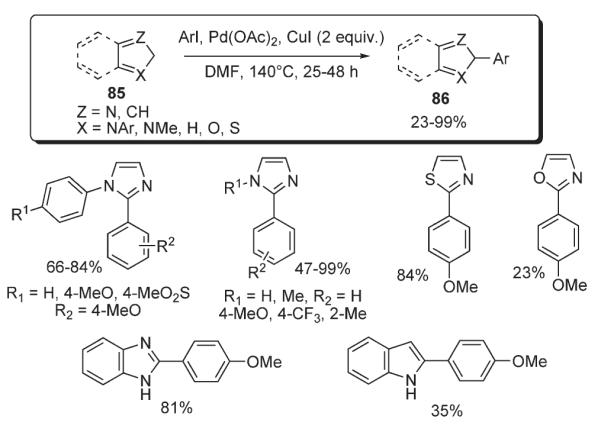
Very recently, Rossi reported developments on C-2-regioselective arylation of a number of different heterocyclic systems including thiazoles, oxazoles, free and N-substituted imidazoles, benzoimidazoles and indoles 85 (Scheme 34).48,49 Under the unprecedented base- and ligand-free conditions, complete regioselectivity has been achieved. The protocol involved catalytic Pd(OAc)2 and stoichiometric CuI in DMF. Remarkably, no products of N-arylation of N-unprotected heterocycles were observed under these conditions. The regioselectivity of arylation was rationalized by a mechanism involving the formation of a 2-copper derivative, followed by transmetalation with aryl palladium(II) halide species with subsequent reductive elimination (Scheme 34),49 analogously to the protocol reported by Mori for C-2 arylation of thiazoles (Scheme 26).41
Scheme 34.
An example of palladium-catalyzed intramolecular cross-coupling of a heterocyclic C–H bond with aryl iodide was reported by Trauner in the context of his efforts in the total synthesis of (±)-Rhazinilam 89 (Scheme 35).50 The key step, C–H derivatization of an unactivated pyrrole ring in 87 afforded a 9-membered ring in 88 with 47% yield. Importantly, the presence of an unprotected amide functionality was shown to be incompatible with this protocol (Scheme 35).50
Scheme 35.
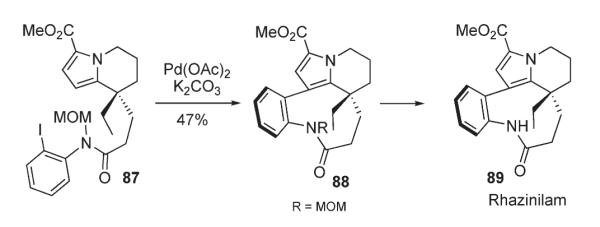
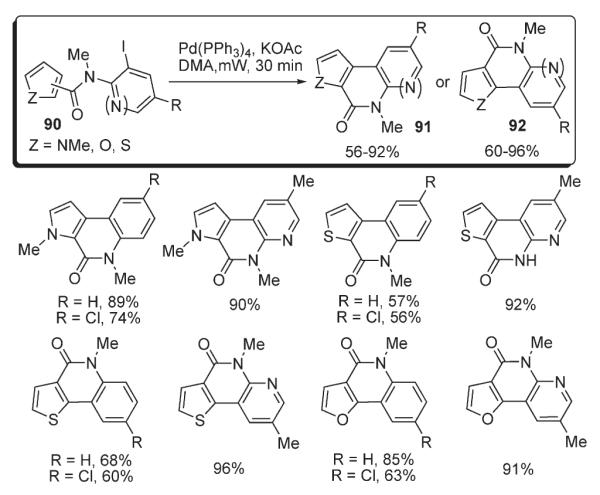
Recently, Beccalli reported a strategy for the preparation of tricyclic fused quinolones 91 and naphthyridones 92 via the palladium-catalyzed intramolecular arylation of 90 (Scheme 36).51 Microwave irradiation was shown to be advantageous over conventional heating, as it accelerates the coupling reactions and leads to better yields.
Scheme 36.
Lautens demonstrated an elegant approach for the formation of six- and seven-membered rings, which allowed for the efficient assembly of polycyclic frameworks. The method involved a norbornene-mediated tandem ortho-alkylation/C–H functionalization between an aryl iodide and a bromoalkyl heterocycle (Scheme 37).56 The key alkylation step of this cascade transformation proceeds via a Catellani reaction.52 Among possible mechanisms for the C–H functionalization step, Lautens names a Heck reaction, a direct nonelectrophilic palladation at C-2 (although requiring a directing group53–55), and electrophilic substitution at C-3 followed by migration of palladium to C-2.16 Thus, a variety of substituted annulated indoles 95 containing six- and seven-membered rings were synthesized in good to excellent yields from bromoalkyl indoles 94 and aryl iodides in the presence of catalytic amounts of Pd(OAc)2, tri-2-furylphosphine, Cs2CO3 and a stoichiometric amount of norbornene (Scheme 38).56
Scheme 37.
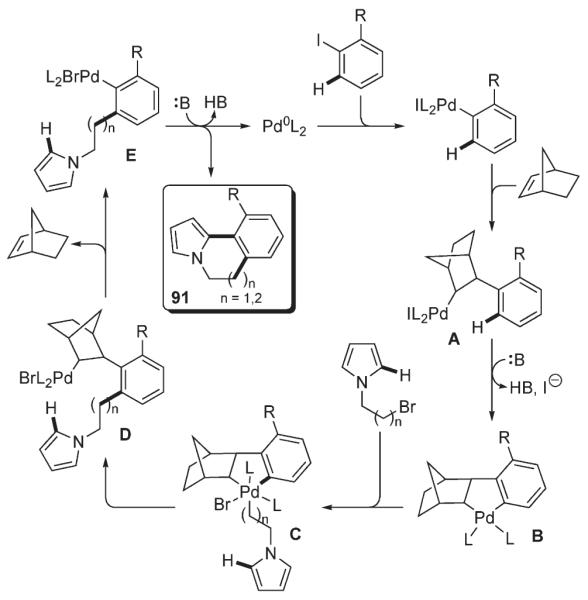
Scheme 38.
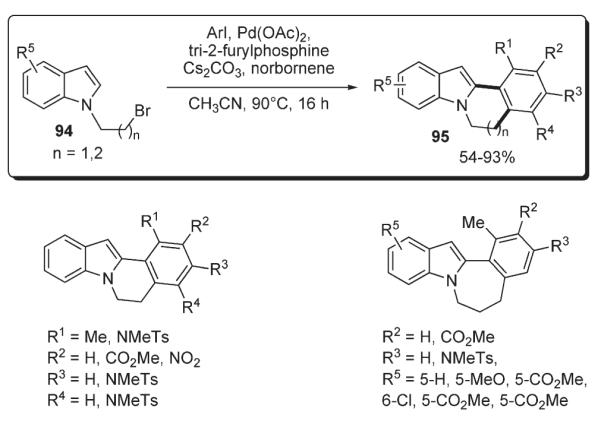
Further, Lautens applied this methodology for annulation of pyrroles and pyrazoles 96 (Scheme 39).57 Similar to the previous example,56 this one-pot protocol was proven to be very effective towards formation of a number of fused six- and seven-membered ring systems.57
Scheme 39.

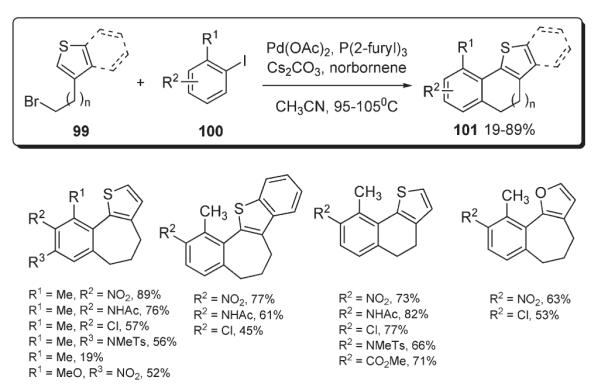
Very recently, Lautens disclosed the synthesis of polycyclic furans and thiophenes 101 via an analogous one-pot alkylation–intramolecular heteroarylation sequence (Scheme 40).58 It was found that the reaction provides good to excellent yields of oxygen and sulfur-containing polycyclic products 101 only when electron-deficient aryl iodides are used. The fact that electron-rich aryl iodides do not work equally well provided additional insight into the mechanistic aspect of this process. Since the arylation step involves an attack at the electron-deficient arylpalladium(II) halide, it was concluded that electron-rich aryl component 100 stabilizes Pd(II) species, reducing its electrophilicity and, consequently, making it less reactive in the arylation step.58
Scheme 40.
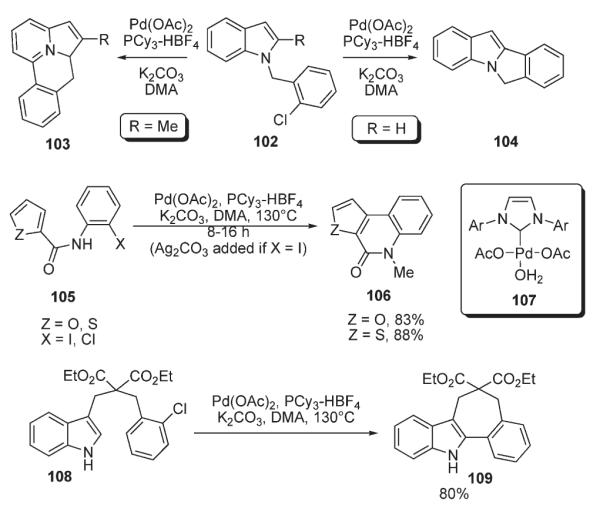
A method for intramolecular arylation of simple arenes and heteroarenes 102 was developed by Fagnou (Scheme 41).59,60,61 The catalytic system consisting of Pd(OAc)2 and PCy3–HBF4 (or palladium complex 107) enabled formation of polycyclic ring systems with indole (103, 104 and 109), furan, and thiophene 106 fragments. Catalyst poisoning during this reaction was observed. It was shown to be associated with the accumulation of KI in the reaction media when aryl iodides are used. Consequently, addition of a stoichiometric amount of silver salts allowed this problem to be overcome. Mechanistic studies revealed a “kinetic importance” of the C–H bond cleavage step, which allowed the mechanism to be rationalized as proceeding via metalation involving either σ-bond methathesis or an SE3 C–H functionalization step.59–61
Scheme 41.
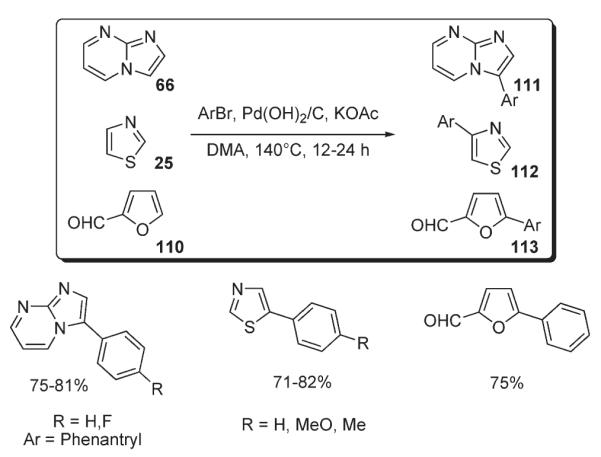
Further, Fagnou demonstrated that Pd(OH)2/C (Pearlman’s catalyst)–KOAc catalytic system effectively works in direct C–H arylation reactions affording high yields of aryl-substituted thiazoles 112, imidazopyrimidines 111 and furans 113 (Scheme 42).62
Scheme 42.
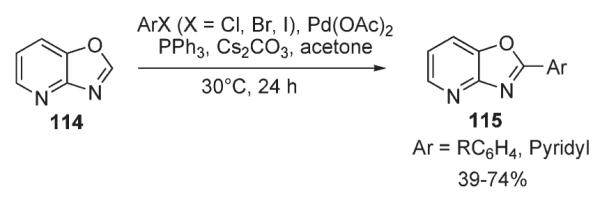
Very recently, Zhuravlev reported a method for Pd-catalyzed arylation of oxazolo[4,5-b]pyridine 114 (Scheme 43).63 Remarkably, the reaction proceeded efficiently at temperatures as low as 30 °C and exhibited good functional group tolerance at the aryl coupling partner, including derivatized amino acids. It was suggested that the combination of the electron-deficiency of the oxazolopyridine system and its high reactivity might indicate that arylation in this ring system proceeds via a non-electrophilic pathway. Additional studies provided certain support for this suggestion: a substantial proton–deuterium exchange was observed in acetone-d6 in the presence of Cs2CO3, supporting the possible involvement of an anionic intermediate, analogous to that proposed by Miura.3
Scheme 43.
1.3. Arylation reactions involving PdII/PdIV manifold
In the last few years an impressive array of publications came from the groups of Sanford,64,65 Yu,66,67 and Daugulis68–72 on oxidative C–H functionalization of arenes. The methods, operating via PdII/PdIV couple, were proposed to be advantageous over those employing Pd0/PdII manifold as they exhibit higher functional group tolerance, and often operate under milder conditions.
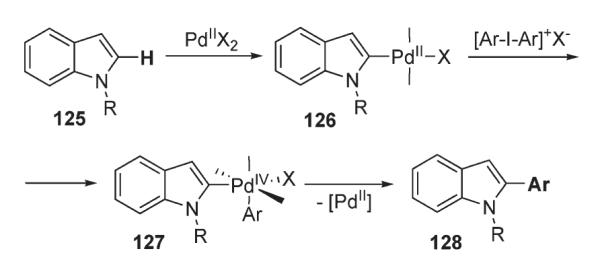
In 2006, Sanford reported an efficient protocol for the regioselective arylation of free and N-substituted indoles and pyrroles (Scheme 44).73 In this method, iodonium salts [Ar–I–Ar]BF4 (115 and 118) were used as arylating agents. Remarkably this mild room temperature method allowed for the efficient synthesis of a variety of C-2-arylated indoles and pyrroles with very good functional group tolerance and high yields (Scheme 44). Interestingly, arylation of N-unprotected and N-methyl indoles was similarly effective. Moreover, as a consequence of a PdII/PdIV mechanism (Scheme 45), this methodology demonstrated not only high functional group tolerance, but appeared to be completely air/moisture insensitive, in contrast to the traditionally used methodology, involving a Pd0/PdII pair.
Scheme 44.
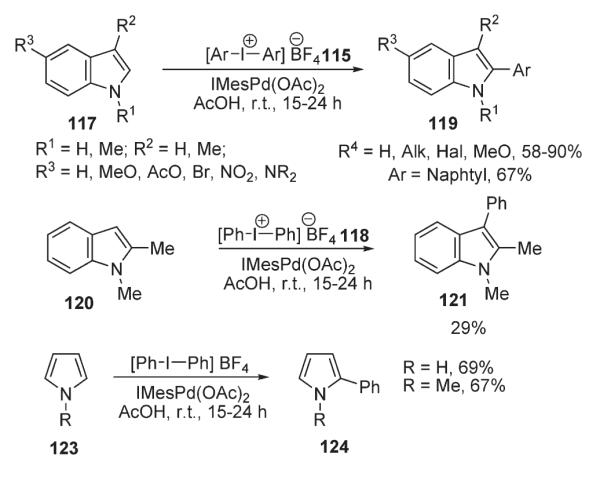
Scheme 45.
1.4. Arylation reactions involving Rh catalysis
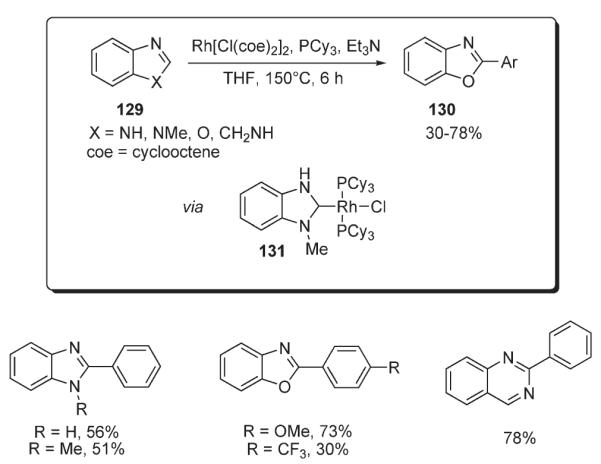
In 2004, Bergman and Ellman reported a method for the rhodium-catalyzed arylation of various heterocyclic systems 129: benzimidazoles, benzothiazoles, quinazolines, dihydroquinazolines, and oxazolines (Scheme 46).74 The protocol utilizes [RhCl(coe)2]2 complex in a combination with PCy3 and Et3N, affording the arylated products 130 in moderate to very good yields. Remarkably, N-unprotected heterocycles were tolerated in this method. Preliminary mechanistic studies revealed the involvement of rhodium N-heterocyclic carbene complex 131, which was isolated and characterized by X-ray analysis (Scheme 46).74,75
Scheme 46.
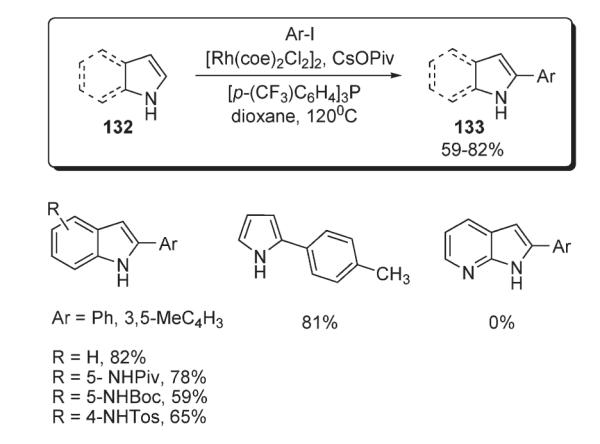
Later, a similar methodology was applied by Sames for arylation of indoles and pyrroles 132 (Scheme 47).76 Application of a modified catalytic system, consisting of [Rh(coe)2Cl2]2, [p-(CF3)C6H4]3P, and CsOPiv, allowed the arylation of free indoles and pyrroles in high yields to be performed (Scheme 47). It was shown that, analogously to the previous example (Scheme 46),74 this catalytic system targets specifically C–H bonds in the presence of more acidic N–H bonds. This selectivity was explained in terms of a greater electrophilicity of the Ar–Rh(III) species (compared to Ar– Pd(II)) which, in conjunction with the electron- deficient phosphine- and pivalate ligands, gain additional reactivity. It was observed that this arylation method is incompatible with pyridine-containing substrates possessing nucleophilic nitrogen, which is a well-known limitation in the chemistry involving rhodium catalysis.77
Scheme 47.
Shortly thereafter, Bergman and Ellman reported a method for Rh-catalyzed microwave-assisted coupling of various azoles 134 with aryl bromides (Scheme 48).78 Employment of the slightly modified procedure ([RhCl(coe)2]2 with trialkylphosphine ligand A, bulkier than PCy3) in combination with microwave irradiation allowed for a dramatic shortening of reaction times (40 minutes vs 6 hours reported earlier,74 Scheme 46).
Scheme 48.
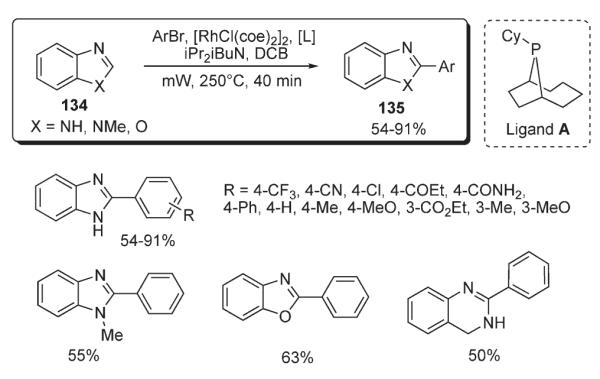
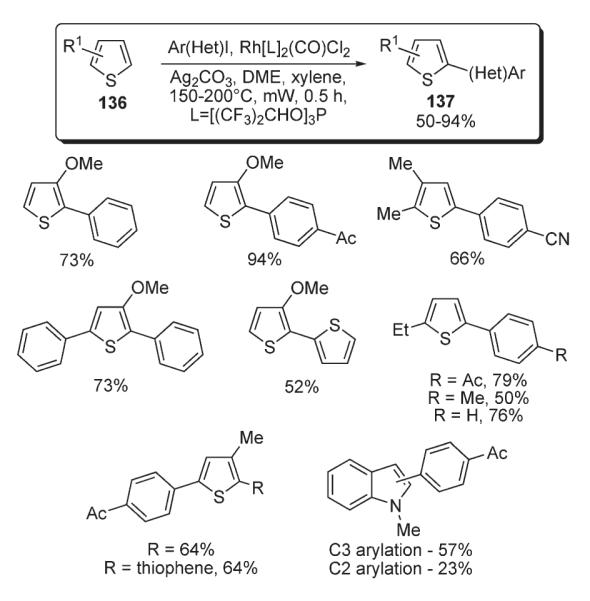
Itami reported studies on rhodium-catalyzed C–H arylation of indoles and thiophenes 136 (Scheme 49).79 The protocol employed RhCl(CO){P[OCH(CF3)2]3}2 complex, bearing π-accepting ligands, in combination with Ag2CO3. An electrophilic metalation of heterocycles with the aryl–Rh(III) species was proposed as a key step for this transformation. This methodology was also efficient for the arylation of moderately electron-rich arenes: such as anisole and 1,3-dimethoxybenzene. The ortho–para selectivity of arylation in this process was entirely consistent with an electrophilic mechanism.79
Scheme 49.
In summary, the employment of Pd0 or PdII catalysts with excess base at elevated temperatures with slight variations in techniques can be considered as a general method for the arylation of electron-rich heteroaromatics. Generally, arylation of pyrroles, furans, thiophenes, indoles, and other substrates with one heteroatom shows strong preference to the position adjacent to the heteroatom. However, arylation of indoles can be redirected to the C-3 position by employing anionic N-magnesium derivatives.16 Additionally, it was demonstrated that arylation of furans and thiophenes bearing an EWG at C-3 can be achieved selectively at C-2 or C-5 by switching reaction conditions to favor either electrophilic or Heck-type mechanisms, leading to different products.5 Arylation of azoles, in most cases, is not selective and leads to the mixtures of C-2 and C-5 arylated products or results in bis-arylation. Nevertheless, excellent C-2 selectivity can be achieved in the presence of stoichiometric amounts of Cu salts.3,41,48
2. C–H arylation of electron-deficient heterocyclic systems
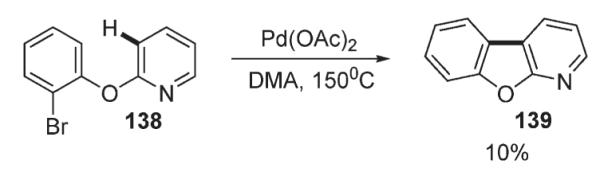
The first example of direct C–H functionalization of an electron-deficient heterocycle was reported by Ames in 1984 (Scheme 50).80 In the context of their studies on intramolecular palladium-catalyzed cyclizations of haloarenes, it was found, that pyridine ring in 138 undergoes intramolecular C–H arylation at the C-3 position by an aryl bromide. However, the reaction was reported to be sluggish and very low yielding, producing 139 in 10% only (Scheme 50).
Scheme 50.
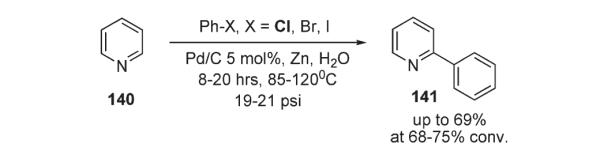
Later, an interesting method for C–H cross-coupling of pyridine 140 with phenylhalides, catalyzed by heterogeneous palladium on carbon in the presence of Zn dust, was reported by Sasson (Scheme 51).81 The mechanism was rationalized as a radical process or, alternatively, as a heterogeneous Heck-type reaction. This method allowed for the regioselective preparation of 2-arylpyridine 141, though it was accompanied by comparable amounts of biaryl side-products.81
Scheme 51.
Recently, Fagnou reported an elegant solution to the arylation of pyridines (Scheme 52).82 He demonstrated that, unlike the pyridine, a pyridine N-oxide 142 can undergo smooth, selective and high-yielding C-2-arylation with a variety of aryl bromides in the presence of Pd(OAc)2/PtBu3–HBF4 catalytic system (Scheme 52). After completion of arylation, pyridine oxide can be efficiently reduced to pyridine derivative 143 in very good overall yield. Importantly, the mechanistic studies pointed out that electrophilic SEAr is not operating in this case. A competition reaction between electron-deficient and electron-rich pyridine oxides (144 vs 145, Scheme 53) was performed. It was suggested, that if an SEAr mechanism operates, then electron-rich substrate 145 would react preferentially. However, electron-deficient pyridine 144 reacted faster, yielding product 146, accompanied by negligible amounts of arylpyridine 147. The kinetic isotope effect studies further disproved the involvement of SEAr mechanism: in the one-pot arylation reaction of pyridine oxide and pyridine oxide-d5, a primary KIE of 4.7 was observed, a value incompatible with the electrophilic mechanism, for which loss of proton is not a rate-limiting step (Scheme 53).15 Although the SEAr mechanism was disproved, no mechanistic rationale to account for the observed transformation has been proposed.82
Scheme 52.
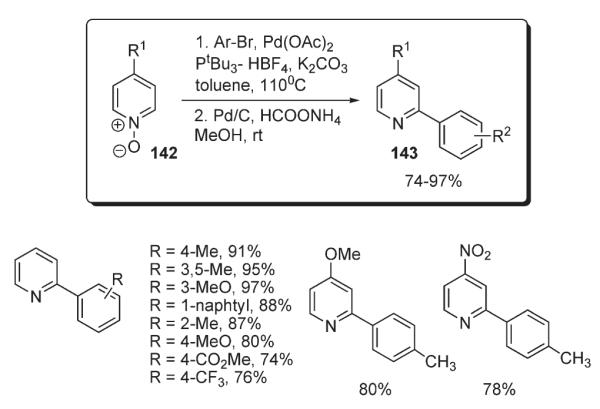
Scheme 53.
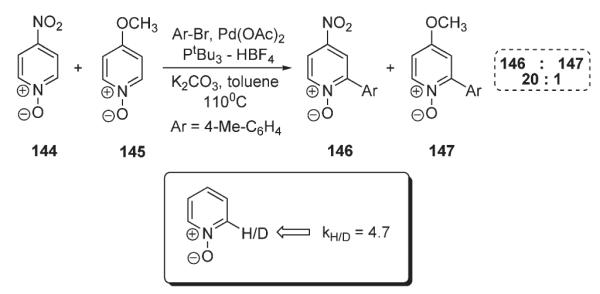
Competitive experiment and kinetic studies of pyridine oxide arylation.
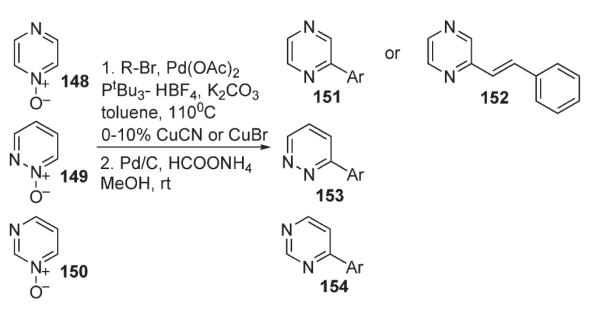
Later, Fagnou extended this methodology for functionalization of diazine N-oxides series (Scheme 54).83 Direct arylation of pyrazine- 148, pyridazine- 149, and pyrimidine-containing 150 substrates was performed with a wide range of both electron-rich and electron-deficient aryl-iodides, -bromides and -chlorides. It was also found that addition of catalytic amounts of Cu(I) salts enhances reactivity of low-reactive substrates like pyrimidine N-oxides 150.
Scheme 54.
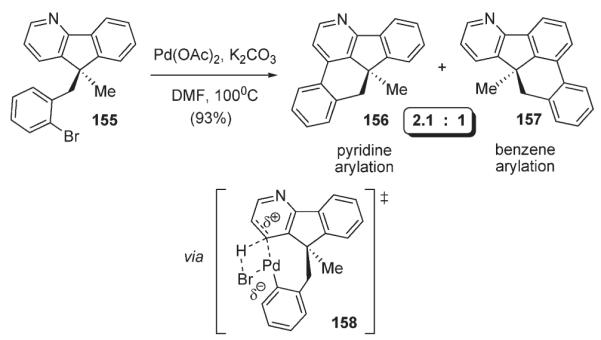
Recently, Echavarren also confirmed, that the Pd-catalyzed arylation of pyridine does not operate via an electrophilic aromatic substitution mechanism.84 In this work, an ambident substrate 155 (Scheme 55), which can be intramolecularly arylated at both pyridine and benzene rings, was tested in the arylation reaction conditions. Notably, the reaction yielded regioisomer 156, resulting from arylation of the pyridine ring as the major product. In other words, substitution mostly occurred at the more electron-deficient pyridine ring, rather than at the more electron-rich benzene. To account for the observed regioselectivity, a concerted proton abstraction mechanism operating via a four-membered transition state (158, Scheme 55) was proposed.84,85
Scheme 55.
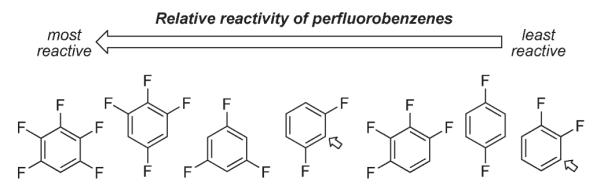
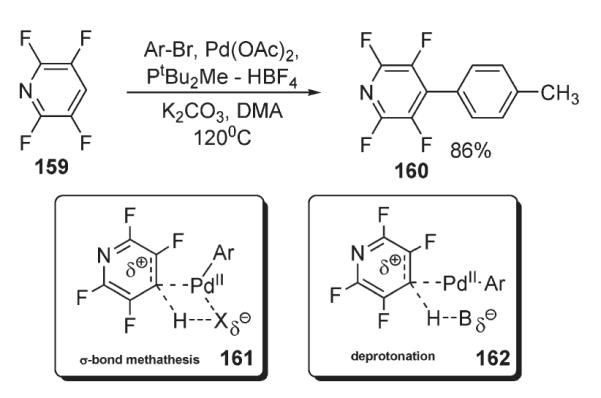
Almost simultaneously, Fagnou disclosed his computational and experimental studies on the arylation of perfluorobenzenes (Schemes 56, 57).86,87 The computational studies indicated that C–H bond cleavage occurs via a concerted mechanism. A complete inversion of the reactivity trend to that expected for the SEAr mechanism was observed in the arylation of a series of perfluorinated aromatics, where more electron-deficient substrates reacted faster (Scheme 56). In addition, in the cases of substrates with more than one C–H bond available, arylation occurred at the most acidic site, which in fluorobenzenes is ortho to a fluorine atom.88 Notably, under these conditions, 2,3,5,6-tetrafluoropyridine 159 was arylated in a very good yield (Scheme 57). Computational studies supported that this selectivity originates not from palladium–fluorine stabilizing interactions, but from the increased C–H acidity. Additionally, primary KIE of 3.0 at C–H bond was observed, indicating the “kinetic significance” of the C–H bond cleavage event. It was suggested that this reaction may proceed via a concerted palladation and proton abstraction by the halogen anion (intermediate 161, Scheme 57) at palladium or by an external base (intermediate 162), analogously to the mechanism proposed by Echavarren84 (intermediate 158, Scheme 55).
Scheme 56.
Scheme 57.
3. Other types of C–H functionalization
3.1 Reactions involving C–C bond formation
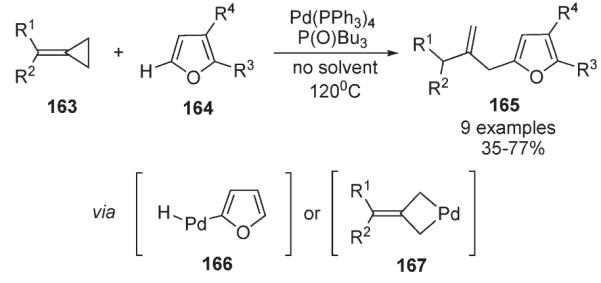
Yamamoto reported a mild and regioselective method for Pdcatalyzed hydrofurylation of alkylidenecyclopropanes, which provides access to 2-allyl furans and benzofurans 165 (Scheme 58).89 It was proposed that this process may operate via two alternative key intermediates; first involving a hydride–palladium intermediate 166, and second, a palladocyclobutane 167, resulting from the insertion of Pd0 into a distal bond of methylenecyclopropane 163 (Scheme 58).89
Scheme 58.
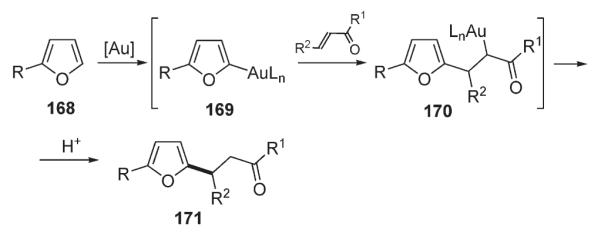
Hashmi found that furans 168 undergo a formal C–H alkylation with α,β-unsaturated ketones in the presence of gold catalyst (Scheme 59).90 It was suggested that the mechanism might involve activation of the enone by gold followed by an electrophilic aromatic substitution at C-5 position of furan to form a new C–C bond. Alternatively, the process might start with a direct electrophilic metalation of furan to form furyl gold intermediate 169 which, subsequently, undergoes a 1,4-addition to the unsaturated system and protonolysis to give the substituted product 171 (Scheme 59).90
Scheme 59.
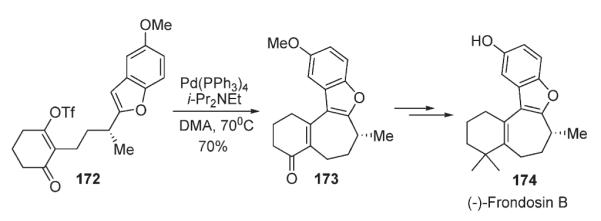
Within studies towards the total synthesis of (−)-Frondosin B 174, Trauner disclosed an intramolecular C–H crosscoupling of a triflate with the benzofuran fragment as a key macrocyclization step. (Scheme 60).91,92
Scheme 60.
The first method for an oxidative palladium-catalyzed intramolecular formal C–H alkylation of indoles 175 and 176 was reported by Stoltz (Scheme 61).93 Remarkably, the method allowed the annulation to be performed with good yields in three possible directions: C-2 to C-3 178, C-3 to C-2 177, and N to C-2 179. This method allows the construction of 5/5 or 5/6 fused, as well as 5/5-spiro skeletons (Scheme 61).83
Scheme 61.

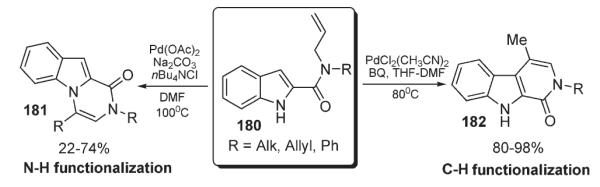
Further, Beccalli developed a protocol for the intramolecular regioselective cyclization of indole carboxamide derivatives 180 into β-carbolinones 182 or pyrazino[1,2-a]indoles 181 (Scheme 62).94,95 It was found that the regiochemistry can be completely controlled by the reaction conditions to allow for either C–H or N–H functionalization selectively or exclusively.
Scheme 62.
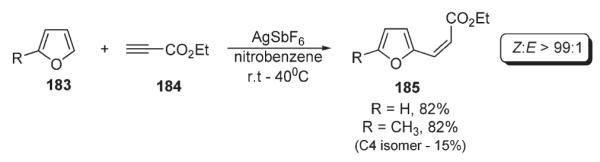
In the course of the investigations of gold- and silver-catalyzed hydroarylation of alkynes 184, Reetz disclosed examples of an alkyne addition to the furan ring of 183 (Scheme 63).96 Under mild conditions furan and methylfuran underwent formal C–H vinylation to afford C-2-functionalized products 185 with good selectivity and high yield.
Scheme 63.
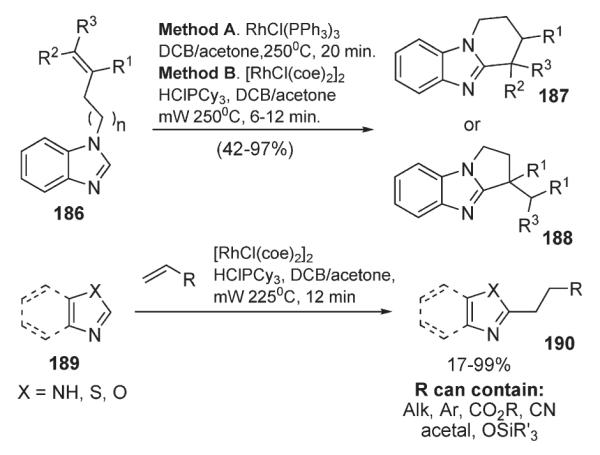
Bergman and Ellman developed a method for Rh-catalyzed intra- and intermolecular alkylation of azoles and their benzoanalogues (Scheme 64).97–99 It was demonstrated that the employment of microwave irradiation for intramolecular cyclizations of 186 allows reaction times of less than 20 minutes and for the tricyclic products 187 and 188 to be obtained in good yields. The intramolecular version of this reaction was shown to have excellent functional group tolerance: silyl ethers, acetals, esters, and nitriles were all compatible with this protocol (Scheme 64).97–99
Scheme 64.
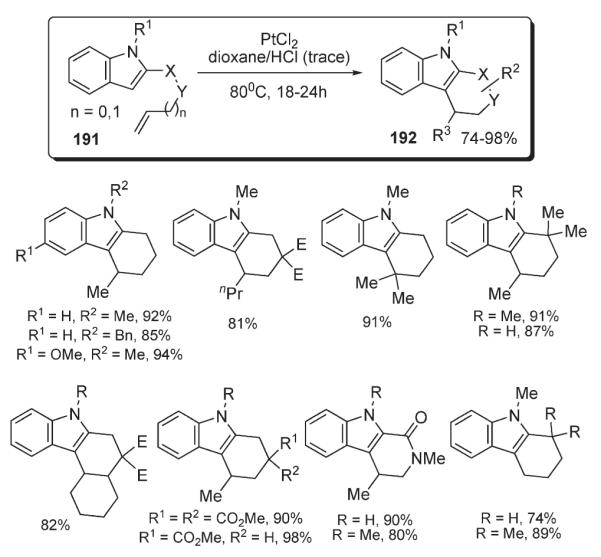
Widenhoefer developed a method for platinum-catalyzed intramolecular alkylation of indoles with tethered unactivated olefins 191 which allowed for construction of tri- and tetracyclic indole-containing structures 192 in excellent yields (Scheme 65).100 The mechanistic rationale (Scheme 66) involved a nucleophilic attack on a platinum-complexed olefin (i) with subsequent protonolysis of the intermediate (ii).100
Scheme 65.
Scheme 66.
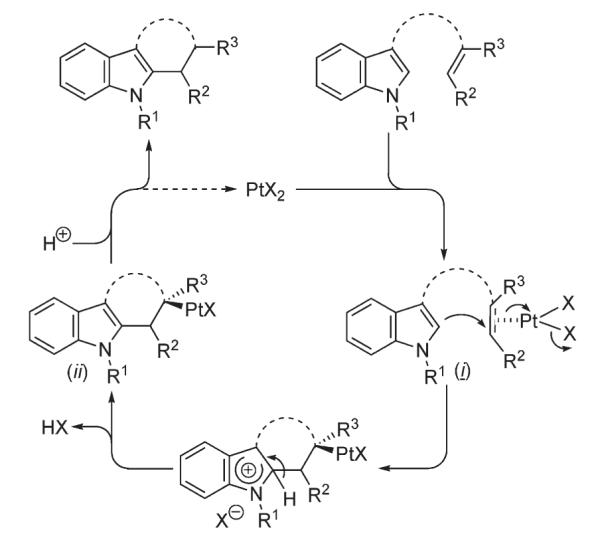
Generalized mechanistic rationale for intramolecular alkylation of indole with olefins.
Further, Widenhoefer disclosed a palladium-catalyzed protocol for the alkylation/carboalkoxylation of alkenyl-indoles 193 (Scheme 67).101 The method utilizes a PdCl2(CH3CN)2–CuCl2 catalytic system and allows for very mild, selective and high-yielding syntheses of functionalized policyclic indoles 194. Remarkably, this method represents the first example of transition metal-catalyzed addition of carbonyl group and carbon nucleophile across the double bond of an olefin (Scheme 67).101
Scheme 67.

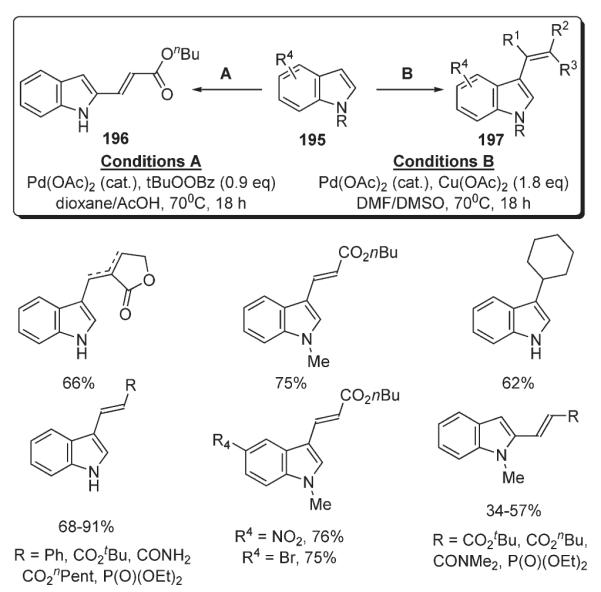
A method for selective and switchable intramolecular alkenylation of indoles 195 was developed by Gaunt (Scheme 68).102 It was found that the choice of solvent governs the regioselectivity of this transformation directing it selectively to C-2 or C-3 positions to produce 196 or 197, respectively. It was rationalized that the mechanism involves an initial electrophilic palladation step at C-3 center with a subsequent solvent-controlled C-3–C-2 migration of the PdX moiety. It was speculated that strongly coordinating solvents such as DMSO or acetonitrile disfavor the migration, thus leading to C-3-regioselectivity, while weakly coordinating solvents, such as 1,4-dioxane, in conjunction with AcOH, facilitate the migration affording the products of C-2 substitution.102
Scheme 68.
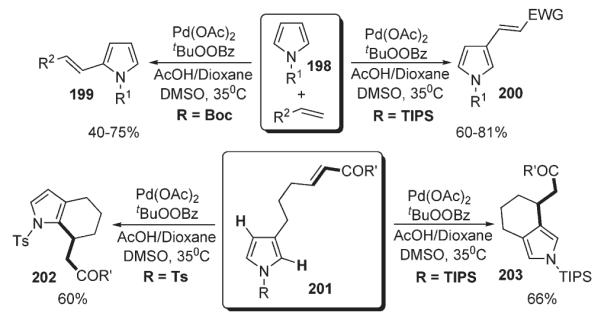
Further, Gaunt disclosed a method for C-2 and C-3 regioselective intermolecular C–H alkenylation and annulation of pyrroles 198 and 199 (Scheme 69).103 Notably, the regiochemistry in this mild and high-yielding aerobic protocol can be completely controlled via sterically and electronically tuned N-protecting groups to obtain either C-2 or C-3 functionalized products 199 and 200 selectively. In intramolecular series, introduction of an electron-withdrawing protecting group at 201, such as N-Ac, N-Boc, or N-Ts results in C-2-substitution to give 202 (electronic control). In contrast, N-TIPS pyrroles afford C-4-substituted products 203 exclusively, which was attributed to the shielding effect of sterically-demanding TIPS group, disfavoring C-2-palladation (steric control) (Scheme 69).103
Scheme 69.
The direct C–H addition of heterocycles to electron-deficient olefins in the presence of Au catalysts was investigated by He (Scheme 70).104 Indoles, furans and benzofurans 204 were shown to undergo the gold(III)-catalyzed formal alkylation with functionalized alkenes and alkynes. Remarkably, this exceptionally mild and high yielding protocol demonstrated excellent functional group tolerance, allowing the production of heterocyclic structures 205 bearing sensitive functionalities, such as aldehyde, carboxylic acid, and nitrile (Scheme 70).104
Scheme 70.
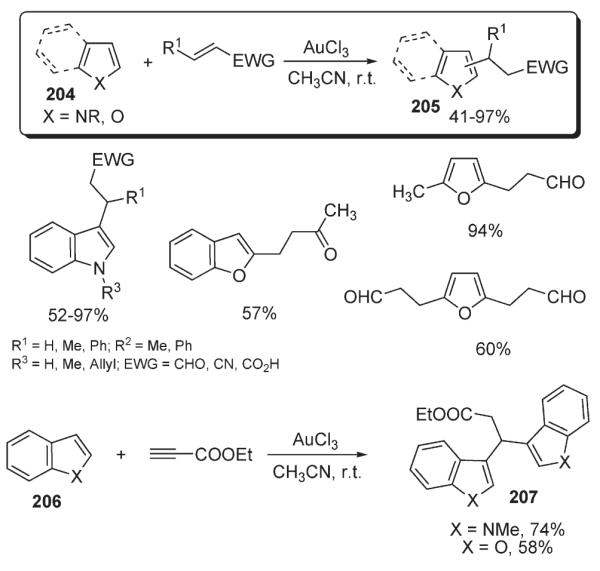
Furstner disclosed a method for platinum- and indium-catalyzed cyclization of 208 leading to the formation of indole, furan and thiophene-containing polycyclic structures 209 in good yields (Scheme 71).105 The mechanism of this reaction was rationalized in terms of the ability of “soft” metals to render an alkyne electrophilic, susceptible to nucleophilic attack by electron-rich heterocycle.
Scheme 71.
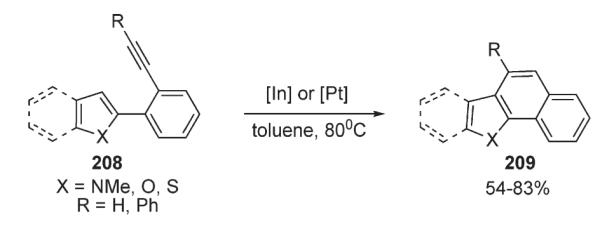
Nelson, in the context of his efforts towards (−)-rhazinilam, reported an asymmetric gold-catalyzed intramolecular pyrrole addition to enantioenriched allenes 210 leading to the formation of a chiral indolizidine fragment 212 (Scheme 72).106 Activation of the π-system of the allene by a metal (intermediate 211) with subsequent intramolecular pyrrole addition resulted in the efficient translation of allene chirality to the quaternary carbon of indolizidine unit (Scheme 72).106
Scheme 72.
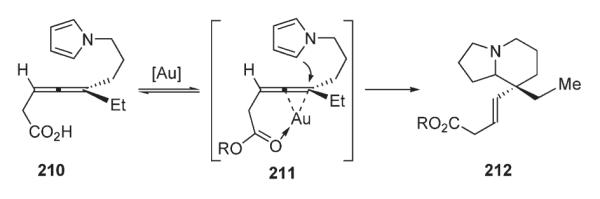
Later, Lu reported a method for the synthesis of carbazoles via the palladium-catalyzed intramolecular oxidative cyclization of 3-(3′-alkenyl)indoles 213 (Scheme 73).107 Carbazole ring system 214 formed as a result of 6-endo-trig-cyclization of the tethered terminal alkene fragment followed by benzoquinone-promoted aromatization. At the same time, substrates with internal double bond were shown to afford onlyexo-cyclization products. The protocol operates under relatively mild conditions in the presence of Pd(OAc)2 and stoichiometric amount of co-oxidant (Scheme 73).107
Scheme 73.
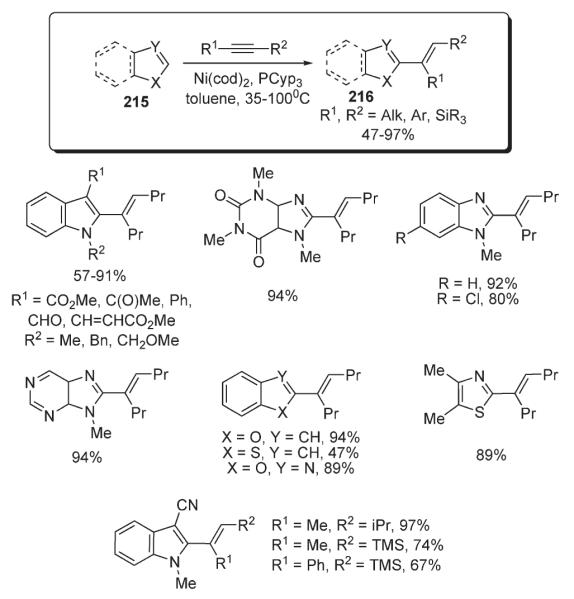
Very recently, Hiyama reported a versatile method for a nickel-catalyzed alkenylation of diverse heterocyclic systems 215 with alkynes (Scheme 74).108 A wide range of hetroarenes were shown to be compatible with this remarkably mild and high-yielding protocol. Exceptional functional group tolerance and chemoselectivity were also demonstrated. For example, it was emphasized, the Ar–H bond reacted exclusively over the formyl C–H bond, which is known to undergo addition across an alkyne under Ni/PR3 catalysis (Scheme 74).109
Scheme 74.
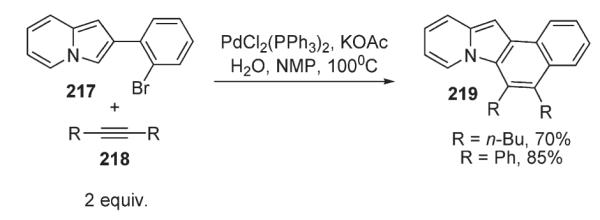
Recently, Gevorgyan developed a series of novel intra- and intermolecular cascade annulations on indole 15 and indolizine substrates 217 and 220, involving sequential arylation and Heck carbo-palladation steps.110 Cascade carbopalladation vinylation reaction was demonstrated on the indolizine substrate 217 (Scheme 75).110 Palladium-catalyzed intramolecular reaction of 2-(2-bromophenyl)indolizine 217 with internal alkynes resulted in aromatic polycyclic indolizines 219 in high yields (Scheme 75).110
Scheme 75.
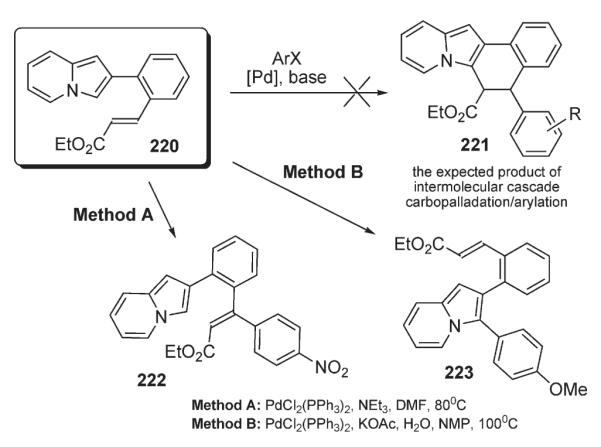
However, the attempts to perform a cascade Heck carbopalladation/formal alkylation on the indolizine substrate 220 to afford the expected tetracyclic indolizine 221 failed (Scheme 76).110 Nonetheless, it was found that employment of different reaction conditions allowed for highly chemoselective Heck reaction at the alkene moiety (222), or arylation of the indolizine ring (223).6
Scheme 76.
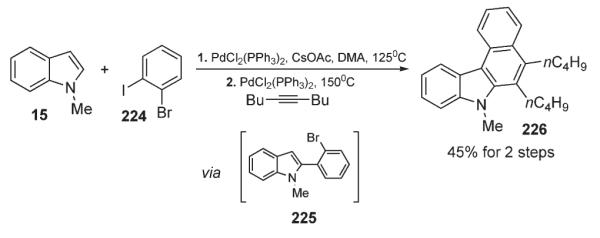
Another novel process involved a one-pot cascade proceeding via a halogen-selective C-2-arylation of indole 15 with 224 to form 226 (Scheme 77), followed by an intramolecular carbopalladation/vinylation sequence to yield benzocarbazole derivative 226 in reasonable overall yield.110
Scheme 77.
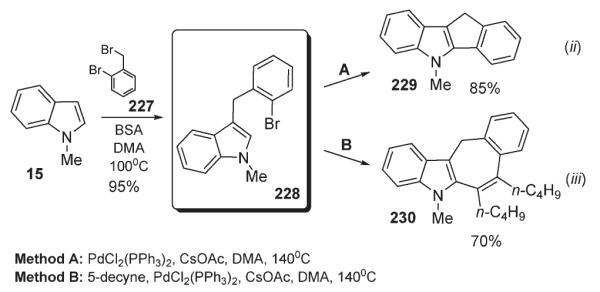
3-Benzylindole 228, readily available from 15 and 227 (Scheme 78), was shown to undergo an intramolecular palladium-catalyzed C-2 arylation forming a tetracyclic indole-containing heterocycle 229 (Scheme 78).110 It was further demonstrated that, under optimized conditions: in the presence of 5-decyne, a cascade carbopalladation/vinylation reaction takes place affording polycyclic indole 230 with high yield. Mechanistically, this one-pot cascade proceeds sequentially through a Heck carbopalladation of the triple bond followed by an intramolecular vinylation of indole ring to form 7-membered ring of 230 (Scheme 78).110
Scheme 78.
3.2 Reactions involving C–N and C–B bond formation
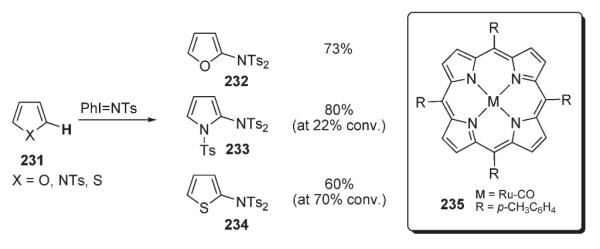
Che developed a method for the intermolecular amination of C–H bond of heterocycles (Scheme 79).111,112 In the presence of ruthenium complex 235, furan, thiophene, N-tosyl-pyrrole, as well as their benzo-analogues, afforded the corresponding amino-derivatives 232–234 in high yields. It was suggested, that these reactions might proceed via either initial direct nitrene insertion into the activated C–H bond or, alternatively, by aziridination with subsequent re-aromatization.111,112
Scheme 79.
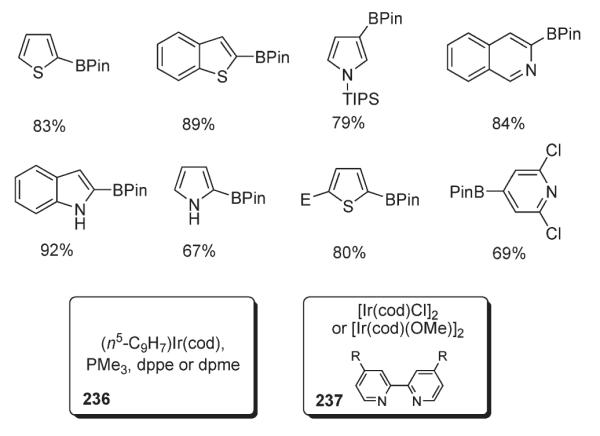
Hartwig demonstrated115,116 that a variety of heterocycles can serve as excellent substrates for C–H activation/borylation reactions catalyzed by irridium complexes 236 and 237114 (Scheme 80).113 Furans, thiophenes, pyrroles and indoles demonstrated very good C-2 selectivity, at the same time, it was shown that C–H borylation of pyrroles and indoles is not regioselective, though it can be redirected to the C-3 position by introducing a sterically demanding N-TIPS group (Scheme 80).111,116,117
Scheme 80.
4. Conclusions
In this review we have outlined the progress made in nearly two decades in the area of direct C–H functionalization of heterocyclic compounds. In spite of the enormous synthetic potential of these methods, most of them still require harsh reaction conditions and exhibit moderate functional group tolerance. In contrast to the well-elaborated methods for functionalization of electron-rich heterocycles, C–H activation of electron-deficient substrates is still in its infancy. However, the latest promising results indicate that this area of chemistry will be rapidly growing. Direct C–H functionalization of heterocycles has already gained widespread acceptance within the synthetic community due to its capacity to utilize simpler and cheaper precursors for construction of complex frameworks. This methodology has already been widely employed in the synthesis of diverse heterocyclic scaffolds, and finds increasing number of applications in the synthesis of complex natural products. Future discoveries promise to add to an already solid methodology of functionalization of electron-rich heterocycles and, we believe, to promote a rapid growth of functionalization of electron-deficient systems.
Acknowledgements
The support of the National Institutes of Health (Grant GM-64444) is gratefully acknowledged.
Biographies
Ilya Seregin was born in Moscow, Russia, in 1979. He received his BSc from Moscow State University in 2001. In 2001–2003 he was a graduate student at Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences. He is currently a PhD student in Professor Gevorgyan’s group at the University of Illinois at Chicago.

Vladimir Gevorgyan was born in Krasnodar, Russia, in 1956. He received his BSc from Kuban State University in 1978 and his PhD from the Latvian Institute of Organic Synthesis in 1984, where he was promoted to Group Leader in 1986. He spent two years (1992–1994) in Tohoku University in Sendai, Japan, the first as a JSPS Postdoctoral Fellow and the second as a Ciba-Geigy International Postdoctoral Fellow. In the following year (1995) he worked as a Visiting Professor at CNR, Bologna, Italy. He returned to Tohoku University in 1996 as an Assistant Professor and was promoted to Associate Professor in 1997. In 1999 he moved to The University of Illinois at Chicago as an Associate Professor. He was promoted to the rank of Full Professor in 2003. Prof. Gevorgyan’s current research interests cover four main areas. The first is concerned with development of highly selective Pd-catalyzed benzannulation reactions. The second area of interest focuses on development of novel transition metal-catalyzed methods for the synthesis of heterocyclic and naturally occurring compounds. The third area of interest covers the development of selective Lewis acid-catalyzed bond formation and cleavage reactions. The fourth deals with the chemistry of strained ring systems.

References
- 1 (a).For reviews on C–H activation, see: Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. Jia CG, Kitamura T, Fujiwara Y. Acc. Chem. Res. 2001;34:844. doi: 10.1021/ar000209h. Ritleng V, Sirlin C, Pfeffer M. Chem. Rev. 2002;102:1731. doi: 10.1021/cr0104330. Kakiuchi F, Murai S. Acc. Chem. Res. 2002;35:826. doi: 10.1021/ar960318p.
- 2.Ames DE, Bull D. Tetrahedron. 1982;38:383. [Google Scholar]
- 3.Pivsa-Art S, Satoh T, Kawamura Y, Miura M, Nomura M. Bull. Chem. Soc. Jpn. 1998;71:467. [Google Scholar]
- 4.Okazawa T, Satoh T, Miura M, Nomura M. J. Am. Chem. Soc. 2002;124:5286. doi: 10.1021/ja0259279. [DOI] [PubMed] [Google Scholar]
- 5.Glover B, Harvey KA, Liu B, Sharp MJ, Tymoschenko M. Org. Lett. 2003;5:301. doi: 10.1021/ol027266q. [DOI] [PubMed] [Google Scholar]
- 6.Park C-H, Ryabova V, Seregin IV, Sromek AW, Gevorgyan V. Org. Lett. 2004;6:1159. doi: 10.1021/ol049866q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For review, see: Behnisch A, Behnisch P, Eggenweiler M, Wallenhorst T. Methoden der Organischen Chemie (Houben-Weyl) Georg. Thieme Verlag; Stuttgart: 1994. Indolizine; pp. 323–450. E6b/1, 2a.
- 8.Brase S, de Meijere A. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. vol. 1. John Wiley and Sons; New York: 2002. pp. 1369–1404. [Google Scholar]
- 9.Brase S, de Meijere A. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. vol. 1. John Wiley and Sons; New York: 2002. pp. 1405–1430. [Google Scholar]
- 10.Okazawa T, Satoh T, Miura M, Nomura M. J. Am. Chem. Soc. 2002;124:5286. doi: 10.1021/ja0259279. [DOI] [PubMed] [Google Scholar]
- 11.Jones WD. Acc. Chem. Res. 2003;36:140. doi: 10.1021/ar020148i. [DOI] [PubMed] [Google Scholar]
- 12.Whisler MC, MacNeil S, Snieckus V, Beak P. Angew. Chem., Int. Ed. 2004;43:2206. doi: 10.1002/anie.200300590. [DOI] [PubMed] [Google Scholar]
- 13.Li W, Nelson DP, Jensen MS, Hoerrner S, Javadi GJ, Cai D, Larsen RD. Org. Lett. 2003;5:4835. doi: 10.1021/ol035878k. [DOI] [PubMed] [Google Scholar]
- 14.Hughes CC, Trauner D. Angew. Chem., Int. Ed. 2002;41:1569. doi: 10.1002/1521-3773(20020503)41:9<1569::aid-anie1569>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; Hughes CC, Trauner D. Angew. Chem., Int. Ed. 2002;41:2227. Erratum: [Google Scholar]
- 15.Taylor R. Electrophilic Aromatic Substitution. John Wiley and Sons; Chichester, UK: 1990. pp. 25–57. [Google Scholar]
- 16.Lane BS, Brown MA, Sames D. J. Am. Chem. Soc. 2005;127:8050. doi: 10.1021/ja043273t. [DOI] [PubMed] [Google Scholar]
- 17.Lautens M, Fang Y-Q. Org. Lett. 2003;5:3679. doi: 10.1021/ol035354k. [DOI] [PubMed] [Google Scholar]
- 18.Ikeda M, El Bialy SAA, Yakura T. Heterocycles. 1999;51:1957. [Google Scholar]
- 19.Takacs JM, Lawson EC, Clement F. J. Am. Chem. Soc. 1997;119:5956. [Google Scholar]
- 20 (a).Holtcamp MW, Henling LM, Day MW, Labinger JA, Bercaw JE. Inorg. Chim. Acta. 1998;270:467. [Google Scholar]; (b) Ho VM, Watson L, Huffman JC, Caulton KG. New J. Chem. 2003;27:1446. [Google Scholar]
- 21.Tollari S, Demartin F, Cenini S, Palmisano G, Raimondi P. J. Organomet. Chem. 1997;527:93. [Google Scholar]
- 22.Nonoyama M, Nakajima K. Polyhedron. 1998;18:533. [Google Scholar]
- 23.Jackson AH, Lynch PP. J. Chem. Soc., Perkin Trans. 2. 1987:1215. [Google Scholar]
- 24 (a).Saulnier MG, Gribble GW. J. Org. Chem. 1982;47:757. [Google Scholar]; (b) Focante F, Camurati I, Nanni D, Leardini R, Resconi L. Organometallics. 2004;23:5135. [Google Scholar]; (c) Yudin LG, Pavlyuchenko AI, Kost AN. Zh. Obshch. Khim. 1969;39:2784. [Google Scholar]; (d) Taylor EC, Kienzle F, Robey RL, McKillop A, Hunt JD. J. Am. Chem. Soc. 1971;93:4845. [Google Scholar]
- 25.Okazawa T, Satoh T, Miura M, Nomura M. J. Am. Chem. Soc. 2002;124:5286. doi: 10.1021/ja0259279. [DOI] [PubMed] [Google Scholar]
- 26.Yokooji A, Okazawa T, Satoh T, Miura M, Nomura M. Tetrahedron. 2003;59:5685. [Google Scholar]
- 27.Akita Y, Itagaki Y, Takizawa S, Ohta A. Chem. Pharm. Bull. 1989;37:1477. [Google Scholar]
- 28.Aoyagi Y, Inoue A, Koizumi I, Hashimoto R, Tokunaga K, Gohma K, Komatsu J, Sekine K, Miyafuji A, Kunoh J, Honma R, Akita Y, Ohta A. Heterocycles. 1992;33:257. [Google Scholar]
- 29.Ohta A, Akita Y, Ohkuwa T, Chiba M, Fukunaga R, Miyafuji A, Nakata T, Tani N, Aoyagi Y. Heterocycles. 1990;31:1951. [Google Scholar]
- 30.Grigg R, Sridharan V, Stevenson P, Sukirthalingam S, Worakun T. Tetrahedron. 1990;46:4003. [Google Scholar]
- 31 (a).For related works by this group, see: Burwood M, Davies B, Diaz I, Grigg R, Molina P, Sridharan V, Hughes M. Tetrahedron Lett. 1995;36:9053. Grigg R, Fretwell P, Meerholtz C, Sridharan V. Tetrahedron. 1991;50:359.
- 32.Kozikowski AP, Ma D. Tetrahedron Lett. 1991;32:3317. [Google Scholar]
- 33.Kuroda T, Suzuki F. Tetrahedron Lett. 1991;47:6915. [Google Scholar]
- 34.Desarbre E, Merour J-Y. Heterocycles. 1995;41:1987. [Google Scholar]
- 35.Jeffery T. Tetrahedron Lett. 1985;26:2667. [Google Scholar]
- 36.Lavenot L, Gozzi C, Ilg K, Orlova I, Penalva V, Lemaire M. J. Organomet. Chem. 1998;567:49. [Google Scholar]
- 37.Gozzi C, Lavenot L, Ilg K, Penalva V, Lemaire M. Tetrahedron Lett. 1997;51:8867. [Google Scholar]
- 38.Chabert JFD, Joucla L, David E, Lemaire M. Tetrahedron. 2004;60:3221. [Google Scholar]
- 39.Kondo Y, Komine T, Sakamoto T. Org. Lett. 2000;2:3111. doi: 10.1021/ol000183u. [DOI] [PubMed] [Google Scholar]
- 40.McClure MS, Glover B, McSorley E, Millar A, Osterhout MH, Roschangar F. Org. Lett. 2001;3:1677. doi: 10.1021/ol0158866. [DOI] [PubMed] [Google Scholar]
- 41.Mori A, Sekiguchi A, Masui K, Shimada T, Horie M, Osakada K, Kawamoto M, Ikeda T. J. Am. Chem. Soc. 2003;125:1700. doi: 10.1021/ja0289189. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi K, Sugie A, Takahashi M, Masui K, Mori A. Org. Lett. 2005;7:5083. doi: 10.1021/ol052063y. [DOI] [PubMed] [Google Scholar]
- 43.Li W, Nelson DP, Jensen MS, Hoerrner RS, Javadi GJ, Cai D, Larsen RD. Org. Lett. 2003;5:4835. doi: 10.1021/ol035878k. [DOI] [PubMed] [Google Scholar]
- 44.Lane BS, Sames D. Org. Lett. 2004;6:2897. doi: 10.1021/ol0490072. [DOI] [PubMed] [Google Scholar]
- 45.Toure BB, Lane BS, Sames D. Org. Lett. 2006;8:1979. doi: 10.1021/ol053021c. [DOI] [PubMed] [Google Scholar]
- 46.Rieth RD, Mankad NP, Calimano E, Sadighi JP. Org. Lett. 2004;6:3981. doi: 10.1021/ol048367m. [DOI] [PubMed] [Google Scholar]
- 47.Bellina F, Cauteruccio S, Mannina L, Rossi R, Viel S. J. Org. Chem. 2005;70:3997. doi: 10.1021/jo050274a. [DOI] [PubMed] [Google Scholar]
- 48.Bellina F, Cauteruccio S, Rossi R. Eur. J. Org. Chem. 2006:1379. doi: 10.1021/jo701496p. [DOI] [PubMed] [Google Scholar]
- 49.Bellina F, Cauteruccio S, Mannina L, Rossi R, Viel S. Eur. J. Org. Chem. 2006:693. doi: 10.1021/jo050274a. [DOI] [PubMed] [Google Scholar]
- 50.Bowie AL, Hughes CC, Trauner D. Org. Lett. 2005;7:5207. doi: 10.1021/ol052033v. [DOI] [PubMed] [Google Scholar]
- 51.Beccalli EM, Broggini G, Martinelli M, Paladino G, Zoni C. Eur. J. Org. Chem. 2005:2091. [Google Scholar]
- 52.Catellani M, Frignani F, Rangoni A. Angew. Chem., Int. Ed. Engl. 1997;36:119. [Google Scholar]
- 53.Tollari S, Demartin F, Cenini S, Palmisano G, Raimondi PJ. J. Organomet. Chem. 1997;527:93. [Google Scholar]
- 54.Motoyama T, Shimazaki Y, Yajima T, Nakabayashi Y, Naruta Y, Yamauchi O. J. Am. Chem. Soc. 2004;126:7378. doi: 10.1021/ja031587v. [DOI] [PubMed] [Google Scholar]
- 55.Capito E, Brown JM, Ricci A. Chem. Commun. 2005;25:1854. doi: 10.1039/b417035k. [DOI] [PubMed] [Google Scholar]
- 56.Bressy C, Alberico D, Lautens M. J. Am. Chem. Soc. 2005;127:13148. doi: 10.1021/ja054472v. [DOI] [PubMed] [Google Scholar]
- 57.Blaszykowski C, Aktoudianakis E, Bressy C, Alberico D, Lautens M. Org. Lett. 2006;8:2043. doi: 10.1021/ol060447y. [DOI] [PubMed] [Google Scholar]
- 58.Martins A, Alberico D, Lautens M. Org. Lett. 2006;8:4827. doi: 10.1021/ol061859+. [DOI] [PubMed] [Google Scholar]
- 59.Campeau L-C, Thansandote P, Fagnou K. Org. Lett. 2005;7:1857. doi: 10.1021/ol050501v. [DOI] [PubMed] [Google Scholar]
- 60.Campeau L-C, Parisien M, Jean A, Fagnou K. J. Am. Chem. Soc. 2006;128:581. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- 61.Campeau L-C, Fagnou K. Chem. Commun. 2006:1253. doi: 10.1039/b515481m. [DOI] [PubMed] [Google Scholar]
- 62.Parisien M, Valette D, Fagnou K. J. Org. Chem. 2005;70:7578. doi: 10.1021/jo051039v. [DOI] [PubMed] [Google Scholar]
- 63.Zhuravlev FA. Tetrahedron Lett. 2006;47:2929. [Google Scholar]
- 64.Kalyani D, Deprez NR, Desai LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]
- 65.Hull KL, Lanni EL, Sanford MS. J. Am. Chem. Soc. 2006;128:14047. doi: 10.1021/ja065718e. [DOI] [PubMed] [Google Scholar]
- 66.Giri R, Chen X, Yu J-Q. Angew. Chem., Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]
- 67.Giri R, Liang J, Lei J-G, Li J-J, Wang D-H, Chen X, Naggar IC, Guo C, Foxman BM, Yu J-Q. Angew. Chem., Int. Ed. 2005;44:7420. doi: 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]
- 68.Zaitsev VG, Daugulis O. J. Am. Chem. Soc. 2005;127:4156. doi: 10.1021/ja050366h. [DOI] [PubMed] [Google Scholar]
- 69.Shabashov D, Daugulis O. Org. Lett. 2005;7:3657. doi: 10.1021/ol051255q. [DOI] [PubMed] [Google Scholar]
- 70.Daugulis O, Zaitsev VG. Angew. Chem., Int. Ed. 2005;44:4046. doi: 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]
- 71.Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 72.Daugulis O, Zaitsev VG, Shabashov D, Pham Q-N, Lazareva A. Synlett. 2006:3382. [Google Scholar]
- 73.Deprez NR, Kalyani D, Krause A, Sanford MS. J. Am. Chem. Soc. 2006;128:4972. doi: 10.1021/ja060809x. [DOI] [PubMed] [Google Scholar]
- 74.Lewis JC, Wiedemann SH, Bergman RG, Ellman JA. Org. Lett. 2004;6:35. doi: 10.1021/ol035985e. [DOI] [PubMed] [Google Scholar]
- 75.For review, see: Fagnou K, Lautens M. Chem. Rev. 2003;103:169. doi: 10.1021/cr020007u.
- 76.Wang X, Lane BS, Sames D. J. Am. Chem. Soc. 2005;127:4996. doi: 10.1021/ja050279p. [DOI] [PubMed] [Google Scholar]
- 77.Davies HM, Townsend RT. J. Org. Chem. 2001;66:6595. doi: 10.1021/jo015617t. [DOI] [PubMed] [Google Scholar]
- 78.Lewis JC, Wu JY, Bergman RG, Ellman JA. Angew. Chem., Int. Ed. 2006;45:1589. doi: 10.1002/anie.200504289. [DOI] [PubMed] [Google Scholar]
- 79.Yanagisawa S, Sudo T, Noyori R, Itami K. J. Am. Chem. Soc. 2006;128:11748. doi: 10.1021/ja064500p. [DOI] [PubMed] [Google Scholar]
- 80.Ames DE, Opalko A. Tetrahedron. 1984;40:1919. [Google Scholar]
- 81.Mukhopadhyay S, Rothenberg G, Gitis D, Baidossi M, Ponde DE, Sasson Y. J. Chem. Soc., Perkin Trans. 2. 2000:1809–1812. [Google Scholar]
- 82.Campeau L-C, Rousseaux S, Fagnou K. J. Am. Chem. Soc. 2005;127:18020. doi: 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]
- 83.Leclerc J-P, Fagnou K. Angew. Chem., Int. Ed. 2006;45:7781. doi: 10.1002/anie.200602773. [DOI] [PubMed] [Google Scholar]
- 84.Garcia-Cuadrado D, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2006;128:1066. doi: 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]
- 85.Mota AJ, Dedieu A, Bour C, Suffert J. J. Am. Chem. Soc. 2005;127:7171. doi: 10.1021/ja050453+. [DOI] [PubMed] [Google Scholar]
- 86.Lefrance M, Rowley CN, Woo TK, Fagnou K. J. Am. Chem. Soc. 2006;128:8754. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]
- 87 (a).For related works on direct arylation by this group, see: Lafrance M, Fagnou K. J. Am. Chem. Soc. 2006;128:16496. doi: 10.1021/ja067144j. Lafrance M, Shore D, Fagnou K. Org. Lett. 2006;8:5097. doi: 10.1021/ol0619967. Leclerc J-P, Andre M, Fagnou K. J. Org. Chem. 2006;71:1711. doi: 10.1021/jo0523619. Campeau L-C, Parisien M, Jean A, Fagnou K. J. Am. Chem. Soc. 2006;128:581. doi: 10.1021/ja055819x.
- 88.Schlosser M, Marzi E. Chem.–Eur. J. 2005;11:3449. doi: 10.1002/chem.200401094. [DOI] [PubMed] [Google Scholar]
- 89.Nakamura I, Saito S, Yamamoto Y. J. Am. Chem. Soc. 2000;122:2661. [Google Scholar]
- 90.Hashmi ASK, Schwarz L, Choi J-H, Frost TM. Angew. Chem., Int. Ed. 2000;39:2285. doi: 10.1002/1521-3773(20000703)39:13<2285::aid-anie2285>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 91.Hughes CC, Trauner D. Angew. Chem., Int. Ed. 2002;41:1569. doi: 10.1002/1521-3773(20020503)41:9<1569::aid-anie1569>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 92.Hughes CC, Trauner D. Tetrahedron. 2004;60:9675. [Google Scholar]
- 93.Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2003;125:9578. doi: 10.1021/ja035054y. [DOI] [PubMed] [Google Scholar]
- 94.Abbiati G, Beccalli EM, Broggini G, Zoni C. J. Org. Chem. 2003;68:7625. doi: 10.1021/jo034636v. [DOI] [PubMed] [Google Scholar]
- 95.Beccalli EM, Broggini G. Tetrahedron Lett. 2003;44:1919. [Google Scholar]
- 96.Reez MT, Sommer K. Eur. J. Chem. 2003:3485. [Google Scholar]
- 97.Tan KL, Vasudevan A, Bergman RG, Ellman JA, Souers AJ. Org. Lett. 2003;5:2131. doi: 10.1021/ol030050j. [DOI] [PubMed] [Google Scholar]
- 98.Tan KL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2002;124:13964. doi: 10.1021/ja0281129. [DOI] [PubMed] [Google Scholar]
- 99.Tan KL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2001;123:2685. doi: 10.1021/ja0058738. [DOI] [PubMed] [Google Scholar]
- 100.Liu C, Han X, Wang X, Widenhoefer RA. J. Am. Chem. Soc. 2004;126:3700. doi: 10.1021/ja031814t. [DOI] [PubMed] [Google Scholar]
- 101.Liu C, Widenhoefer RA. J. Am. Chem. Soc. 2004;126:10250. doi: 10.1021/ja046810i. [DOI] [PubMed] [Google Scholar]
- 102.Grimster NP, Gauntlett C, Godfrey CRA, Gaunt MJ. Angew. Chem., Int. Ed. 2005;44:3125. doi: 10.1002/anie.200500468. [DOI] [PubMed] [Google Scholar]
- 103.Beck EM, Grimster NP, Hatley R, Gaunt MJ. J. Am. Chem. Soc. 2005;128:2528. doi: 10.1021/ja058141u. [DOI] [PubMed] [Google Scholar]
- 104.Li Z, Shi Z, He C. J. Organomet. Chem. 2005;690:5049. [Google Scholar]
- 105.Furstner A, Mamane V, Seidel G, Laurich D. Org. Synth. 2006;83:103. [Google Scholar]
- 106.Liu Z, Wasmuth AS, Nelson SG. J. Am. Chem. Soc. 2006;128:10352. doi: 10.1021/ja0629110. [DOI] [PubMed] [Google Scholar]
- 107.Kong A, Han X, Lu X. Org. Lett. 2006;8:1339. doi: 10.1021/ol060039u. [DOI] [PubMed] [Google Scholar]
- 108.Nakao Y, Kanyiva KS, Oda S, Hiyama T. J. Am. Chem. Soc. 2006;128:8146. doi: 10.1021/ja0623459. [DOI] [PubMed] [Google Scholar]
- 109.Tsuda T, Kiyoi T, Saegusa T. J. Org. Chem. 1990;55:2554. [Google Scholar]
- 110.Tilly D, Gevorgyan V. unpublished results.
- 111.For review, see: Dick AR, Sanford MS. Tetrahedron. 2006;62:2439.
- 112.He L, Chan PWH, Tsui W-M, Yu W-Y, Che C-M. Org. Lett. 2004;6:2405. doi: 10.1021/ol049232j. [DOI] [PubMed] [Google Scholar]
- 113.For related direct borylation of arenes see: Cho J, Tse MK, Holmes D, Maleczka RE, Jr., Smith MR., III Science. 2002;295:305. doi: 10.1126/science.1067074.
- 114.Mertins K, Zapf A, Beller M. J. Mol. Catal. A: Chem. 2004;207:21. [Google Scholar]
- 115.Ishiyama T, Takagi J, Yonekawa Y, Hartwig JF, Miyaura N. Adv. Synth. Catal. 2003;345:1103. [Google Scholar]
- 116.Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N. Tetrahedron Lett. 2002;43:5649. [Google Scholar]
- 117.Tse MK, Cho J-Y, Smith MR., III J. Am. Chem. Soc. 2001;3:2831. [Google Scholar]