

Abstract
Otitis media with effusion (OME) is the most common cause of hearing loss in children and tympanostomy to alleviate the condition remains the commonest surgical intervention in children in the developed world. Chronic and recurrent forms of OM are known to have a very significant genetic component, however, until recently little was known of the underlying genes involved. The identification of mouse models of chronic OM has indicated a role of transforming growth factor beta (TGFβ) signalling and its impact on responses to hypoxia in the inflamed middle ear. We have, therefore, investigated the role of TGFβ signalling and identified and characterized a new model of chronic OM carrying a mutation in the gene for transforming growth interacting factor 1 (Tgif1). Tgif1 homozygous mutant mice have significantly raised auditory thresholds due to a conductive deafness arising from a chronic effusion starting at around 3 weeks of age. The OM is accompanied by a significant thickening of the middle ear mucosa lining, expansion of mucin-secreting goblet cell populations and raised levels of vascular endothelial growth factor, TNF-α and IL-1β in ear fluids. We also identified downstream effects on TGFβ signalling in middle ear epithelia at the time of development of chronic OM. Both phosphorylated SMAD2 and p21 levels were lowered in the homozygous mutant, demonstrating a suppression of the TGFβ pathway. The identification and characterization of the Tgif mutant supports the role of TGFβ signalling in the development of chronic OM and provides an important candidate gene for genetic studies in the human population.
INTRODUCTION
Otitis media with effusion (OME), inflammation of the middle ear, is the commonest cause of hearing impairment in children and the commonest reason for surgery in children. Both chronic and recurrent forms of OM are known to have a significant genetic component (1,2), but until recently little was known about the genes or pathways involved (3,4). Several mouse models of OM have been reported and some of them, including the mutants Jeff and Junbo, implicate the transforming growth factor beta (TGFβ) signalling pathway in susceptibility to OM (5–7). Both mutants were identified from a deafness screen as part of a large-scale phenotype-driven mouse ENU mutagenesis programme (8). Mice heterozygous for the Jeff mutation develop chronic proliferative OM (9) and the gene mutated in Jeff was identified as Fbxo11 (5). We found that Fbxo11 is involved in the regulation of the TGFβ signalling pathway by regulating the levels of pSmad2 in the epithelial cells of different embryonic mouse tissues. Furthermore, we identified a genetic interaction between Fbxo11 and Smad2 (7). Junbo mice display chronic suppurative OM and carry a mutation in the transcription factor Evi1 (6). Evi1 can repress the TGFβ signalling pathway by interacting with Smad3 and can antagonize the growth-inhibitory effect of the pathway (10). The identification of these mutants has focussed studies on the role of TGFβ signalling in the development of chronic OM and the interaction of this pathway with other molecular and cellular changes occurring in the inflamed middle ear (11).
The TGFβ signalling pathway is involved in a variety of cellular processes such as proliferation, differentiation and apoptosis (12,13). TGFβ pathway members act by linking membrane receptors to specific target genes. TGFβ ligands initiate the signalling by binding to a type II receptor on the cell surface that in turn binds and phosphorylates a type I receptor. The intracellular mediators, R-Smads, Smad2 and Smad3, become phosphorylated by the TGFβ receptor I. The activated Smads form a complex with the co-mediator Smad4. The Smad complex then translocates into the nucleus and in conjunction with other nuclear cofactors regulates the transcription of different target genes (12). The phosphorylation of the R-Smads is regulated by the Smad anchor for receptor activation (SARA) protein, cytoplasmic promyelocytic leukaemia (cPML) protein and transforming growth interacting factor (TGIF). SARA has a Smad binding domain and a domain interacting with the TGFβ receptor and recruits Smad2 and/or Smad3 to the receptor (14). In the cytoplasm, cPML physically interacts with Smad2/3 and SARA and is required for the association of Smad2/3 with SARA (15).
TGIF1, also known as TGIF, belongs to a three amino acid loop extension (TALE) subgroup of atypical homeodomain proteins (16,17). It has been found that TGIF functions through several routes as a negative regulator of the TGFβ signalling pathway. TGIF was initially identified as a Smad2-binding protein and a co-repressor of TGFβ-induced transcription. This repression is mediated by the ability of TGIF to recruit to Smad2 a co-repressor containing histone deacetylases (HDACs) (18). TGIF can also inhibit Smad2 phosphorylation by an alternative mechanism to its association with Smad2, acting in partnership with c-Jun to sequester cPML in the nucleus. This prevents the formation of the cPML-SARA complex that is required for the phosphorylation of Smad2 (19). In addition, TGIF can restrict TGFβ signalling by recruiting TGIF-interacting ubiquitin ligase 1-WW-containing protein. Tiul1 is an E3 ubiquitin ligase that interacts with TGIF to induce the degradation of Smad2 (20). Recently, PML competitor for TGIF association (PCTA) has been found to compete with cPML for binding to TGIF, resulting in the accumulation of cPML in the cytoplasm and inducing the phosphorylation of R-Smads by the TGFβ type I receptor. PCTA can reverse the inhibitory activity of TGIF (21,22). TGIF2 is also a member of the TALE subclass of homeobox proteins and shows distinct homology with TGIF1, especially in its DNA-binding domain (23). It was reported that TGIF2 can perform many of the same functions as TGIF1 (24). Similarly to TGIF1, TGIF2 binds to Smad and HDAC, acting as a repressor in the TGFβ signalling pathway (24).
Human TGIF1 maps to 18p11.3 (25). This chromosomal region contains a locus associated with holoprosencephaly (HPE), a common developmental defect affecting the forebrain and the face (26). Mutations in the human TGIF1 gene have been associated with HPE (27), but Tgif1 knockout mice on a mixed background were reported to be indistinguishable from wild-type mice possibly due to a functional redundancy with Tgif2 (28,29). Recently, it has been demonstrated that the Tgif1 mutation in mice can cause HPE when combined with a mutation in the closely related Tgif2 gene (30). In humans, TGIF1 was found to be highly expressed in adult tissues such as placenta, liver, kidney, prostate, ovary and testis (16). Mouse Tgif1 was found to be widely expressed in the E9.5 embryo, with the highest expression in the forebrain, the branchial arches, the otic pit and the limb buds (28). It was also detected at E10.5 and E12.5 in the ventricular neuroepithelium. The expression was found to decline by E14.5 (28).
Given the role of TGIF as a regulator of the TGFβ signalling pathway, we decided to investigate whether Tgif knockout mice display a similar phenotype to Jeff and Junbo mice. We report here the Tgif mutant mouse as a novel model of OM. Tgif mutants exhibit auditory deficits due to the development of OM by weaning age. Our findings further elaborate the role of the TGFβ pathway in genetic predisposition to OM.
RESULTS
A targeted knockout of Tgif1 was used to examine potential auditory phenotypes associated with the gene, including OM. Tgif mutant mice had originally been generated and maintained on a mixed background and were reported to be viable, fertile and with no obvious phenotype in the major organ systems (28). We maintained the colony on a C57BL/6J background by recurrent backcrossing. To generate mice homozygous for Tgif, heterozygous animals were intercrossed and the progeny were genotyped by PCR assay as previously described (28). The surviving homozygotes at weaning age comprised 10.84% (31 of 286) of the mice from this cross that was less than the expected 25% (χ2 = 0.999857458, df1, P = 0.000000032).
To produce additional homozygous mice for this study, we crossed heterozygote Tgif females with Tgif/Tgif males. The surviving homozygotes at weaning age were 27.86% (39 of 140) that was less than the expected 50% (χ2 = 0.999680234, df1, P = 0.00000016). About 50% of the homozygous mice from both crosses were missing by weaning (postnatal day 21, P21), suggesting embryonic or neonatal lethality (Table 1). Some of the mutant mice (4 of 199 heterozygotes and 11 of 67 homozygotes) developed hydrocephalus at the age of 1 month, which may be one of the reasons why some of the mutant mice do not survive (Fig. 1A). No wild-type littermate mice (0 of 68) in this study developed hydrocephalus, in line with the low (0.029%) spontaneous rate of this condition in the background C57BL/6J strain (http://jaxmice.jax.org/jaxnotes/archive/490f.html). We also set up a number of homozygote intercrosses. Out of five plugged females, only one (20%) gave birth to seven pups, suggesting that the null mothers may have a placental defect and lose their embryos before birth (Table 1).
Table 1.
Genotyping data from Tgif mutant crosses
| M × F | Stage | Wt | Het | Hom | Total/% alive | Resorbed/found dead | Total | Litters | |
|---|---|---|---|---|---|---|---|---|---|
| hom × hom | E9.5 | 7 | 7 of 87.5% | 1 | 8 | 1 | |||
| hom × hom | E11.5 | 7 | 7 of 87.5% | 1 | 8 | 1 | |||
| hom × hom | E13.5 | 9 | 9 of 56.25% | 7 | 16 | 2 | |||
| hom × hom | E15.5 | 5 | 5 | 0 | 5 | 1 | |||
| hom × hom | E18.5 | 5 | 5 | 0 | 5 | 1 | |||
| hom × het | E13.5 | 5 | 9 | 14 of 73.68% | 5 | 19 | 2 | ||
| het × het | E13.5 | 5 | 8 | 9 | 22 of 81.48% | 5 | 27 | 3 | |
| wt × het | E13.5 | 6 | 8 | 14 of 93.33% | 1 | 15 | 2 | ||
| hom × hom | P21 | 7 | 7 | 2 | 9 | 5 | |||
| het × hom | P21 | 77 | 39 | 140 of 80.92% | 33 | 173 | 23 | ||
| het × het | P21 | 68 | 133 | 31 | 286 of 84.86% | 51 | 337 | 41 |
The genotype of the mother (M) and the father (F) are indicated in the table. The number of the embryos and mice were analysed at different embryonic days (E) and at weaning age, postnatal day 21 (P21).
Figure 1.

Hydrocephalus and placental defect in Tgif mutant mice. (A) One-month-old Tgif/Tgif homozygous mutant mouse with hydrocephalus (indicated with an arrow). (B). Placentas from wild-type and mutant E13.5 embryos. No differences in size or morphology were observed. (C). Representative images of H&E stained sections through the centre of the placenta for each genotype combination. Eight M+/+E+/−, 10 M+/− E+/− and 9 M−/− E+/− placentas were analysed. Arrows and arrowheads indicate fetal and maternal blood spaces. The genotype of the mother (M) and the embryo (E) are indicated in the pictures and the sections.
Placental defects in Tgif null mothers
Because of the low number of pups born from homozygote intercrosses, we searched for defects in the placenta of the Tgif mutants. It has been described previously that Tgif null mice (29) on a relatively pure C57BL/6J background have severe placental defects, primarily due to absence of TGIF function in the mother (31). We studied the placental phenotype of the mice from homozygote intercrosses at E9.5, 11.5, 13.5, 15.5 and 18.5. At E9.5 and 11.5, out of eight embryos from each litter, seven looked normal and only one was resorbed (Table 1). By E13.5, the number of the resorbed embryos had increased and the number of embryos scored as normal decreased. Out of 16 embryos from 2 litters, only 9 (56.25%) appeared normal, and we, therefore, proceeded to study the homozygote placental phenotype at E13.5 and compare it with the phenotype of the placenta from heterozygote and wild-type mothers. In scoring, 93.33 and 81.48% of the embryos from wild-type or heterozygous females, respectively, were scored as normal which was higher than that observed for homozygous females at this stage (73.68%); however, this did not achieve statistical significance (P = 0.3299 2 × 3 Fisher's exact test) (Table 1). We examined placentas from heterozygous embryos for each maternal genotype at E13.5. We did not detect a size difference in the placentas from mice with different genotypes (Fig. 1B), but after sectioning and staining with haematoxylin and eosin (H&E), we observed a placental phenotype similar to that previously described for Tgif null mice on a similar background to our mice (31). For both heterozygous and homozygous mutants, the central regions of the placental labyrinths were less well developed. There was a reduction in maternal blood spaces in mutant placentas, and the maternal blood spaces appeared larger when compared with wild types with fewer surrounding fetal vessels (Fig. 1C).
Craniofacial phenotype in Tgif mice
Tgif homozygous mice are smaller and appeared to have a shorter face when compared with their wild-type littermates (Fig. 2A). To investigate the craniofacial phenotype of the mice, we took dorsoventral X-ray images of the skulls and measured the length of the nasal bone. These measurements were expressed relative to the full length of the skull to normalize for differences in body size (Fig. 2B, C and D). Although the results showed a significant difference between homozygous Tgif mice and wild-type mice (P = 0.000436), no significant difference between the heterozygous Tgif mice and wild-type mice was observed (P = 0.5617), and the magnitude of the difference between homozygotes and wild types was small (<2%).
Figure 2.

Craniofacial defect of Tgif mutant mice. (A) Size comparison of a male Tgif/Tgif mutant (bottom) and a wild-type mouse (top). (B) X-ray analysis of 2-month-old skulls from homozygous (hom, Tgif/Tgif), heterozygous (het, Tgif/+) and wild-type (wt, +/+) mice. Lateral X-rays show the craniofacial defect of the mutant mice. (C) Dorsoventral view of a 2-month-old wild-type mouse skull showing the measurements used in this study. Scale bar = 2 cm. (D) Graphic comparison of the ratio of the nasal bone length (NL) to the full dorsal length of the skull (FL) for each Tgif genotype. N, number of mice used in the measurements from each genotype. Bars: standard error of mean. *P < 0.05. (E) Ventral view of the middle ear and dissected bulla showing the ET of 2-month-old homozygous (hom) and wild-type (wt) mice. ET, Eustachian tube; solid double-ended arrows: length of the tube; open double-ended arrows: width of the tube. (F) Dissected and stained skulls of wild-type and homozygous mutant 2-month-old mice. ET angle measurements are indicated by white lines. R, right ear; L, left ear. (G) Mean length and width of wild-type (wt) and homozygous mutant (hom) ETs along with mean angle between the midline of the skull and the bony part of the left and the right Eustachian tubes in wild-type and homozygous mutants. Bars: standard error of mean.
To investigate orientation and morphology of the Eustachian tube (ET) in mutant mice compared with wild type, the skulls were stained and the angle between the midline of the skull and the bony part of the left and the right ETs was measured (Fig. 2F and G). We did not detect statistically significant differences in ET measurements between Tgif homozygotes (n = 3) and wild-type mice (n = 5). In addition, we measured the length and width of the bony part of the ET (Fig. 2E and G). The mean length in wild-type mice (n = 4) was 1.03 mm, when compared with 1.14 mm in mutant homozygotes (n = 4), and mean width for wild-type mice was 1.43 mm, when compared with 1.46 mm in the homozygous mutant. These differences were not statistically significant.
Hearing deficiency in Tgif mutants
We proceeded to assess auditory function in Tgif mutant mice. A clickbox, that generates a brief 20 kHz soundburst at 90 dB sound pressure levels (SPL), was used initially to assess the hearing ability of mice through the presence or absence of a Preyer's reflex (32). When 1 month old, most of the Tgif homozygote mice (86%, n = 14) showed a reduced Preyer's reflex. When 2 months old, all the homozygote mice demonstrated reduced hearing, but to a variable degree. Five mice (36%, n = 14) were scored deaf by clickbox as they did not display a Preyer's reflex, seven mice (50%, n = 14) had reduced hearing and two mice (14%, n = 14) had almost normal hearing, but were still assessed as reduced when compared with wild-type mice. At the same age, heterozygous mice had normal or slightly reduced hearing. Three of them (27%, n = 11) had reduced hearing and eight mice (73%, n = 11) had normal hearing. By comparison, 10 wild-type mice all had normal hearing by clickbox.
An auditory-evoked brainstem response (ABR) test was used to assess hearing thresholds in mutant mice (32). We tested 14 homozygous, 11 heterozygous and 10 wild-type mice at the age of 2 months for ABR response at 4 frequencies 8, 12, 20 and 26 kHz. We showed that the average ABR thresholds were elevated by about 30 dB SPL in homozygotes and by about 20 dB SPL in heterozygotes, when compared with wild-type mice, suggesting a conductive hearing loss (Fig. 3). At all four frequencies, the ABR thresholds of the homozygote mice were significantly different from the wild types (at 8 kHz P = 0.000447; 12 kHz P = 0.003297; 20 kHz P = 0.000486; 26 kHz P = 0.004445). Significant differences were also seen between heterozygotes and wild types at two frequencies (8 kHz P = 0.003229 and 20 kHz P = 0.023927).
Figure 3.

Deafness phenotype by ABR. ABR thresholds in the right ears of 2-month-old Tgif/Tgif homozygote (hom), Tgif/+ heterozygote (het) and wild-type (wt) mice. n, number of mice in ABR testing for each genotype. Bars: standard error of mean. *P < 0.05.
We assessed whether there was a sensorineural element to the hearing loss by dissecting inner ears from two 1-month-old homozygous, heterozygous and wild-type mice to examine the structure of the organ of Corti and the sensory cells. We did not detect any abnormalities in the inner hair cell (IHC) or outer hair cell (OHC) stereocilia bundles. Scanning electron micrographs showed normal cell and bundle morphology in the mid (Fig. 4A), apical and basal (data not shown) turns of the cochlea in both mutants and wild types. In all the samples, we could see well-organized rows of OHCs in the organ of Corti and normal-sized IHCs. The histological examination of sagittal sections from the cochlea of 2-month-old mice also confirms that there were no obvious abnormalities in the cochlea or organ of Corti of homozygote Tgif mice. We also observed no loss of spiral ganglion neurons (Fig. 4B).
Figure 4.

Normal bundle morphology of the hair cells. (A) Scanning electron micrographs showing hair cell morphology in the mid turn of the cochlea of wild-type (on the left) and homozygous Tgiif/Tgif (on the right) mice at two magnifications, age 1 month. Three rows of OHCs and a row of IHCs are observed along the length of the organ of Corti in both wild-type and mutant mice. Normal bundle morphology for both OHCs and IHCs is observed in wild-type and mutants. Scale bars = 10 μm for the top panel and 1 μm for other panels; OHC, outer hair cell; IHC, inner hair cell. (B) Histological analysis of the hair cells in wild-type (left panels) and homozygous Tgif/Tgif (right panels) mice, aged 2 months. Scale bars = 1 mm for the top panel and 100 μm for other panels; OHC, outer hair cell; IHC, inner hair cell; OC, organ of Corti; SG, spiral ganglion, RM, Reissner's membrane.
Tgif mice display OM by 21 days after birth (DAB)
Five-, thirteen- and twenty-one-day old as well as 1-, 2- and 4-month-old adult heads were sectioned to investigate the middle ear phenotype. Five- and thirteen-day-old Tgif/Tgif middle ears were indistinguishable from wild types (Fig. 5A–D). However, we detected an OM phenotype in the middle ear of homozygote mice at 3 weeks. At that time point, most of the homozygote mice (67%, n = 6) had fluid in the middle ear cavity and a thickened epithelial lining in comparison with wild-type (n = 16) controls. There was only one wild type that demonstrated some fluid in the ear. The ear effusions were cellular, with the presence of polymorphonuclear cells (PMNs) that are the first responding inflammatory cells that migrate towards the site of inflammation (Fig. 5E–H). By comparison, three of the heterozygote mice at 21 days of age had fluid in one ear and thickened lining (19%, n = 16), and some of them had a thickened epithelial lining only (37%, n = 16) (Supplementary Material, Fig. S1).
Figure 5.

Middle ear histology in Tgif homozygous mice. Each group of panels consists of representative images (transverse sections) of the middle ears of homozygous (Tgif/Tgif) and wild-type (+/+) mice at different time points. (A and B) Five DAB (mutants analysed n = 8, controls analysed n = 5); (C and D) 14 DAB (mutants n = 8, controls n = 10); (E–H) 21 DAB (mutants n = 6, controls n = 16); (I–L) 1-month old (mutants n = 11, controls n = 7); (M–T) 2 months old (mutants n = 12, controls n = 7); (U–X) 4 months old (mutants n = 10, controls n = 10). (F and G) enlarged views of E; (J and K) enlarged views of I; (N and P) enlarged views of M; (R and S) enlarged views of Q; (V and W) enlarged views of U. (F) Inset viable and apoptotic PMNs. (O) Immunohistochemical staining of a 2-month-old homozygous mouse ear section using an F4/80 antibody. Macrophages are indicated with an arrow. Where indicated, the thickness of the epithelial lining is shown with a double arrow. The presence of cellular aggregates is indicated by an asterisk (P) and an inflamed polyp by an arrowhead (V). A–E, I, M, Q, U scale bar = 4 mm; F, J, N, O, R scale bar = 100 μm; G, H, K, L, P, S, T, V, W, X scale bar = 200 μm. Pie charts represent percentage of homozygote mice from different time points with no effusion and with effusion in one or in both ears.
At 1 month, four homozygote mice had fluid in one ear (37%, n = 11) and only two had some fluid in both ears (18%, n = 11). Five mice did not have any fluid, but some of these had a thickened epithelial lining (27% of mice). Surprisingly, the effusions at 1 month did not appear cellular (Fig. 5I–L). Two of the heterozygote mice had ear fluid and a thickened lining (16%, n = 13) and five had only a thickened lining (38%, n = 13). None of the wild type mice had OM at this stage (n = 7).
In 2-month-old homozygote ears, the inflammation had progressed to a chronic inflammation with effusion, as evidenced by fluid in the middle ear cavity along with a thickened epithelial lining. The effusion content was variable. In some of the homozygote mice, the middle ear fluid was more liquid and in some thickened, more viscous (Fig. 5M, N, Q and R). At 2 months, five homozygote mice (42%, n = 12) had unilateral OM, five (42%, n = 12) in both ears in each case associated with thickening of the mucoperiosteum. Two homozygotes (17%, n = 12) did not have effusions, nor thickened epithelial lining. Both of these mice showed relatively normal ABR thresholds. By comparison, two of the heterozygote mice had ear fluid (20%, n = 10) and three had their epithelial lining thickened (30%, n = 10). Heterozygotes with ear fluid and heterozygotes with thickened lining had elevated ABR thresholds. None of the wild-type mice (n = 7) had OM. In homozygotes, the effusion was again cellular, and staining with F4/80 antibody confirmed the presence of macrophages in the ear fluid at this stage (Fig. 5O). We also studied the OM phenotype of 4-month-old homozygote mice and observed that six of them (60%, n = 10) had unilateral OM, two (20%, n = 10) had effusion in both ears and two (20%, n = 10) did not have any fluid in the ears. Three of the Tgif/+ mice had fluid in the ears and thickened lining (25%, n = 12) and three had thickened lining only (25%, n = 12). At both 2 and 4 months of age, we observed the appearance of cellular debris-like aggregates in the ear effusions of the homozygote (Fig. 5P, W). In addition, at 4 months we observed polyps arising from the middle ear mucosa of the homozygote mice (Fig. 5V). The wild-type mice (n = 10) at this stage did not display OM.
To quantify the mucosal thickness of the middle ears at different stages, we examined the histological sections and took measurements at five equidistant points along a 1 mm length of the mucosa. Four to five homozygous and wild-type mice were analysed at each stage. There were significant differences between mutant and wild type in the thickness of the epithelial lining at each time point [21 days after birth (DAB) P = 0.004713728; 1 month P = 0.000598192; 2 months P = 0.0000443631; 4 months P = 0.004381456; see Fig. 6A].
Figure 6.

Thickened epithelial lining and cytology of Tgif homozygous middle ears. (A) Measurements of mucosal thickness of the middle ears of homozygous (hom, Tgif/Tgif) and wild-type (wt, +/+) mice at different stages: 21 DAB (mutant ears measured n = 6, controls measured n = 7); 1-month-old (mutants n = 6, controls n = 6); 2 months old (mutants n = 7, controls n = 7); 4 months old (mutants n = 7, controls n = 6). Five measurements of the epithelial lining of each ear were taken at a spacing of 250 μm. (B) FACS analysis of the ear effusions of 21 DAB, 1- and 2-month-old mice. Two antibodies were used: Gr1 that recognizes granulocytes (neutrophils and eosinophils) and monocytes and F4/80 that recognizes macrophages. n, number of mice used in the analysis for each time point. Bars: standard error of mean. *P < 0.05.
In addition to histological analysis of the middle ear fluid, we analysed the cell types within the ear fluids by fluorescence-activated cell sorting analysis (FACS) (Fig. 6B). Two antibodies were used for the study. Gr-1 recognizes granulocytes (PMNs) and monocytes; the other, F4/80, is a marker for macrophages. The results from the FACS analysis confirmed the results obtained from the histological study. At 21DAB, 33.2% of cells were Gr-1 positive and only 1.71% of cells were F4/80 positive. Examination of ear fluids in histological sections did not reveal other populations of cells such as lymphocytes so the >60% shortfall in stainable cells in the FACS analysis is probably attributable to high proportion of apoptotic (Fig. 5F inset) and necrotic cells. At 1 month, the ear fluids contained few identifiable cells (Fig. 5J), and the percentage of cells positive for Gr-1 (4.4%) and F4/80 (0.6%) were low (Fig. 6B). The proportion of F4/80 positive cells by FACS was significantly lower at 1 month compared with 21 DAB (P = 0.028). Although there was a very large reduction in Gr-1 cells at 1 month compared with 21 DAB, this difference was not significant. At 2 months, there were again more cells positive for Gr-1 (10.7%), but also more for F4/80 (2.3%), confirming the result from the immunohistochemistry (Fig. 5O, Fig. 6B). The proportion of F4/80 positive cells by FACS was significantly higher at 2 months compared with 1 month (P = 0.026).
Tgif homozygous mice have mucous effusions and elevated levels of cytokines in ear fluids
To study the type of the effusion in the middle ear of the homozygote mice, we looked for the expression of matrix metalloproteinase-2 (MMP-2). The expression profile of MMPs has been found to be specific to the type of middle ear effusion (33), with the active form of MMP-2 only found in mucous effusions of human patients with OM (33). We looked for the expression of MMP-2 using western blot and gelatine zymography and detected both pro- and active MMP-2 in ear effusions of 2-month-old homozygote mice (Fig. 7A and B). The zymography demonstrated high levels of MMP-9 as well, which is specific to mucous effusions (33). The result suggested that the homozygote mice have a mucous-type ear effusion at 2 months.
Figure 7.
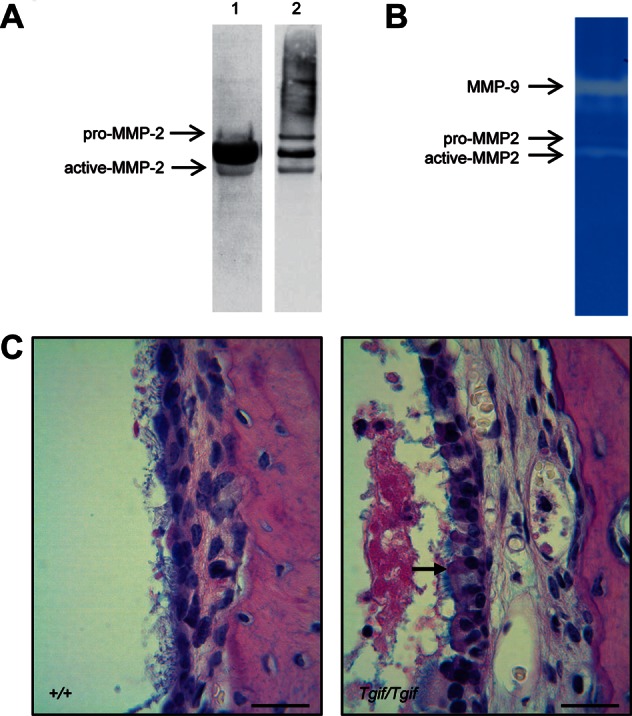
OM phenotype of the 2-month-old Tgif mutants. (A) Western blot analysis of total protein from the ear fluid of a 2-month-old homozygous (Tgif/Tgif) mouse before (1) and after (2) removing the albumin from the sample. The blot was probed with a MMP-2 antibody that detected both pro- and active forms of MMP-2. (B) Gelatine zymography demonstrating enzyme activity of MMP-2 and MMP-9 in Tgif/Tgif ear effusions. (C) Histological images of middle ear sections stained with AB-PAS, showing mucus-producing goblet cells, stained in pink, in the epithelium of the middle ear cavity (arrow) of a homozygous (right panel) and a wild-type (left panel) mouse. Scale bar = 20 μm.
This result was also confirmed by histological analysis of middle ears performed with a combined Alcian blue/Periodic acid-Schiff staining method (AB-PAS). Goblet cells are epithelial cells whose function is to secrete mucin that dissolves in water to form mucus. We detected a high density of goblet cells, stained in purple by AB-PAS, among other cells in the epithelium of the middle ear cavity of Tgif homozygote mice (Fig. 7C).
Vascular endothelial growth factor A (VEGF-A) is a member of the vascular permeability factor/VEGF family of cytokines that are critical for vasculogenesis and pathological and physiologic angiogenesis. The low level of VEGF expression in normal tissues and the overexpression in pathological angiogenesis are regulated by many factors, including hypoxia (34). Occurrence of hypoxia and elevated levels of the VEGF protein has recently been reported for Jeff and Junbo mice (11). For this reason, we assessed the level of VEGF protein in ear fluids, comparing it to serum levels. VEGF protein was detected in all serum samples and there was no significant difference between genotypes (Fig. 8A). Levels are very similar to the previously reported concentration of VEGF in sera of Jeff heterozygote mice (78 pg/ml). However, the level of the protein in ear fluids of Tgif homozygote mice was significantly higher (between 356.7 and 25 923.1 pg/ml) and was detected in 8 samples out of 10 (2 of the samples had levels below the detectable range). VEGF protein levels in Tgif/Tgif ear effusions were 66.6-fold elevated when compared with Tgif/Tgif serum (Fig. 8A), similar to Jeff mice (74-fold), although less than Junbo mice (335-fold) (11).
Figure 8.

Protein composition of middle ear fluids in Tgif mutant mice. Levels of mouse VEGF (A), TNF-α (B) and IL-1β (C) in serum samples and ear fluid from wild-type (wt, +/+), heterozygote (het, Tgif/+) and homozygote (hom, Tgif/Tgif) mice determined by Quantikine Immunoassays. n, number of mice used in the analysis from each genotype. 25% quartile, median and 75% quartile are indicated on the box plots and the whiskers represent the data range.
TNF-α and Il-1β are cytokines involved in mediating inflammatory reactions. In wild-type, heterozygote and homozygote Tgif mice, TNF-α protein was detectable in all serum samples (except one) in very low concentrations (n = 28, range 1.2–18.3 pg/ml). The average concentration of TNF-α in serum of homozygote mice was found to be 5.8 pg/ml, heterozygote mice 5 pg/ml and wild-type mice 5.2 pg/ml. Elevated concentrations of TNF-α were detected in ear fluids of homozygote mice (n = 11, range between 480.9 and 3425.6 pg/ml) that were 262-fold elevated when compared with Tgif/Tgif serum (Fig. 8B). TNF-α levels are also highly raised in the ear fluids of Jeff (50-fold) and Junbo mice (78-fold).
Il-1β protein was detectable in all blood samples (n= 28, range 1.8–24.6 pg/ml) with no significant difference between the genotypes and was also elevated (78-fold) in the ear effusions of Tgif/Tgif homozygtoes (n = 10, range between 91.8 and 1748.7 pg/ml) (Fig. 8C). Il-1β levels are raised in Jeff (3-fold) and Junbo mice (26-fold).
The mutation in Tgif affects the TGFβ signalling pathway in the epithelial cells of the middle ears
Finally, we proceeded to investigate the action of TGIF on TGFβ signalling in the epithelial cells of the mouse middle ear at the time when Tgif mutant mice start to develop OM. We performed immunohistochemical staining using TGIF, pSmad2, p21 and PML antibodies on ear sections from 3-week-old mice. TGIF protein was localized in the nucleus of middle ear cavity epithelial cells in wild-type ears, but as expected not in homozygous mutant ears. Phosphorylated Smad2 was also localized in the epithelial cells. We detected a clear difference between the number of epithelial cells positive for the pSmad2 antibody in wild-type and homozygous mutant middle ears (Fig. 9). We counted epithelial cells in eight mutant and four wild-type ears and found a significant difference between the percentage of cells positive for pSmad2 (P = 0.00029036). In wild-type middle ear, activated Smad2 was localized in 34.87% of the epithelial cells when compared with 15.58% in the homozygous middle ear. We also performed immunohistochemistry with a p21 antibody to examine localization of p21 in wild-type and mutant middle ear epithelial cells and, consistent with raised levels of pSmad2, detected more cells positive for p21 and much stronger staining in wild-type cells (Fig. 9). Finally, we examined labelling patterns of cPML, an activator of TGFβ signalling that is sequestered in the nucleus, by binding to TGIF. In wild type, the bulk of the labelling was observed in the cytoplasm and was unaffected in the homozygous mutant. We conclude from these observations that TGIF induces TGFβ-mediated transcriptional regulation in the epithelial cells of the mouse middle ear at time when the mutant mice develop OM and that this regulation does not involve significant changes in the distribution of the cPML activator protein.
Figure 9.

Protein expression in middle ear epithelia in Tgif mutant mice. (A) Immunohistochemistry of middle ear sections of wild-type (wt, +/+) and homozygous (hom, Tgif/Tgif) mice, age 21 days after birth, stained with TGIF, phosphoSmad2 and p21 and cPML antibodies. Arrows indicate nuclear localization in the epithelial cells. cPML was localized in the cytoplasm of the cells. No difference in the localization between wild-type and homozygous cells was observed with the cPML antibody. MEC, middle ear cavity. Scale bars = 1 mm and 20 μm. (B) Graphic comparison of the percentage of epithelial cells in the middle ear positive for pSmad2. Bars: standard error of mean. *P < 0.05.
DISCUSSION
Our study has identified the Tgif knockout mouse as a novel model of OM, providing further insights into the genetic basis for chronic OM. Tgif mutants have a craniofacial defect and reduced hearing by weaning age. Mice homozygous for Tgif do not have any obvious inner ear abnormalities, but demonstrate raised auditory thresholds detected by ABR. OM is the major cause of hearing impairment in Tgif homozygotes. The histological analysis revealed that they develop OM by 21 DAB. At that stage, inflammatory cells are detectable in the middle ear cavity of the mutant mice. By 2 months, the inflammation had progressed to a chronic state accompanied by a thickened middle ear epithelial lining and the presence of inflammatory cells and macrophages within the middle ear space. The inflammatory cell populations varied over time, and this may reflect episodes of bacterial infection and clearance. ET angle and morphology in homozygote mutants was normal ruling out ET dysfunction as a contributory factor to the OM that develops. Whereas Tgif homozygotes demonstrate a highly penetrant OM, our analysis of heterozygotes reveals chronic OM, but at a markedly lower frequency. Thus, Tgif is a semi-dominant mutation of OM.
Previously, we have identified Fbxo11 and Evi1 as the genes underlying two other mouse models of OM: Jeff and Junbo (5,6). Both of these models are also associated with defects in the regulation of TGFβ signalling. Jeff has a chronic proliferative OM (9), and it was shown that Fbxo11 affects TGFβ signalling by regulating the levels of phosphoSmad2 in the epithelial cells of palatal shelves, eyelids and airways of the lungs (7). Mice heterozygous for Evi1 have a hearing loss due to chronic suppurative OM (6). Evi1 is a zinc-finger protein, that represses TGFβ signalling by interacting with the MH2 domain of Smad3 (10). However, for the first time, we have identified OM arising from a lesion in a protein within the TGFβ signalling pathway and directly involved in its regulation and control.
Recently, the role of hypoxia and HIF-mediated VEGF has been highlighted as playing a key role in the pathogenesis of OM in the Junbo and Jeff mutants (11). Protein levels of VEGF, as well as the inflammatory cytokines, modulators of HIF-1α, are significantly elevated in middle ear fluids in the Junbo and Jeff mutants. There is considerable cross-talk between TGFβ and HIF-1α pathways and, for example, Smad3 and HIF-1α cooperate with TGFβ to induce VEGF expression (35,36). We might, therefore, expect TGFβ pathway mutants to impact upon the middle ear's response to the hypoxia that develops in chronic OM. It is striking, therefore, that the elevation of protein titres of VEGF, IL-1β and TNFα that were observed in the Junbo and Jeff mutants are also found in the Tgif mutant. Overall, both the OM phenotype in Tgif, along with the molecular pathogenesis observed in the middle ear, provide considerable support to the view that defects in TGFβ signalling may impact generally on susceptibility to chronic OM.
TGIF is implicated in the regulation of TGFβ signalling through a number of routes. Initially, it was found that TGIF can inhibit the signalling pathway, but recently, there is evidence that it can also induce the pathway. TGIF can repress transcription by recruiting to SMAD2 a co-repressor containing HDACs (37), or alternatively carboxyl terminus binding protein (38) to an activated Smad complex. TGIF may also inhibit TGF-β signalling by participating in the degradation of Smad2. TGIF can bind to TI1UL, and this interaction allows the ubiquitin ligase to target Smad2 for degradation (20). Recently, it has been shown that TGIF may inhibit TGFβ signalling independent of its association with Smads. The inhibition involves interaction with cPML in the nucleus, thereby, suppressing the phosphorylation of Smad2 by the TGFβ receptor (19). The interaction between TGIF and cPML can be influenced by PCTA as PCTA competes with cPML for binding with TGIF and reverses the inhibitory activity of TGIF (21,22). PCTA binding to TGIF causes the release of cPML from the nucleus to the cytoplasm, where it binds to the TGFβ receptors and promotes the phosphorylation of Smad2. Induction of PCTA can enhance the ability of TGFβ to induce expression of endogenous ADAM12 and p21, two TGFβ target genes. In addition, PCTA depletion suppresses their expression and can also blunt the growth inhibitory action of TGFβ (21).
We have shown that TGIF is localized in the epithelial cells of the middle ear of wild-type mice at the time when the Tgif mutant begins to develop OM. Moreover, pSmad2 was also localized in the same cells. In the Tgif homozygous mutant, as expected there are no epithelial cells positive for TGIF and significantly fewer cells positive for pSmad2. This suggests that TGIF is regulating the TGFβ signalling pathway by promoting the phosphorylation of Smad2 in middle ear epithelia. Indeed, consistent with this, we have observed down regulation of the TGFβ target gene, p21, in the homozygous mutant. It is not clear that these effects in the mutant are mediated by cPML as we have not observed any changes in levels of cPML that would account for the observed down regulation of p21 in the Tgif mutant. It is possible that changes to TGFβ pathway activity occur via another route not involving PCTA and cPML.
In conclusion, the discovery of an OM mouse model with a mutation in a gene regulating the TGFβ signalling pathway highlights the role of this pathway in the genetic predisposition to OM. Indeed, two candidate gene studies (3,39) have demonstrated that FBXO11, a regulator of TGFβ signalling, is significantly associated with chronic and recurrent OM in three independent cohorts. Recently, a GWAS analysis of association with OM in the Western Australian Pregnancy Cohort (Raine) study has identified 32 regions with significant evidence of association, and pathway analysis demonstrated a connection between top candidates and the TGFβ pathway (40). The identification of the Tgif mutant provides support for the role of TGFβ signalling and its effects on responses to hypoxia in the chronically inflamed middle ear. Moreover, it provides a new candidate gene to explore the genetic contributions to chronic and recurrent OM in the human population.
MATERIALS AND METHODS
Mice
Tgif mutant mice were imported from The Jackson Laboratory. They were backcrossed onto a C57BL/6J background for at least eight generations. The colony was then maintained on a C57BL/6J background and genotyped as previously described (28). Mice of all three possible genotypes were produced by intercrossing heterozygous Tgif-deficient mice.
Histology
Mouse adult 5-, 13- and 21-day old; 1-, 2- and 4-month-old heads from wild-type, heterozygote and homozygote Tgif mice were fixed in 10% buffered formaldehyde, decalcified in Kristensen's fluid for 4r days and embedded in paraffin following routine procedures. Three-micrometre-thick sections were obtained, de-paraffinized in xylene substitute and rehydrated via a graded ethanol. For morphological observations, sections were stained with H&E stain. Goblet cells were identified by a combined AB-PAS staining method.
Clickbox
One-month- and two-month-old mice were tested for a hearing defect using a clickbox (Institute of hearing Research, Nottingham, UK) that generates a brief 20 kHz soundburst at 90 dB SPL, for a presence or a lack of Preyer's reflex as previously described (32).
ABR analysis
Two-month-old wild-type, heterozygote and homozygote mice, at least 10 from each group, were used for this hearing test. The mice were anaesthetized and placed in an audiometric chamber. The acoustic stimuli were delivered to the right ear, and the test was analysed as previously described (32). After the test, the mice were sacrificed by overdose of anaesthetic, the heads were skinned and used for further analysis.
X-ray analysis
Radiography was performed using a Faxitron Mx-20 DC-4 specimen X-ray System. Image analysis program ImageJ was used to conduct measurements of the 2-month-old skulls. To study the craniofacial defect of the Tgif mutant mice, we compared the ratio between the full dorsal length of the skull and the dorsal length of the nasal bone of 10 wild-type, heterozygote and homozygote mice.
Scanning electron microscopy
Inner ears, dissected from two 1-month-old wild-type, heterozygote and homozygote mice, were fixed, washed and decalcified as previously described (41). After the decalcification, the organ of Corti was exposed, the ears were dehydrated in ethanol, dried, sputter coated with gold and then viewed on a JEOL 6010 LV scanning electron microscope under high vacuum conditions.
Western blot
Total protein was extracted from the ear effusions of 2-month-old Tgif homozygote mice. Briefly, the effusions were collected in phosphate-buffered saline (PBS), containing a cocktail of protease inhibitors (cOmplate mini, Roche), centrifuged at 10 000g for 30 min at 4°C. The albumin was removed from the supernatants using Qproteome Murine Albumin depletion kit (QIAGEN). The samples were subjected to 4–12% SDS NuPAGE (Invitrogen) and immunoblotted onto nitrocellulose membrane (Invitrogen). The antibody for MMP2 (Abcam) was used in 1:500 dilution. ECL Plus (GE Healthcare) was used as detection system. We have to repeat the western blotting with a new antibody!
Zymography
The enzyme activity of MMP-2 was assessed by zymography using gelatine impregnated gels. Total protein from ear effusion, prepared the same way as for the western blots, was loaded onto 10% SDS PAGE gels containing gelatine (Invitrogen). After electrophoresis, the gels were incubated in 1X Zymogram Renaturing Buffer (Invitrogen) for 30 min, followed by two incubations in 1X Zymogram Developing Buffer (Invitrogen)— the first one for 30 min at room temperature and the second overnight at 37°C. The gels were stained with SimplyBlue Safestain (Invitrogen) and destained in water.
Blood and ear effusion collection for Vegf, TNF-α and Il-1β protein assays
Blood from 8–10 wild-type, heterozygote and homozygote mice was collected into serum-gel clotting activator tubes (Sarstedt) from the retro-orbital sinus of mice under terminal anaesthesia. The samples were left to clot for 2 h at room temperature before centrifuging for 20 min at 2000g. The serum was removed, aliquoted and stored at −20°C until required.
Measured volumes of ear fluids from 10 homozygote mice were transferred in cold PBS, vortexed and centrifuged for 10 min at 500g, 4°C. Supernatant ear fluids were stored at −80°C until required.
Quantikine mouse VEGF, IL-1β and TNF-α ELISA kits (R&D System) were used to compare the levels of these three cytokines in the blood and ear fluid of the Tgif mutant mice.
Ear effusion collection for FACS analysis
Ear effusions from homozygote mice of age 21 DAB, 1 and 2 months were collected in 100 µl cold FACS buffer (1% BSA in PBS) and half of the amount was processed for the analysis. The samples were washed, blocked and incubated with a mixture of the two antibodies Gr1 (PerCP-Cy 5.5 Rat anti-mouse Ly6G and Ly-6C, BD Pharminogen) and F4/80 (Rat anti mouse mAb APC conjugated, Invitrogen) in 1:400 dilution for 20 min. After washing, the samples were processed on BD FACS Cantoll and set to acquire 20 000 events in FSC versus SSC gate with a flow rate of 0.5–1 µl/s.
Immunohistochemistry
For immunohistochemical analysis, the avidin–biotin complex (ABC) method was used to look for the localization of Tgif, pSmad2 and p21 in wild-type and mutant mouse middle ears. The sections through the ears of the 3-week-old mice were de-paraffinized, and endogenous peroxidase activity was quenched with 3% hydrogen peroxide in isopropanol for 20 min. Vectastain Elite ABC kit (Vector Laboratories, PK 6101) was used to perform the immunohistochemistry. The antibodies were as follows: rabbit polyclonal TGIF (H-172; sc-9084 Santa Cruz Biotechnology), rabbit polyclonal anti-phospho-Smad2 (Ser465/467) (AB3849 Chemicon International) and rabbit polyclonal p21 (C-19; sc-397 Santa Cruz Biotechnology). The sections were incubated with the antibodies in 1:200 dilutions over night at 4°C. DAB+ chromogen system (DAKO K3468) was used to develop the specific signals. The slides were counterstained with haematoxylin.
Data analysis
We used the Chi-squared test to compare the difference between the observed and the expected number of the mutant mice from crosses and Fisher's exact tests to test proportions of genotypes. To evaluate the probability of the calculated Chi-squared value, the CHIDIST function in Excel was used. Two-tailed t-test was used for comparing mean ABR and FACS thresholds. A value of P < 0.05 was considered significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work was funded by the Medical Research Council, UK. Funding to pay the Open Access publication charges for this article was provided by the MRC.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Caroline Barker, Jennifer Corrigan, Adele Seymour and Elizabeth Darley for histology services, David Shipston, Kate Vowell and Jim Humphreys for necropsy skills, Paras Pathak, Helen Natukunda and Tertius Hough for the protein assays and the FACS analysis, Sarah Carter, Andrew Hinton, Lisa Ireson and Lucie Vizor for the technical support and Steve Thomas and Kevin Glover for preparing the figures. The authors are also grateful to Christopher Walsh and Jun Shen for the Tgif knockout mice.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Casselbrant M.L., Mandel E.M., Rockette H.E., Kurs-Lasky M., Fall P.A., Bluestone C.D., Ferrell R.E. The genetic component of middle ear disease in the first 5 years of life. Arch. Otolaryngol. Head Neck Surg. 2004;130:273–278. doi: 10.1001/archotol.130.3.273. doi:10.1001/archotol.130.3.273. [DOI] [PubMed] [Google Scholar]
- 2.Daly K.A., Brown W.M., Segade F., Bowden D.W., Keats B.J., Lindgren B.R., Levine S.C., Rich S.S. Chronic and recurrent otitis media: a genome scan for susceptibility loci. Am. J. Hum. Genet. 2004;75:988–997. doi: 10.1086/426061. doi:10.1086/426061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Segade F., Daly K.A., Allred D., Hicks P.J., Cox M., Brown M., Hardisty-Hughes R.E., Brown S.D., Rich S.S., Bowden D.W. Association of the FBXO11 gene with chronic otitis media with effusion and recurrent otitis media: the Minnesota COME/ROM Family Study. Arch. Otolaryngol. Head Neck Surg. 2006;132:729–733. doi: 10.1001/archotol.132.7.729. doi:10.1001/archotol.132.7.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rye M.S., Bhutta M.F., Cheeseman M.T., Burgner D., Blackwell J.M., Brown S.D., Jamieson S.E. Unraveling the genetics of otitis media: from mouse to human and back again. Mamm. Genome. 2011;22:66–82. doi: 10.1007/s00335-010-9295-1. doi:10.1007/s00335-010-9295-1. [DOI] [PubMed] [Google Scholar]
- 5.Hardisty-Hughes R.E., Tateossian H., Morse S.A., Romero M.R., Middleton A., Tymowska-Lalanne Z., Hunter A.J., Cheeseman M., Brown S.D. A mutation in the F-box gene, Fbxo11, causes otitis media in the Jeff mouse. Hum. Mol. Genet. 2006;15:3273–3279. doi: 10.1093/hmg/ddl403. doi:10.1093/hmg/ddl403. [DOI] [PubMed] [Google Scholar]
- 6.Parkinson N., Hardisty-Hughes R.E., Tateossian H., Tsai H.T., Brooker D., Morse S., Lalane Z., MacKenzie F., Fray M., Glenister P., et al. Mutation at the Evi1 locus in Junbo mice causes susceptibility to otitis media. PLoS Genet. 2006;2:e149. doi: 10.1371/journal.pgen.0020149. doi:10.1371/journal.pgen.0020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tateossian H., Hardisty-Hughes R.E., Morse S., Romero M.R., Hilton H., Dean C., Brown S.D. Regulation of TGF-beta signalling by Fbxo11, the gene mutated in the Jeff otitis media mouse mutant. Pathogenetics. 2009;2:5. doi: 10.1186/1755-8417-2-5. doi:10.1186/1755-8417-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nolan P.M., Peters J., Strivens M., Rogers D., Hagan J., Spurr N., Gray I.C., Vizor L., Brooker D., Whitehill E., et al. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat. Genet. 2000;25:440–443. doi: 10.1038/78140. doi:10.1038/78140. [DOI] [PubMed] [Google Scholar]
- 9.Hardisty R.E., Erven A., Logan K., Morse S., Guionaud S., Sancho-Oliver S., Hunter A.J., Brown S.D., Steel K.P. The deaf mouse mutant Jeff (Jf) is a single gene model of otitis media. J. Assoc. Res. Otolaryngol. 2003;4:130–138. doi: 10.1007/s10162-002-3015-9. doi:10.1007/s10162-002-3015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurokawa M., Mitani K., Irie K., Matsuyama T., Takahashi T., Chiba S., Yazaki Y., Matsumoto K., Hirai H. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature. 1998;394:92–96. doi: 10.1038/27945. doi:10.1038/27945. [DOI] [PubMed] [Google Scholar]
- 11.Cheeseman M.T., Tyrer H.E., Williams D., Hough T.A., Pathak P., Romero M.R., Hilton H., Bali S., Parker A., Vizor L., et al. HIF-VEGF pathways are critical for chronic otitis media in Junbo and Jeff mouse mutants. PLoS Genet. 2011;7:e1002336. doi: 10.1371/journal.pgen.1002336. doi:10.1371/journal.pgen.1002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi Y., Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. doi:10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- 13.Massague J., Chen Y.G. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- 14.Tsukazaki T., Chiang T.A., Davison A.F., Attisano L., Wrana J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. doi:10.1016/S0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 15.Lin H.K., Bergmann S., Pandolfi P.P. Cytoplasmic PML function in TGF-beta signalling. Nature. 2004;431:205–211. doi: 10.1038/nature02783. doi:10.1038/nature02783. [DOI] [PubMed] [Google Scholar]
- 16.Bertolino E., Reimund B., Wildt-Perinic D., Clerc R.G. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J. Biol. Chem. 1995;270:31178–31188. doi: 10.1074/jbc.270.52.31178. doi:10.1074/jbc.270.52.31178. [DOI] [PubMed] [Google Scholar]
- 17.Holland P.W., Booth H.A., Bruford E.A. Classification and nomenclature of all human homeobox genes. BMC Biol. 2007;5:47. doi: 10.1186/1741-7007-5-47. doi:10.1186/1741-7007-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wotton D., Lo R.S., Lee S., Massague J. A Smad transcriptional corepressor. Cell. 1999;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. doi:10.1016/S0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- 19.Seo S.R., Ferrand N., Faresse N., Prunier C., Abecassis L., Pessah M., Bourgeade M.F., Atfi A. Nuclear retention of the tumor suppressor cPML by the homeodomain protein TGIF restricts TGF-beta signaling. Mol. Cell. 2006;23:547–559. doi: 10.1016/j.molcel.2006.06.018. doi:10.1016/j.molcel.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Seo S.R., Lallemand F., Ferrand N., Pessah M., L'Hoste S., Camonis J., Atfi A. The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J. 2004;23:3780–3792. doi: 10.1038/sj.emboj.7600398. doi:10.1038/sj.emboj.7600398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faresse N., Colland F., Ferrand N., Prunier C., Bourgeade M.F., Atfi A. Identification of PCTA, a TGIF antagonist that promotes PML function in TGF-beta signalling. EMBO J. 2008;27:1804–1815. doi: 10.1038/emboj.2008.109. doi:10.1038/emboj.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu F. PCTA: a new player in TGF-beta signaling. Sci. Signal. 2008;1:pe49. doi: 10.1126/scisignal.146pe49. doi:10.1126/scisignal.146pe49. [DOI] [PubMed] [Google Scholar]
- 23.Imoto I., Pimkhaokham A., Watanabe T., Saito-Ohara F., Soeda E., Inazawa J. Amplification and overexpression of TGIF2, a novel homeobox gene of the TALE superclass, in ovarian cancer cell lines. Biochem. Biophys. Res. Commun. 2000;276:264–270. doi: 10.1006/bbrc.2000.3449. doi:10.1006/bbrc.2000.3449. [DOI] [PubMed] [Google Scholar]
- 24.Melhuish T.A., Gallo C.M., Wotton D. TGIF2 interacts with histone deacetylase 1 and represses transcription. J. Biol. Chem. 2001;276:32109–32114. doi: 10.1074/jbc.M103377200. doi:10.1074/jbc.M103377200. [DOI] [PubMed] [Google Scholar]
- 25.Edwards M.C., Liegeois N., Horecka J., DePinho R.A., Sprague G.F., Jr, Tyers M., Elledge S.J. Human CPR (cell cycle progression restoration) genes impart a Far- phenotype on yeast cells. Genetics. 1997;147:1063–1076. doi: 10.1093/genetics/147.3.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overhauser J., Mitchell H.F., Zackai E.H., Tick D.B., Rojas K., Muenke M. Physical mapping of the holoprosencephaly critical region in 18p11.3. Am. J. Hum. Genet. 1995;57:1080–1085. [PMC free article] [PubMed] [Google Scholar]
- 27.Gripp K.W., Wotton D., Edwards M.C., Roessler E., Ades L., Meinecke P., Richieri-Costa A., Zackai E.H., Massague J., Muenke M., et al. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat. Genet. 2000;25:205–208. doi: 10.1038/76074. doi:10.1038/76074. [DOI] [PubMed] [Google Scholar]
- 28.Shen J., Walsh C.A. Targeted disruption of Tgif, the mouse ortholog of a human holoprosencephaly gene, does not result in holoprosencephaly in mice. Mol. Cell. Biol. 2005;25:3639–3647. doi: 10.1128/MCB.25.9.3639-3647.2005. doi:10.1128/MCB.25.9.3639-3647.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartholin L., Powers S.E., Melhuish T.A., Lasse S., Weinstein M., Wotton D. TGIF inhibits retinoid signaling. Mol. Cell. Biol. 2006;26:990–1001. doi: 10.1128/MCB.26.3.990-1001.2006. doi:10.1128/MCB.26.3.990-1001.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taniguchi K., Anderson A.E., Sutherland A.E., Wotton D. Loss of Tgif function causes holoprosencephaly by disrupting the SHH signaling pathway. PLoS Genet. 2012;8:e1002524. doi: 10.1371/journal.pgen.1002524. doi:10.1371/journal.pgen.1002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartholin L., Melhuish T.A., Powers S.E., Goddard-Leon S., Treilleux I., Sutherland A.E., Wotton D. Maternal Tgif is required for vascularization of the embryonic placenta. Dev. Biol. 2008;319:285–297. doi: 10.1016/j.ydbio.2008.04.027. doi:10.1016/j.ydbio.2008.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardisty-Hughes R.E., Parker A., Brown S.D. A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat. Protoc. 2010;5:177–190. doi: 10.1038/nprot.2009.204. doi:10.1038/nprot.2009.204. [DOI] [PubMed] [Google Scholar]
- 33.Moon S.K., Linthicum F.H., Jr, Yang H.D., Lee S.J., Park K. Activities of matrix metalloproteinases and tissue inhibitor of metalloproteinase-2 in idiopathic hemotympanum and otitis media with effusion. Acta Otolaryngol. 2008;128:144–150. doi: 10.1080/00016480701477610. doi:10.1080/00016480701477610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levy A.P., Levy N.S., Iliopoulos O., Jiang C., Kaplin W.G., Jr, Goldberg M.A. Regulation of vascular endothelial growth factor by hypoxia and its modulation by the von Hippel-Lindau tumor suppressor gene. Kidney Int. 1997;51:575–578. doi: 10.1038/ki.1997.82. doi:10.1038/ki.1997.82. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez-Elsner T., Botella L.M., Velasco B., Corbi A., Attisano L., Bernabeu C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. doi:10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- 36.Jeon S.H., Chae B.C., Kim H.A., Seo G.Y., Seo D.W., Chun G.T., Kim N.S., Yie S.W., Byeon W.H., Eom S.H., et al. Mechanisms underlying TGF-beta1-induced expression of VEGF and Flk-1 in mouse macrophages and their implications for angiogenesis. J. Leukoc. Biol. 2007;81:557–566. doi: 10.1189/jlb.0806517. doi:10.1189/jlb.0806517. [DOI] [PubMed] [Google Scholar]
- 37.Wotton D., Lo R.S., Swaby L.A., Massague J. Multiple modes of repression by the Smad transcriptional corepressor TGIF. J. Biol. Chem. 1999;274:37105–37110. doi: 10.1074/jbc.274.52.37105. doi:10.1074/jbc.274.52.37105. [DOI] [PubMed] [Google Scholar]
- 38.Melhuish T.A., Wotton D. The interaction of the carboxyl terminus-binding protein with the Smad corepressor TGIF is disrupted by a holoprosencephaly mutation in TGIF. J. Biol. Chem. 2000;275:39762–39766. doi: 10.1074/jbc.C000416200. doi:10.1074/jbc.C000416200. [DOI] [PubMed] [Google Scholar]
- 39.Rye M.S., Wiertsema S.P., Scaman E.S., Oommen J., Sun W., Francis R.W., Ang W., Pennell C.E., Burgner D., Richmond P., et al. FBXO11, a regulator of the TGFbeta pathway, is associated with severe otitis media in Western Australian children. Genes Immun. 2011;12:352–359. doi: 10.1038/gene.2011.2. doi:10.1038/gene.2011.2. [DOI] [PubMed] [Google Scholar]
- 40.Rye M.S., Warrington N.M., Scaman E.S., Vijayasekaran S., Coates H.L., Anderson D., Pennell C.E., Blackwell J.M., Jamieson S.E. Genome-wide association study to identify the genetic determinants of otitis media susceptibility in childhood. PLoS One. 2012;7:e48215. doi: 10.1371/journal.pone.0048215. doi:10.1371/journal.pone.0048215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mburu P., Romero M.R., Hilton H., Parker A., Townsend S., Kikkawa Y., Brown S.D. Gelsolin plays a role in the actin polymerization complex of hair cell stereocilia. PLoS One. 2010;5:e11627. doi: 10.1371/journal.pone.0011627. doi:10.1371/journal.pone.0011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.