

Abstract
Rett syndrome (RTT), an X-linked postnatal disorder, results from mutations in Methyl CpG-binding protein 2 (MECP2). Survival and breathing in Mecp2NULL/Y animals are improved by an N-terminal tripeptide of insulin-like growth factor I (IGF-I) treatment. We determined that Mecp2NULL/Y animals also have a metabolic syndrome and investigated whether IGF-I treatment might improve this phenotype. Mecp2NULL/Y mice were treated with a full-length IGF-I modified with the addition of polyethylene glycol (PEG-IGF-I), which improves pharmacological properties. Low-dose PEG-IGF-I treatment slightly improved lifespan and heart rate in Mecp2NULL/Y mice; however, high-dose PEG-IGF-I decreased lifespan. To determine whether insulinotropic off-target effects of PEG-IGF-I caused the detrimental effect, we treated Mecp2NULL/Y mice with insulin, which also decreased lifespan. Thus, the clinical benefit of IGF-I treatment in RTT may critically depend on the dose used, and caution should be taken when initiating clinical trials with these compounds because the beneficial therapeutic window is narrow.
INTRODUCTION
Rett syndrome (RTT) is an X-linked post-natal neurological disorder affecting ∼1 in 10 000 live female births (1), which is characterized by loss of spoken language, loss of hand use, repetitive hand movements, abnormal gait (2) and a variety of autonomic abnormalities such as cardiorespiratory dysfunction (3). Most people with RTT have mutations in Methyl-CpG-binding Protein 2 (MECP2) (4,5), a gene which encodes the transcriptional regulator, MeCP2 (6).
Daily administration of the N-terminal tripeptide of insulin-like growth factor 1 ([1–3]IGF-I) to male mice with a mutant Mecp2 allele (7,8) resulted in improved survival, dendritic spine density, and breathing. These results led to a trial in humans with RTT to investigate the therapeutic potential of recombinant human full-length IGF-I (rhIGF-I, ClinicalTrials.gov #NCT01253317).
The possible mechanism of action for the beneficial effect of [1–3]IGF-I in RTT is unknown, but it has been speculated that IGF-I and molecules derived from IGF-I might be beneficial in RTT by providing neurotrophic activity similar to Brain derived neurotrophic factor (Bdnf), a neurotrophic factor that is markedly downregulated in Mecp2 knockout mouse brains (9). Exogenous expression of Bdnf ameliorates synaptic abnormalities in MeCP2-deficient neurons (9,10) and genetically increasing Bdnf partially rescues phenotypic problems and survival in Mecp2NULL/Y animals (11).
Additional possible beneficial mechanisms for IGF-I on RTT should be considered. First, IGF-I utilizes murine thymoma viral oncogene homolog 1 (AKT) for signal transduction. AKT signaling is reduced in mice lacking Mecp2 (12) and thus increasing AKT signaling via IGF-I may be beneficial to RTT. Another consideration is the effects IGF-I can have on metabolic abnormalities. Mice lacking Mecp2 are overweight and have glucose intolerance (13), and increasing insulin signaling by the addition of IGF-I might also modify this metabolic phenotype.
To determine whether increasing signaling through IGF-I receptors might improve the RTT-like phenotypes observed in the mouse model, we treated mice with a null Mecp2 allele with a version of IGF-I with site-specific addition of polyethylene glycol at lysine 68 of IGF-I (PEG-IGF-I). PEG-IGF-I has a slower rate of receptor association (14) and longer persistence in the brain (15) in animals compared with IGF-I, but exerts identical signaling activity (14). The IGF-I receptor (IGF-IR) shares functional homology with the insulin receptor (InsR) and treatment with rhIGF-I thus can exhibit fast, insulinotropic signaling activation via the IGF-IR. Previous work has indicated that PEG-IGF-I has lower potency on fast IGF-IR-mediated functions than rhIGF-I (14), suggesting that insulinotropic side effects such as hypoglycemia and edema are reduced.
Here, we report that in addition to neurological problems, mice with a Mecp2 null allele develop a metabolic syndrome characterized by insulin resistance, hypertriglyceridemia and obesity (13) which is not ameliorated but rather aggravated by high doses of PEG-IGF-I or insulin treatment. Our results indicate that metabolic syndrome in mice with a null Mecp2 allele severely impacts the therapeutic value of IGF-I and insulin, as the induction of insulin resistance is deleterious. Our data suggest a tight therapeutic window for IGF-I therapy in RTT patients with metabolic complications.
RESULTS
Mecp2NULL/Y mice have a metabolic syndrome characterized by severe hyperinsulinemia
Previously, we noted that Mecp2NULL/Y mice on a 129S6B6F1 background are heavier than littermate controls and perform poorly on a glucose tolerance test (13). We wondered whether these mice might have a metabolic syndrome with insulin resistance, obesity, and altered serum lipids. To test this, we measured insulin levels during a glucose tolerance test (Fig. 1A). After fasting, insulin in Mecp2NULL/Y mice was markedly elevated compared with wild-type control animals, and the insulin level did not further increase after exposure to glucose (Fig. 1A). Similarly, blood glucose levels do not appropriately respond to insulin in Mecp2NULL/Y mice in an insulin tolerance test (Fig. 1B), and in fact glucose increased after the insulin injection rather than decreased (Fig. 1B). Thus, Mecp2NULL/Y mice show severe insulin resistance.
Figure 1.

Mecp2NULL/Y mice have a metabolic syndrome. (A) At 6 weeks of age, Mecp2NULL/Y mice (NULL, n = 4) have increased serum insulin throughout the glucose tolerance test, compared with wild-type (WT, n = 7). (B) When injected with insulin for an insulin tolerance test, NULL mice (n = 4) show an unusual increase in blood glucose at 15 min after insulin injection, and blood glucose remained markedly elevated compared with WT (n = 7) animals similarly treated with insulin. (C) NULL animals (n = 4) have increased whole body and fat mass, compared with wild-type (n = 7). (D, solid lines) NULL animals have increased serum triglycerides compared with WT at 4 to 5 weeks (WT n = 4, NULL n = 5) and after 6 weeks of age (WT n = 7, NULL n = 14). (D, dashed lines) NULL (n= 5) and WT (n = 4) have comparable serum cholesterol levels at 4 to 5 weeks (WT n = 4, NULL n = 5), but NULL mice have increased serum cholesterol compared to WT (n = 15) after 6 weeks age (WT n = 15, NULL n = 8). ***P < 0.001, **P < 0.01, *P < 0.05. Error bars represent SEM.
To determine whether the increased weight in Mecp2NULL/Y mice represents obesity, we assessed body composition using magnetic resonance imaging. The increased weight found in Mecp2NULL/Y mice was due to increased fat mass with no change in lean body mass (Fig. 1C). Characterization of sera from Mecp2NULL/Y mice revealed markedly increased triglycerides (Fig. 1D) throughout life and increased cholesterol after 6 weeks of age (Fig. 1D), supportive of dyslipidemia seen in metabolic syndrome. Mecp2NULL/Y mice also had decreased insulin-like growth factor binding-protein 2 (IGFBP-2) compared with wild-type animals, a feature found in metabolic syndrome (16), but levels of IGF-I and IGFBP-3 were not different (Supplementary Material, Fig. S1A–C). Because Mecp2NULL/Y mice have insulin resistance, hyperinsulinemia, high serum triglycerides and obesity, we conclude that Mecp2NULL/Y mice in this genetic background have a metabolic syndrome.
Mecp2NULL/Y animals show PEG-IGF-I dose-dependent alterations in weight and heart rate
To determine whether PEG-IGF-I has an effect on RTT-like phenotypes and the metabolic syndrome in the mouse model, we treated Mecp2NULL/Y mice with PEG-IGF-I from 28 days of life and then throughout their life. Because PEG-IGF-I has the same pharmacokinetic properties when injected subcutaneously into Mecp2NULL/Y mice as observed in other mouse strains and in rat (15) (Supplementary Material, Table S1), we choose to use the dose range we have previously found to be effective for treating neurological disease with PEG-IGF-I in mice (14,15,17).
Treatment with PEG-IGF-I resulted in either worsening or improvement in body weight in Mecp2NULL/Y mice depending on the dose administered (Fig. 2A). The highest dose, 1 mg/kg PEG-IGF-I, accelerated weight gain starting at 5 weeks age compared with vehicle-treated wild-type animals which developed increased weight at 6 weeks of age (Fig. 2A). In contrast, animals treated with the lowest dose, 0.1 mg/kg PEG-IGF-I, did not show increased weight compared with wild-type until after 7 weeks of age.
Figure 2.
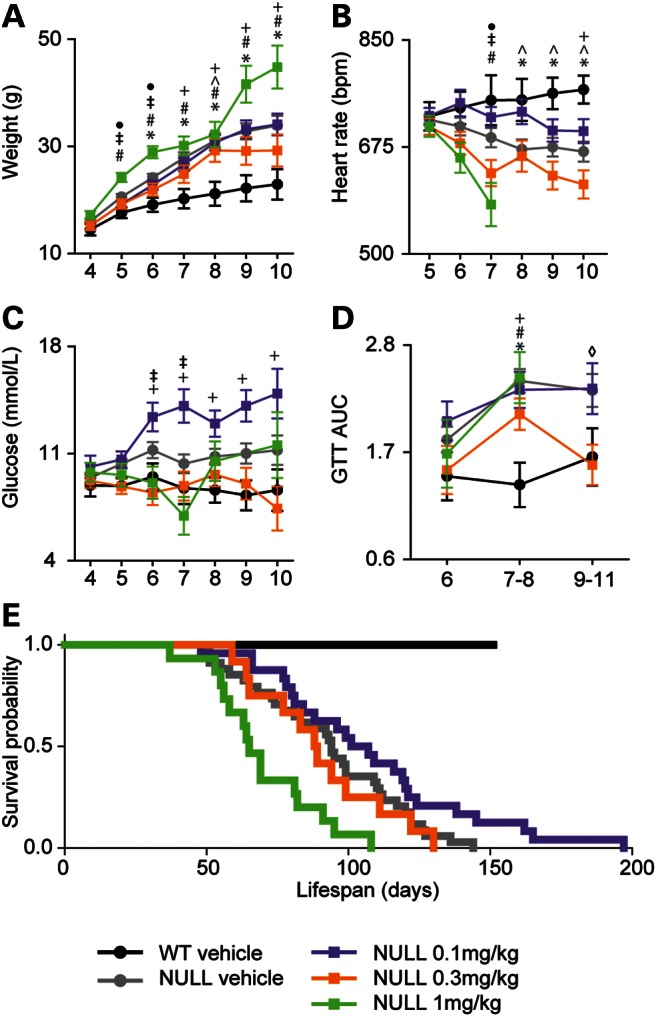
Narrow therapeutic window for PEG-IGF-I treatment on Mecp2NULL/Y mice on weight, heart rate, blood glucose and survival. (A) Vehicle-treated NULL mice show increased weight compared with littermate WT control animals starting at 6 weeks of life. Treatment with PEG-IGF-I did not improve the increased weight phenotype and at the highest dose tested, 1 mg/kg, mice show accelerated development of increased weight beginning at 5 weeks of life compared with vehicle-treated animals. (Symbols represent P < 0.05 for the following comparisons, *WT vehicle and NULL vehicle; #NULL vehicle and NULL 1 mg/kg; ^WT vehicle and NULL 0.3 mg/kg; ‡NULL 0.1 mg/kg and NULL 1 mg/kg; •NULL vehicle, NULL 1 mg/kg; +WT vehicle and NULL 0.1 mg/kg.) (B) Compared with vehicle-treated WT, the heart rate decreased in vehicle-treated NULL mice at 8–10 weeks of age and in NULL 1 mg/kg at 7 weeks of age. The sharp decline seen in heart rate in the 1 mg/kg group is because some mice have a progressive decline in the heart rate in the weeks preceding death. (C) Serum glucose is increased in vehicle-treated NULL beginning at 5 weeks of life. Treatment with 0.1 mg/kg causes an increase in baseline glucose compared with placebo-treated NULL. (D) NULL mice have abnormal glucose tolerance as shown by the area under the curve for glucose tolerance testing after 7 to 8 weeks of life (diamond indicates P < 0.05 NULL vehicle versus NULL 0.3 mg/kg). Treatment with PEG-IGF-I did not improve the glucose tolerance, except at 0.3 mg/kg at 9–11 weeks of life. (E) As expected, untreated NULL mice show early lethality (n = 34, median age of death = 94 days). The highest dose of PEG-IGF-I, 1 mg/kg, shortened lifespan (n = 15, median age of death = 65 days, P < 0.001). The medium dose, 0.3 mg/kg, had no effect on lifespan (n = 12, median age of death = 88 days, P = 0.207). There was a trend toward increased lifespan with the lowest dose of PEG-IGF-I, 0.1 mg/kg (n = 24, median age of death = 107 days, P = 0.060). For (A–D), errors bars represent SEM. See Supplementary Material, Table S2 for full details regarding the number of animals in each group at each age and full statistical comparisons for (A–D).
MecpNULL/Y mice have a decreased heart rate, observable after 8 weeks of age (Fig. 2B). Treatment with PEG-IGF-I at the lowest dose (0.1 mg/kg) delayed the heart rate decline by 2 weeks to 10 weeks of age (Fig. 2B), whereas the highest dose (1 mg/kg) led to a more rapid decline in the heart rate, observable at 7 weeks of age (Fig. 2B). Thus, treatment of Mecp2NULL/Y with a low dose of PEG-IGF-I slightly delayed the onset of obesity and heart rate decline, whereas high-dose PEG-IGF-I accelerated the onset of both.
Treatment with the highest dose of PEG-IGF-I had little effect on elevated resting glucose observed in Mecp2NULL/Y mice, except at one time point (Fig. 2C). In contrast, the 0.3 mg/kg PEG-IGF-I normalized resting glucose (Fig. 2C). Unexpectedly, the 0.1 mg/kg dose caused an increase in baseline glucose at 7 weeks of age (Fig. 2C) compared with vehicle-treated Mecp2NULL/Y. Glucose intolerance was not improved in MecpNULL/Y mice by PEG-IGF-I treatment (Fig. 2D), except at one time point (9–11 weeks old) with only one dose (0.3 mg/kg).
Low-dose PEG-IGF-I treatment increases lifespan while high-dose treatment shortens lifespan
A strong phenotype in Mecp2NULL/Y mice is decreased lifespan (18). The lowest dose of PEG-IGF-I (0.1 mg/kg) showed a trend (P = 0.06) to increase lifespan compared with vehicle-treated Mecp2NULL/Y mice (Fig. 2E), similar to the mild improvement we observed in the heart rate and delayed onset of obesity we observed at this dose (Fig. 2A and B). The intermediate dose (0.3 mg/kg) showed no effect on survival, while the highest dose (1.0 mg/kg) had a detrimental effect on survival (Fig. 2E), in agreement with the negative effects on the heart rate and the increased weight observed at this dose (Fig. 2A and B).
Low-dose PEG-IGF-I treatment did not improve RTT-like breathing or behavioral abnormalities
Because treatment with the 0.1 mg/kg dose of PEG-IGF-I delayed the heart rate decline and obesity observed in Mecp2NULL/Y mice (Fig. 2A and B) and had a trend toward a positive effect on survival (Fig. 2E), we explored other phenotypes to determine whether this low dose improved behavior or physiological functioning. Breathing, characterized by using whole-body plethysmography, was assessed at 5 and 8 weeks of life both at baseline and during exposure to a hypoxic gas challenge. Treatment had no effect on the abnormal breathing response observed in Mecp2NULL/Y mice to hypoxia at either 5 weeks (Supplementary Material, Fig. S2A and B) or 8 weeks (Fig. 3A and Supplementary Material, Fig. S2C). Additionally, treatment did not improve the baseline hyperventilation (Fig. 3A) and decreased tidal volume (Supplementary Material, Fig. S2C) observed in Mecp2NULL/Y mice at 8 weeks.
Figure 3.
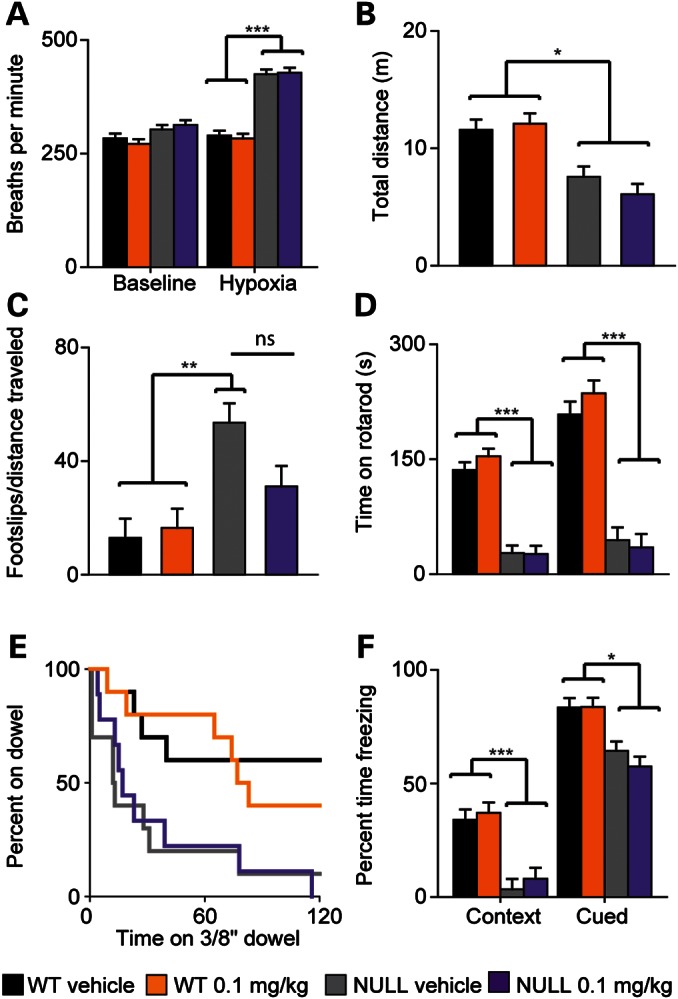
Low-dose PEG-IGF-I treatment does not improve breathing or behavior in Mecp2NULL/Y mice. (A) Untreated and treated (0.1 mg/kg PEG-IGF-I) NULL mice have significantly increased the breathing rate during the hypoxia challenge compared with WT at 8 weeks age. (B) NULL animals have decreased distance traveled in an open field, and no effect of treatment was observed. (C) NULL animals have more footslips per distance traveled compared with WT animals in the parallel rod walking task and this phenotype is not improved with PEG-IGF-I treatment. (D) In other tasks of motor learning and motor coordination, the accelerating rotating rod (E) and the dowel walking task, PEG-IGF-I treatment had no effect. (F) NULL mice perform poorly in both the context and the cue tasks in the conditioned fear task, and this performance is not improved by treatment. NULL 0.1 mg/kg n ≥ 9, all other WT and NULL groups n = 10. ***P < 0.001, **P < 0.01, *P < 0.05. Error bars represent SEM.
By 6 weeks of age, Mecp2NULL/Y mice exhibit a variety of abnormal behaviors, including hypoactivity in an open field (18), impaired motor ability (18) and motor learning (19), and impaired memory (19). Treatment with PEG-IGF-I did not improve total distance traveled (Fig. 3B) or the center distance to total distance ratio (Supplementary Material, Fig. S2D) of Mecp2NULL/Y mice in the open field. Treatment caused a trend toward improved ability of Mecp2NULL/Y mice to walk without slipping on the parallel rod (Fig. 3C, P = 0.124), but did not improve performance on the accelerating rotating rod (Fig. 3D), dowel walking (Fig. 3E) or on the wire hanging task (Supplementary Material, Fig. S2E). Finally, treatment had no effect on learning in and contextual fear or cued fear learning task (Fig. 3F) or normal acoustic startle or pre-pulse inhibition (Supplementary Material, Fig. S2F and G). Thus, treatment with PEG-IGF-I caused no meaningful behavioral or breathing improvement in Mecp2NULL/Y mice. In agreement with this, we also did not observe any improvement in dendritic spine density in hippocampal slice cultures from Mecp2NULL/Y mice when treated with full-length recombinant human IGF-I (Supplementary Material, Fig. S3), in contrast to the improvement observed in the spine phenotype in pyramidal neurons of the visual cortex after treatment with [1–3]IGF-I(7).
Insulin is detrimental to survival of Mecp2NULL/Y mice
The significant reduction in lifespan caused by 1 mg/kg of PEG-IGF-I in Mecp2NULL/Yanimals was unexpected because PEG-IGF-I has been used safely in doses of up to 3 mg/kg in wild-type mice (15) and the [1–3]IGF-I is safe up to doses of 10 mg/kg in Mecp2 mutant mice (7). One possible explanation is that Mecp2NULL/Y mice might be hypersensitive to insulinotropic effects that may occur with high doses of PEG-IGF-I. If high-dose PEG-IGF-I was detrimental to Mecp2NULL/Y mice because of the insulinotropic effects through IGF-IR, we speculated that increasing InsR activity by directly treating Mecp2NULL/Y mice with insulin would also show the same detrimental effect. To test this hypothesis, we treated Mecp2NULL/Y mice with either 0.01 mg/kg insulin (human recombinant) or vehicle throughout life. This insulin dosing schedule was selected because it allows sufficient blood–brain penetration of insulin but does not cause acute hypoglycemia (20).
Chronic treatment with insulin slightly accelerated the onset of obesity in Mecp2NULL/Y(Fig. 4A), with treated animals becoming significantly heavier than vehicle-treated wild-type animals at 6 weeks of age, whereas vehicle-treated Mecp2NULL/Y animals were not observably heavier than vehicle-treated wild-type animals until 7 weeks of age. Insulin treatment hastened the decline in heart rate of Mecp2NULL/Y mice, with treatment causing a decreased heart rate at 7 weeks of age, whereas a comparable decline in heart rate was not observed in vehicle-treated Mecp2NULL/Y animals until 9 weeks of age (Fig. 4B). Both insulin- and placebo-treated Mecp2NULL/Y mice became hyperglycemic compared with wild-type mice at 6–7 weeks of age (Fig. 4C), but the insulin-treated Mecp2NULL/Y mice trended (P = 0.053) toward increased blood glucose at this age compared with vehicle-treated Mecp2NULL/Y mice (Fig. 4C). After 8 weeks of age, there was no observable difference between insulin and vehicle-treated Mecp2NULL/Y mice. While chronic insulin treatment did not affect the baseline breathing rate at either 5–6 weeks or 8 weeks (Fig. 4D, Supplementary Material, Fig. S4A), insulin treatment did cause an exaggerated breathing rate during the hypoxia challenge at both ages in the mutant and trended toward increasing breathing rate in the wild-type animals (P = 0.07), demonstrating an unexpected detrimental drug effect (Fig. 4D, Supplementary Material, Fig. S4A). At both ages tested, there were no significant differences in tidal volume (Supplementary Material, Fig. S4B and C). Chronic insulin treatment was detrimental to Mecp2NULL/Y survival with treated animals dying 40 days earlier than untreated animals (Fig. 4E). Interestingly, we observed increased longevity of vehicle-treated Mecp2NULL/Y animals in the insulin cohort (median survival 126 days, Fig. 4E) compared with vehicle-treated Mecp2NULL/Y animals in the PEG-IGF-I cohorts (median survival 94 days, Fig. 1E). There is a distinct difference in the vehicle treatments between these two cohorts in terms of the frequency of dosing. In the PEG-IGF-I treatment group, both drug- and vehicle-treated animals were injected twice weekly, whereas in the insulin-treated group both drug and vehicle treatments were administered twice daily. We have previously observed that increased handling of Mecp2NULL/Y mice improves lifespan (data not shown). The differences we observed in the survival between these two cohorts of vehicle-treated animals is likely due to environmental enrichment provided due to the twice daily handling required for injections (21).
Figure 4.
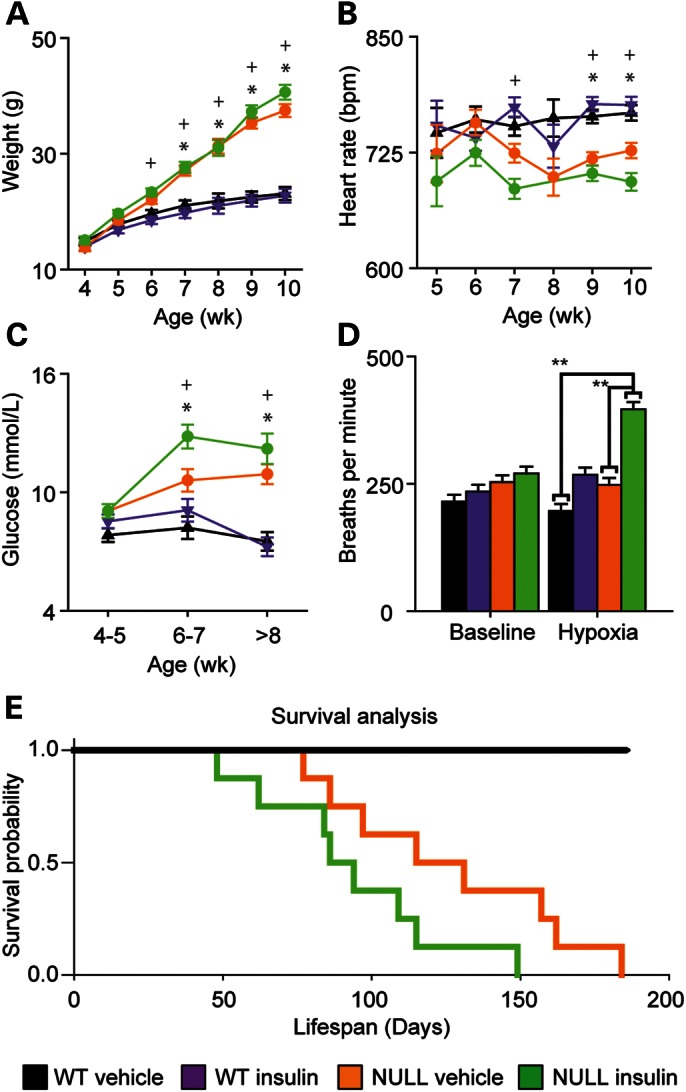
Chronic treatment of Mecp2NULL/Y mice with insulin shortens lifespan. (A) Vehicle-treated NULL show an increased body weight compared with WT starting at 7 weeks of age, while NULL treated with insulin begin showing an increased body weight at 6 weeks of life. (Symbols represent P < 0.05 for the following comparisons, *WT vehicle and NULL vehicle; +NULL vehicle and NULL insulin). (B) Heart rate is decreased in both NULL groups at 9–10 weeks of life, and insulin treatment additionally causes a decline in heart rate compared with vehicle in NULL animals at 7 weeks of age. (C) Insulin-treated NULL mice become markedly hyperglycemic at 6 to 7 weeks age compared with vehicle-treated NULL mice. (D) Insulin treatment had no effect on breathing at baseline at 8 weeks. Exposure to hypoxia caused the expected exaggerated breathing response to hypoxia in the NULL animals (n = 4), unexpectedly treatment with insulin further exaggerated this response in NULL animals (E). As expected, vehicle-treated NULL mice show early lethality (n = 8, median age of death = 126 days). Insulin treatment has a detrimental effect on survival (n = 8, median age of death = 86 days, P = 0.0582) when compared with untreated Mecp2NULL/Y. ***P < 0.001, **P < 0.01, *P < 0.05. Error bars represent SEM. See Supplementary Material, Table S3 for full details regarding the number of animals in each group at each age and full statistical comparisons for (A–C).
DISCUSSION
Based on our previous findings of insulin resistance and obesity in Mecp2NULL/Y mice, we speculated that they might have a metabolic syndrome. To confirm this hypothesis, we characterized blood serum markers in this mouse model and our findings indicate that loss of Mecp2 contributes to a metabolic syndrome. Previous work found improvement in phenotypes in a mutant mouse model of RTT after treatment with [1–3]IGF-I (7). We wanted to see whether treatment with PEG-IGFI would additionally improve this newly described metabolic phenotype. Here, we investigated the ability of PEG-IGF-I to improve phenotypes seen in Mecp2NULL/Y mice. In contrast to the improvement observed after treatment with [1–3]IGF-I, we found only a modest improvement with a low dose of PEG-IGF-I and a markedly detrimental effect on survival when Mecp2NULL/Y mice were treated with a high dose of PEG-IGF-I. We speculated that the insulinotropic effect of high-dose PEG-IGF-I is detrimental to these animals because they acquire a metabolic syndrome with insulin resistance, high serum triglycerides, obesity (22) and decreased serum levels of IGFBP2, which is similarly decreased in humans with metabolic syndrome (16). These data suggest that the detrimental effects of PEG-IGF-I may be due to increased activation of insulin signaling pathways in the setting of reduced response to insulin. This negative effect of increased insulinotropic signaling may mask any beneficial effects present from IGF-I signaling. In support of this idea, we found that treatment of Mecp2NULL/Y mice with insulin resulted in a dramatically shortened lifespan.
Why do these mice get a metabolic syndrome and how is the insulinotropic effect leading to a shortened lifespan? MeCP2 has been shown to transcriptionally regulate insulin expression by binding to a tissue-specific methylated cytosine in the promoter of the Insulin-2 gene and preventing the binding of CREB (23). This prevents the expression of insulin, and thus in the absence of MeCP2, insulin will be overexpressed and potentially expressed from non-pancreatic tissue. Constant, unregulated exposure to insulin will eventually lead to insulin resistance. Future work should focus on understanding the exact mechanism of this process as it may provide general insight into the development of metabolic syndrome.
Additionally, disruption of MeCP2 function also affects downstream signaling from the InsR. mTOR and AKT signaling is reduced in cells lacking MeCP2 (12). Therefore, a loss of MeCP2 function leads to both increased insulin and decreased insulin signaling and a complete dissociation between insulin levels and the appropriate metabolic effects of insulin on glucose regulation. The addition of more insulinotropic signals from either high doses of PEG-IGF-I or insulin would be expected to further exacerbate the situation leading to even more dramatic insulin resistance. This worsening insulin resistance will then lead to hyperglycemia and hyperinsulinemia, which is known to have detrimental effects on a variety of organs. Notably, hyperglycemia in the face of hyperinsulinemia has been shown to cause cardiac arrhythmia and specifically increase the corrected Q to T interval QTc (24–26). As the mouse models of RTT have prolonged QTc intervals and are susceptible to fatal cardiac arrhythmias (27), this hyperglycemia with hyperinsulinemia may lead to cardiac dysfunction and cardiac death. This might be the underlying cause of the shortened lifespan we observed with high-dose PEG-IGF-I and insulin.
Thus, caution is needed when utilizing IGF-I treatment in people with RTT, as there is the potential for similar adverse effects. Importantly, a subset of people with RTT has evidence for carbohydrate intolerance (K. Motil, personal communication). Identifying those people with RTT who have carbohydrate intolerance or other features of a metabolic syndrome may be critical to determine who might be susceptible to adverse events when treated with IGF-I versus those who may show improvements without problems when treated. Future work should focus on systematic metabolic characterization of people with RTT. One group of people with RTT that may deserve special consideration is those people who have pre-existing QTc interval prolongation (27), as QTc interval prolongation can predispose to the development of fatal cardiac arrhythmias and IGF-I treatment may exacerbate this risk.
There are a number of possible explanations for the observed difference in the efficacy of PEG-IGF-I and [1–3]IGF-I in the treatment of animals lacking MeCP2 function. First, [1–3]IGF-I has different pharmacological properties than full-length IGF-I (28,29) and [1–3]IGF-I does not bind either the IGF-I receptor or the InsR. Another possible explanation for the discrepancy between beneficial effects of [1–3]IGF-I treatment versus the detrimental effects of PEG-IGF-I treatment is the different effects of the two molecules on signaling within distinct cell types in the central nervous system. Recent work demonstrated that full-length IGF-I causes increased MAP kinase and phosphatidylinositol 3-kinase (PI3K) signaling within neurons, whereas [1–3]IGF-I reduced PI3K signaling in neurons and increased PI3K signaling in glia (28). This is very interesting because it has recently been demonstrated that restoration of wild-type MeCP2 into glial cells in a mouse model of RTT eliminates RTT-like phenotypes and increases survival (30). Thus, the positive effects observed from [1–3]IGF-I treatment may be due to the ability of this compound to stimulate glia, whereas the full-length IGF-I compound may not be able to provide the same amount of glia stimulation and thus lack this beneficial effect.
In addition to the ability of [1–3]IGF-I to signal through PI3K in glia, [1–3]IGF-I has been shown to have a number of additional pharmacological effects that could contribute to a beneficial effect on RTT. For example, [1–3]IGF-I stimulates both dopamine and acetylcholine release in brain slice cultures independent of IGF-I receptors (29). Both of these neurotransmitters are decreased in RTT (31,32), and increasing dopamine and norepinephrine production improves the phenotypes in mice lacking MeCP2 (33). Thus, the ability of [1–3]IGF-I to increase dopamine and acetylcholine release may contribute to the improved phenotypes in RTT mice after treatment with [1–3]IGF-I. Although full-length IGF-I may also be able to increase the release of these neurotransmitters, it does so at higher concentrations (29).
Finally, [1–3]IGF-I can interact with the N-methyl-d-aspartate (NMDA) receptor and interfere with glutamate signaling. Elevated glutamate levels in the spinal fluid of people with RTT have been reported (34,35) and this has been proposed to contribute to the pathogenesis and the increased susceptibility to seizures in this disorder. Recent work has demonstrated that microglia lacking MeCP2 release excessive amounts of glutamate (36), and that restoring MeCP2 function within microglia via bone marrow transplantation can improve phenotypes in animals lacking MeCP2 (37). Therefore, it may be that the NMDA antagonism provided by [1–3]IGF-I contributes to the beneficial effects of treatment with this compound, and effects that may not be present with full-length IGF-I treatment.
One unexpected finding from this work is the ability of different dosing regimens of vehicle injections to alter survival, at an effect size equal to or exceeding that seen with drug treatment. More frequent handling and injections increase survival, which is consistent with the improved survival seen when Mecp2NULL/Yanimals are raised in an enriched environment (38). We believe that the additional handling required for twice daily injections provides a similar stimulation of neurotrophic factors, such as Bdnf, as exposure to an enriched environment. This is an extremely important consideration when conducting preclinical trials in this animal model, as it demonstrates the absolute requirement for concurrent and identical vehicle controls for all studies to accurately assess the treatment effect. Historical controls are unacceptable, as likely are controls performed at a different time.
MATERIALS AND METHODS
Mice
All methods and animal care procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Animals used for whole-animal drug treatment, behavior and physiological characterizations were housed in AAALAC approved facilities at Baylor College of Medicine and at The University of Alabama at Birmingham. Mice for whole-animal drug treatment, behavior and physiological characterization were Mecp2NULL (Mecp2TM1.1Bird, JAX #003890), and genotyping was performed as per Jackson Lab instructions. The stock is maintained on a 129S6 background. Experimental mice were in a F1 isogenic strain background (B6129S6F1) generated by mating heterozygous Mecp2NULL/+female mice to C57Bl6 (Jackson Laboratories) male mice. All experiments were performed with littermate wild-type animals as controls.
Drug treatments
In all cases, animals were randomly assigned to a treatment group and the drug and vehicle administered and all behavioral and physiological characterizations were performed by an experimenter blinded to both treatment and genotype groups. For all experiments, treatment began at 28 days of life or when the animal reached 10 g weight. PEG-IGF-I or vehicle-containing PEG and dilutant was dissolved in filter-sterilized 0.9% NaCl and administered subcutaneously twice a week at 0.1, 0.3 or 1 mg/kg (10 ml/kg). Insulin (Sigma 91077C) or vehicle was dissolved in 0.9% NaCl, injected subcutaneously twice daily at 0.01 mg/kg (10 ml/kg).
Animal assessment
Twice weekly weight and temperature and weekly heart rate via pulse oximetry using MouseOx (STARR Life Sciences) was recorded, as described previously (13). In the presentation of weight and heart rate, data within 7 days of death are removed because we previously showed that all of these measures have a marked decline during the week before death (13). Glucose tolerance testing was performed as previously described (13). Briefly, mice were fasted overnight for 16 h and for the insulin tolerance test mice were fasted for 4 h. A fasting measurement was taken before intraperitoneal administration of either 1995 mg/kg glucose or 6.9 pmol/l insulin. Glucose was measured using either the Accu-Chek Aviva (Roche) or OneTouch UltraMini (LifeScan) system. Insulin was measured using enzyme-linked immunosorbent assay. Body composition analysis was performed by MRI using the EchoMRI-100 (Echo Medical system, Houston, TX, USA) which measures whole-body fat mass, lean tissue mass, free fluid as per published methods (39).
Breathing was assessed at 5 to 6, or 8 weeks of age using unrestrained whole-body plethysmography as previously described (13). Data were filtered for artifacts from excessive movement and sniffing behavior as described previously (13). Open field, dowel walking, wire hanging, accelerating rotating rod, fear conditioning, pre-pulse inhibition were performed as described previously (31). For parallel rod walking, mice were placed in a chamber with a square-shaped floor (19.8 × 19.8 × 29 cm) with parallel metal bars, spaced at 8 cm apart, suspended 1 cm above a metal floor. Mice were allowed to walk on the parallel rods in this chamber for 10 min. When a foot touched the metal floor beneath the parallel rods, a circuit was completed and a footslip was recorded. Locomotor activity was recorded using ANY-maze software and an overhead camera. Data were analyzed using a two-way ANOVA with genotype and drug treatment as factors, followed by Tukey t-test post hoc pairwise comparisons between all groups.
Serum biochemistry
Mice were anesthetized with isoflurane and blood was collected via retro-orbital bleed or mice were deeply anesthetized with ketamine/xylazine and blood was collected via cardiac puncture. All tissue was immediately frozen in liquid nitrogen. Serum was separated via centrifugation and frozen. IGFBPs were analyzed by quantitative western blotting, as descried previously (14). In brief, serum samples were diluted 1:5 in phosphate buffered saline (pH 7.4), separated by 12% SDS-polyacrylamide-gel electrophoresis and blotted to PVDF membranes (Millipore, Darmstadt, Germany). Bioactive IGFBPs were detected by human recombinant 125I-IGF-II and the concentrations of IGFBP-2 and -3 were calculated by using serial dilutions of human recombinant IGFBP-2 and -3 included in the gels, respectively. Data were analyzed using an unpaired t-test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by funding from F. Hoffman-La Roche Ltd., the Cynthia and Anthony Petrello Scholar fund at the Jan and Dan Duncan Neurological Research Institute, Texas Children's Hospital (J.L.N.), US National Institutes of Health grants HD062553 (J.L.N.), NS066601 (C.S.W.), HD24064 (Baylor IDDRC), P30 DK079638 (Mouse Metabolism Core at Baylor under Diabetes Research Center), NS065027 (L.P.-M.), IRSF-2806 HeART award (L.P.-M.) and supported in part by a grant to Baylor College of Medicine from the Howard Hughes Medical Institute through the Med Into Grad Initiative. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the National Institutes of Health.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Ekaette Shammah and Diana Parra for technical assistance and Huda Zoghbi for helpful comments on the manuscript. We also thank the Baylor IDDRC Neurobehavioral Core (Director Richard Paylor and Corinne Spencer) for the use of the facility and advice in behavioral experiments. We thank Karin Weber and Prof. Gehard Binder for their generous gift of radiolabeled human recombinant IGF-II.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Laurvick C.L., de Klerk N., Bower C., Christodoulou J., Ravine D., Ellaway C., Williamson S., Leonard H. Rett syndrome in Australia: a review of the epidemiology. J. Pediatr. 2006;148:347–352. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 2.Neul J.L., Kaufmann W.E., Glaze D.G., Christodoulou J., Clarke A.J., Bahi-Buisson N., Leonard H., Bailey M.E., Schanen N.C., Zappella M., et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann. Neurol. 2010;68:944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glaze D.G. Neurophysiology of Rett Syndrome. J. Child Neurol. 2005;20:740–746. doi: 10.1177/08830738050200090801. [DOI] [PubMed] [Google Scholar]
- 4.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 5.Neul J.L., Fang P., Barrish J., Lane J., Caeg E.B., Smith E.O., Zoghbi H., Percy A., Glaze D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett Syndrome. Neurology. 2008;70:1313–1321. doi: 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chahrour M., Jung S.Y., Shaw C., Zhou X., Wong S.T., Qin J., Zoghbi H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tropea D., Giacometti E., Wilson N.R., Beard C., McCurry C., Fu D.D., Flannery R., Jaenisch R., Sur M. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. PNAS. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen R.Z., Akbarian S., Tudor M., Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 9.Kline D.D., Ogier M., Kunze D.L., Katz D.M. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J. Neurosci. 2010;30:5303–5310. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larimore J.L., Chapleau C.A., Kudo S., Theibert A., Percy A.K., Pozzo-Miller L. Bdnf overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobio. of Disease. 2009;34:199–211. doi: 10.1016/j.nbd.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang Q., Khare G., Dani V., Nelson S., Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 12.Ricciardi S., Boggio E.M., Grosso S., Lonetti G., Forlani G., Stefanelli G., Calcagno E., Morello N., Landsberger N., Biffo S., et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Gen. 2011;20:1182–1196. doi: 10.1093/hmg/ddq563. [DOI] [PubMed] [Google Scholar]
- 13.Ward C.S., Arvide E.M., Huang T.W., Yoo J., Noebels J.L., Neul J.L. MeCP2 is critical within HoxB1-derived tissues of mice for normal lifespan. J. Neurosci. 2011;31:10359–10370. doi: 10.1523/JNEUROSCI.0057-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metzger F., Sajid W., Saenger S., Staudenmaier C., van der Poel C., Sobottka B., Schuler A., Sawitzky M., Poirier R., Tuerck D., et al. Separation of fast from slow anabolism by site-specific PEGylation of insulin-like growth factor I (IGF-I) J. Bio. Chem. 2011;286:19501–19510. doi: 10.1074/jbc.M110.172189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saenger S., Goeldner C., Frey J.R., Ozmen L., Ostrowitzki S., Spooren W., Ballard T.M., Prinssen E., Borroni E., Metzger F. PEGylation enhances the therapeutic potential for insulin-like growth factor I in central nervous system disorders. Growth Horm. IGF Res. 2011;21:292–303. doi: 10.1016/j.ghir.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Heald A.H., Kaushal K., Siddals K.W., Rudenski A.S., Anderson S.G., Gibson J.M. Insulin-like growth factor binding protein-2 (IGFBP-2) is a marker for the metabolic syndrome. Exp. Clin. Endocrinol. Diabetes. 2006;114:371–376. doi: 10.1055/s-2006-924320. [DOI] [PubMed] [Google Scholar]
- 17.Jablonka S., Holtmann B., Sendtner M., Metzger F. Therapeutic effects of PEGylated insulin-like growth factor I in the pmn mouse model of motorneuron disease. Exp. Neurol. 2011;232:261–269. doi: 10.1016/j.expneurol.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 18.Guy J., Hendrich B., Holmes M., Martin J.E., Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 19.Stearns N.A., Schaevitz L.R., Bowling H., Nag N., Berger U.V., Berger-Sweeney J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neurosci. 2007;146:907–921. doi: 10.1016/j.neuroscience.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Banks W.A., Jaspan J.B., Kastin A.J. Selective, physiological transport of insulin across the blood-brain barrier: novel demonstration by species-specific radioimmunoassays. Peptides. 1997;18:1257–1262. doi: 10.1016/s0196-9781(97)00198-8. [DOI] [PubMed] [Google Scholar]
- 21.Lonetti G., Angelucci A., Morando L., Boggio E.M., Giustetto M., Pizzorusso T. Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biol. Psychiatry. 2010;67:657–665. doi: 10.1016/j.biopsych.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 22.Kassi E., Pervanidou P., Kaltsas G., Chrousos G. Metabolic syndrome: definitions and controversies. BMC Med. 2011;9:13–48. doi: 10.1186/1741-7015-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuroda A., Rauch T.A., Todorov I., Ku H.T., Al-Abdullah I.H., Kandeel F., Mullen Y., Pfeifer G.P., Ferreri K. Insulin gene expression is regulated by DNA methylation. PloS One. 2009;4:e6953. doi: 10.1371/journal.pone.0006953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiorentini A., Perciaccante A., Valente R., Paris A., Serra P., Tubani L. The correlation among QTc interval, hyperglycaemia and the impaired autonomic activity. Auton. Neurosci. 2010;154:94–98. doi: 10.1016/j.autneu.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Pickham D., Helfenbein E., Shinn J.A., Chan G., Funk M., Weinacker A., Liu J.N., Drew B.J. High prevalence of corrected QT interval prolongation in acutely ill patients is associated with mortality: results of the QT in Practice (QTIP) Study. Critical Care Med. 2012;40:394–399. doi: 10.1097/CCM.0b013e318232db4a. [DOI] [PubMed] [Google Scholar]
- 26.van Noord C., Sturkenboom M.C., Straus S.M., Hofman A., Kors J.A., Witteman J.C., Stricker B.H. Serum glucose and insulin are associated with QTc and RR intervals in nondiabetic elderly. Eur. J. Endocrinol. 2010;162:241–248. doi: 10.1530/EJE-09-0878. [DOI] [PubMed] [Google Scholar]
- 27.McCauley M.D., Wang T., Mike E., Herrera J., Beavers D.L., Huang T.W., Ward C.S., Skinner S., Percy A.K., Glaze D.G., et al. Pathogenesis of lethal cardia arrhythmias in Mecp2 mutant mice: implication for therapy in Rett Syndrome. Sci. Transl. Med. 2011;3:113ra125. doi: 10.1126/scitranslmed.3002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corvin A.P., Molinos I., Little G., Donohoe G., Gill M., Morris D.W., Tropea D. Insulin-like growth factor 1 (IGF1) and its active peptide (1–3) IGF1 enhance the expression of synaptic markers in neuronal circuits through different cellular mechanisms. Neurosci. Lett. 2012;520:51–56. doi: 10.1016/j.neulet.2012.05.029. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson-Hakansson L., Civalero I., Zhang X., Carlsson-Skwirut C., Sara V.R., Nordberg A. Effects of IGF-1, truncated IGF-1 and the tripeptide Gly-Pro-Glu on acetylcholine release from parietal cortex of rat brain. Neuroreport. 1993;4:1111–1114. [PubMed] [Google Scholar]
- 30.Lioy D., Garg S., Monaghan C., Raber J., Foust K., Kaspar B., Hirrlinger P., Kirchhoff F., Bissonnette J., Ballas N., Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samaco R.C., Fryer J.D., Ren J., Fyffe S., Chao H.T., Sun Y., Greer J.J., Zoghbi H.Y., Neul J.L. A partial loss of function allele of Methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum. Mol. Gen. 2008;17:1718–1727. doi: 10.1093/hmg/ddn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wenk G.L., Hauss-Wegrzyniak B. Altered cholinergic function in the basal forebrain of girls with Rett syndrome. Neuropediatrics. 1999;30:125–129. doi: 10.1055/s-2007-973476. [DOI] [PubMed] [Google Scholar]
- 33.Panayotis N., Pratte M., Borges-Correia A., Ghata A., Villard L., Roux J.C. Morphological and functional alterations in the substantia nigra pars compacta of the Mecp2-null mouse. Neurobio. Dis. 2011;41:385–397. doi: 10.1016/j.nbd.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Hamberger A., Gillberg C., Palm A., Hagberg B. Elevated CSF glutamate in Rett syndrome. Neuropediatrics. 1992;23:212–213. doi: 10.1055/s-2008-1071344. [DOI] [PubMed] [Google Scholar]
- 35.Lappalainen R., Riikonen R.S. High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr. Neurol. 1996;15:213–216. doi: 10.1016/s0887-8994(96)00218-4. [DOI] [PubMed] [Google Scholar]
- 36.Maezawa I., Jin L.W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derecki N.C., Cronk J.C., Lu Z., Xu E., Abbott S.B., Guyenet P.G., Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kondo M., Gray L.J., Pelka G.J., Christodoulou J., Tam P.P., Hannan A.J. Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndrome—Mecp2 gene dosage effects and BDNF expression. European J. Neurosci. 2008;27:3342–3350. doi: 10.1111/j.1460-9568.2008.06305.x. [DOI] [PubMed] [Google Scholar]
- 39.Lacaria M., Saha P., Potocki L., Bi W., Yan J., Girirajan S., Burns B., Elsea S., Walz K., Chan L., et al. A duplication CNV that conveys traits reciprocal to metabolic syndrome and protects against diet-induced obesity in mice and men. PLoS Genet. 2012;8:e1002713. doi: 10.1371/journal.pgen.1002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.