

Abstract
Mutations in the gene encoding Fused in Sarcoma (FUS) cause amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disorder. FUS is a predominantly nuclear DNA- and RNA-binding protein that is involved in RNA processing. Large FUS-immunoreactive inclusions fill the perikaryon of surviving motor neurons of ALS patients carrying mutations at post-mortem. This sequestration of FUS is predicted to disrupt RNA processing and initiate neurodegeneration. Here, we demonstrate that C-terminal ALS mutations disrupt the nuclear localizing signal (NLS) of FUS resulting in cytoplasmic accumulation in transfected cells and patient fibroblasts. FUS mislocalization is rescued by the addition of the wild-type FUS NLS to mutant proteins. We also show that oxidative stress recruits mutant FUS to cytoplasmic stress granules where it is able to bind and sequester wild-type FUS. While FUS interacts with itself directly by protein–protein interaction, the recruitment of FUS to stress granules and interaction with PABP are RNA dependent. These findings support a two-hit hypothesis, whereby cytoplasmic mislocalization of FUS protein, followed by cellular stress, contributes to the formation of cytoplasmic aggregates that may sequester FUS, disrupt RNA processing and initiate motor neuron degeneration.
Introduction
Amyotrophic lateral sclerosis (ALS, also known as motor neuron disease) is an adult-onset disorder characterized by progressive muscular weakness due to motor neuron degeneration. Death from respiratory failure occurs, on average, 3 years after symptom onset. In 5–10% of cases, there is a family history of ALS and/or frontotemporal lobar dementia (FTLD), usually with an autosomal-dominant pattern of inheritance (1). We, and others, have recently identified mutations in the gene encoding the RNA-binding protein Fused in Sarcoma (FUS) as a cause of familial ALS (2–16). Large FUS-immunoreactive inclusions were detected in the anterior horn of the spinal cord in FUS mutant patients at post-mortem. Cytoplasmic FUS inclusions are also detected in tau- and TDP-43-negative FTLD cases, but they are not associated with FUS mutations (17).
FUS is a ubiquitously expressed protein involved in DNA repair, the regulation of RNA transcription, splicing and export to the cytoplasm (18,19). It belongs to the FET (FUS, EWS and TAF15) family of proteins, which also includes Ewing's sarcoma (EWS) and TATA box-binding protein (TBP)-associated factor (TAF15). FUS has 526 amino acids and contains an N-terminal serine, tyrosine, glycine and glutamine-rich domain, followed by an RNA-recognition motif (RRM) and a zinc finger domain. Three RGG-repeat regions are found interspersed between these domains. The RNA-binding properties are conferred by the RRM, zinc finger and the RGG domains (20).
In ALS, a cluster of non-synonymous mutations lie at the extreme C-terminus of FUS, with the R521C and R521H mutations being the most common (Fig. 1; 2–16,21–28). In cellular transfection studies, mutation of FUS increased the cytoplasmic localization of this predominantly nuclear protein (12,14).
Figure 1.

The majority of ALS mutations are located in the C-terminus. Schematic diagram showing published ALS mutations in FUS demonstrating that the majority are located within the last 30 amino acids of the protein. As well as point mutations that affect single amino acids, there are nonsense and frameshift mutations which result in the loss and disruption of the C-terminus. The different functional domains of the protein are indicated in bold letters. RRM, (RNA-recognition motif); Zn, (zinc finger domain); NLS (nuclear localizing signal).
Here, we demonstrate that (i) ALS-linked mutations in FUS disrupt the nuclear localizing signal (NLS), (ii) the addition of a wild-type C-terminus to a mutant FUS protein rescues mislocalization, (iii) mutant FUS is associated with stress granules in the cytoplasm and that this is downstream of FUS mislocalization, (iv) mutant FUS interacts with the wild-type and mislocalizes it to the cytoplasm, (v) the interaction between the stress granule protein PABP and FUS occurs in an RNA-dependent manner and (vi) the accumulation of FUS in stress granules is recapitulated in patient fibroblasts following oxidative stress. We propose that ALS-linked FUS mutations result in increased cytoplasmic localization. Following stress, FUS is recruited to stress granules which initiate aggregate formation and the sequestration of wild-type FUS.
RESULTS
C-terminal ALS FUS mutants form cytoplasmic inclusions due to the disruption of the NLS, an effect abolished by the addition of a wild-type NLS to mutant proteins
We generated N-terminally GFP-tagged full-length wild-type and ALS mutant FUS (GFP-FUSWT GFP-FUSR521C, GFP-FUSR521H and GFP-FUSR514G) and a C-terminal truncated form (unpublished ALS mutant, GFP-FUSK510X), deleting the C-terminal 17 amino acids after the last RGG region (Fig. 2A). In transiently transfected CV-1 cells, the GFP-FUSWT protein was predominantly nuclear, while the ALS mutants (GFP-FUSR521C, GFP-FUSR521H and GFP-FUSR514G) showed increased cytoplasmic localization in a high percentage of cells (Fig. 2B and C). The truncated FUS protein GFP-FUSK510X showed almost exclusive cytoplasmic localization, confirming that the C-terminal region contains the dominant NLS for FUS (Fig. 2B and C).
Figure 2.

The C-terminus of FUS contains a nuclear localizing signal (A) Schematic of GFP-FUS constructs showing the wild-type, truncation mutation, the ALS-linked mutants and the extended proteins with the wild-type C-terminal 17 amino acids added to the C-terminus. The position of the mutations is marked with a red bar. (B) The percentage of transfected CV-1 cells with cytoplasmic FUS is significantly increased for mutant constructs compared with wild-type. The addition of the wild-type 17 amino acids completely rescued FUS mislocalization. (***P < 0.001; minimum of 200 cells counted per transfection). (C) The subcellular localization of the green fluorescent protein tagged-FUS constructs. GFP fluorescence (green) and the merged image of GFP and DAPI stained nuclei (blue) of CV-1 cells are shown. Scale bar 25 μm.
In order to determine whether the ALS-linked mutants were mislocalized due to a specific disruption of the NLS, we added the 17 C-terminal amino acids containing the wild-type NLS to the C-terminus of the three familial ALS mutants (GFP-FUSR521C, GFP-FUSR521H and GFP-FUSR514G) and to wild-type FUS (GFP-FUSWT) as a control (Fig. 2A). Following the addition of the wild-type NLS to the wild-type protein FUS remained in the nucleus while its addition to each of the ALS mutant proteins (mutant + NLS) restored their nuclear localization (one-way ANOVA, P < 0.001; Bonferroni post-hoc, all P < 0.001). This supports the hypothesis that the ALS-linked mutations directly disrupt the NLS resulting in mislocalization and accumulation in the cytoplasm (Fig. 2B and C).
Cytoplasmic mislocalized FUS associates with stress granule markers
Because FUS is involved in regulating RNA transcription, splicing and export, and the appearance of the cytoplasmic FUS was granular and has previously been shown to be associated with stress granules, we sought to determine whether cytoplasmic mutant FUS was associated with all types of RNA granules. CV-1 cells were transfected with GFP-FUSWT, GFP-FUSR514G and GFP-FUSK510X and immunostained for a range of RNA granule markers. As previously shown in Fig. 2B, GFP-FUSR514G and GFP-FUSK510X formed cytoplasmic granules. Antibodies to the proteins XRN1 (a marker for P bodies), Staufen (RNA transport granules) and SC-35 (nuclear speckles) did not co-localize with these granules (Supplementary Material, Fig. S1A, B and C). However, striking co-localization was seen for GFP-FUS mutants and two stress granule markers, poly A-binding protein 1 (PABP) and T-cell-restricted intracellular antigen-1 (TIA-1), in the cytoplasm of CV-1 cells (Fig. 3A and B) and rat cortical neurons (Fig. 3C and D and Supplementary Material, Fig. S2). In contrast, GFP-FUSWT and GFP-FUSR514G+NLS remained nuclear, and did not form either PABP or TIA-1-positive cytoplasmic granules.
Figure 3.

Mutant FUS co-localizes with stress granule markers in the cytoplasm. Immunofluorescent staining of CV-1 cells (A and B) and rat primary neurons (C and D). CV1 cells and primary neurons were transfected with GFP-FUSWT, GFP-FUSR514G, GFP-FUSK510X and GFP-FUSR514G+NLS (green) and immunostained for the stress granule markers, PABP (red, A and C) and TIA-1 (red, B and D). GFP-FUSR514Gand GFP-FUSK510X co-localized with PABP and TIA-1 in the cytoplasm, while GFP-FUSWT and GFP-FUSR514G+NLS remained in the nucleus and did not affect the subcellular distribution of the stress granule markers. Nuclei are stained blue with DAPI in the merged images (right hand panel). Scale bar 25 μm (A and B), 50 μm (C and D).
While this incorporation into stress granules has previously been reported, there has been some conflicting data as to whether this is a spontaneous event or requires an additional cellular stress for it to occur. This led us to try to establish whether the formation of stress granules in these cells was a natural consequence of mutant FUS localizing to the cytoplasm, or whether stress granule formation could be linked to the protein over-expression associated with transient transfections. To explore this question and to also rule out any aggregation effect mediated by the large GFP epitope (29), we generated stable SH-SY5Y cells expressing doxycycline (DOX)-inducible FUS, where the N-terminal GFP tag was replaced by a HA tag. Following induction of expression of HA-FUS with DOX, both immunocytochemistry (ICC) and biochemical fractionation showed a significant increase in cytoplasmic localization of the HA-FUSR514G and HA-FUSK510X compared with HA-FUSWT (one-way ANOVA, HA-FUSR514G (P < 0.05) and HA-FUSK510X (P < 0.005), Fig. 4A, C and D). This amount of HA-FUSWT in the cytoplasmic FUS was similar to that seen when endogenous HEK293T cells were fractionated (Supplementary Material, Fig. S3). However, there was no formation of stress granules, as measured by ICC, and PABP remained diffuse throughout the cytoplasm, indicating that in this cellular model mutant FUS in the cytoplasm does not directly induce, or become recruited to, stress granules.
Figure 4.

Mutant FUS is mislocalized in stable cell lines but requires oxidative stress to form cytoplasmic granules. (A) Immunofluorescence of SH-SY5Y cell lines stably transfected with HA-FUSWT, HA-FUSR514G or HA-FUSK510X. Staining for HA (green) shows HA-FUSWT localized entirely to the nucleus, while HA-FUSR514G is predominantly nuclear but also partially diffuse in the cytoplasm, and HA-FUSK510X has much more diffuse FUS in the cytoplasm. PABP (red) remained diffuse in the cytoplasm in all the three cell lines. Nuclei are stained blue in the merged image. (B) Immunofluorescence following treatment with 1 mm arsenite showing that the PABP (red) has formed distinct granules in the cytoplasm. HA-FUS (green) remains entirely nuclear in the wild-type cell line, while cytoplasmic HA-FUS in the mutant cell lines formed granules that co-localize with the PABP signal. Nuclei are stained blue in the merged image. (C and D) Subcellular fractionation of cell lines expressing HA-FUSWT, HA-FUSR514G or HA-FUSK510X showing a significant increase in the proportion of cytoplasmic HA-FUS with the R514G mutation (* P < 0.05) and the truncation mutation (** P < 0.005; n = 3). GAPDH is used to mark the cytoplasmic fraction, while Lamin B1 is used to mark the nuclear fraction. Scale bar 25 μm.
Sodium arsenite (hereafter referred to as arsenite) is widely used to model oxidative stress in cell culture, and has been shown to induce PABP-positive stress granules (30). Oxidative stress has previously been implicated in the pathogenesis of ALS (21). Addition of arsenite to HA-FUS-expressing cells resulted in the formation of PABP-positive stress granules within an hour of exposure (Fig. 4B). In cells expressing HA-FUSR514G and HA-FUSK510X, HA-FUS was recruited to PABP-positive stress granules in the cytoplasm. However, there was no alteration in the distribution or appearance of HA-FUSWT following arsenite treatment, despite the formation of PABP-positive stress granules.
To further characterize the interaction between FUS and PABP, we used the proximity ligation assay (PLA). The PLA uses antibodies directed against the two proteins of interest, in combination with DNA probe-linked secondary antibodies, to determine whether the two proteins lie within 40 nm from one another. Through polymerase amplification of the DNA probes, this method allows detection of proximity and co-localization that is often below the detection level of ICC. Using the inducible SH-SY5Y cells, this assay showed that HA-FUSWT and PABP were located close to each other in the cytoplasm under basal conditions (Fig. 5A and C). This is consistent with the fact that FUS is known to shuttle between the nucleus and the cytoplasm and indeed a small proportion of wild-type FUS was seen in the cytoplasmic fraction by western blot (Fig. 4C and D). This interaction between HA-FUS and PABP was slightly increased in HA-FUSR514G and significantly increased in HA-FUSK510X-expressing cells (two-way ANOVA, P < 0.005, Fig. 5A and C). Treatment with arsenite resulted in a significant increase in HA-FUS and PABP co-localization for HA-FUSR514G and HA-FUSK510X but not for HA-FUSWT, which remained at a low level of proximity to PABP (two-way ANOVA, P < 0.05, Fig. 5B and C). A no primary-antibody control was performed which showed a weak diffuse cytoplasmic signal with no distinct puncta (Supplementary Material, Fig. S4).
Figure 5.

FUS and PABP interact in the cytoplasm without stress. (A) PLA showing the interaction between PABP and HA-FUS in all three stable cell lines expressing wild-type or mutant FUS under basal conditions and (B) following induction of stress granules with the addition of arsenite. The red dots indicate points of FUS and PABP interaction (40 nm). Nuclei are stained blue with DAPI in the merged images (right-hand panels). (C) Quantification of the size and number of granules showed that there was a significant increase in the interaction between HA-FUSK510X and PABP compared to HA-FUSWT (**P < 0.005). The treatment with arsenite significantly increased the interaction between HA-FUS and PABP for both R514G and K510X (*P < 0.05, minimum of 70 nuclei counted per condition). Scale bar 25 μm.
Mutant FUS binds to wild-type FUS and both are recruited to cytoplasmic stress granules
In spinal cord tissue from an ALS patient with a FUSR521H mutation, we observed that in motor neurons containing cytoplasmic inclusions there is a near absence of nuclear FUS (Fig. 6A). This suggests that both mutant and wild-type FUS can be sequestered within cytoplasmic inclusions and that levels of nuclear FUS are greatly reduced, as is observed with TDP-43 inclusions in FTLD (31). To explore this experimentally, we co-transfected CV-1 cells with N-terminally Myc-tagged wild-type FUS (Myc-FUSWT) with an N-terminally GFP-tagged ALS mutant (GFP-FUSR514G), the truncation mutation (GFP-FUSK510X) or a wild-type control (GFP-FUSWT). We observed that when wild-type FUS was co-transfected with mutant or truncated FUS, the wild-type protein was now localized to both the nucleus and the cytoplasm (Fig. 6B). Thus, the accumulation of mutant FUS in the cytoplasm contributes to the mislocalization of wild-type FUS and may later result in inclusion formation.
Figure 6.
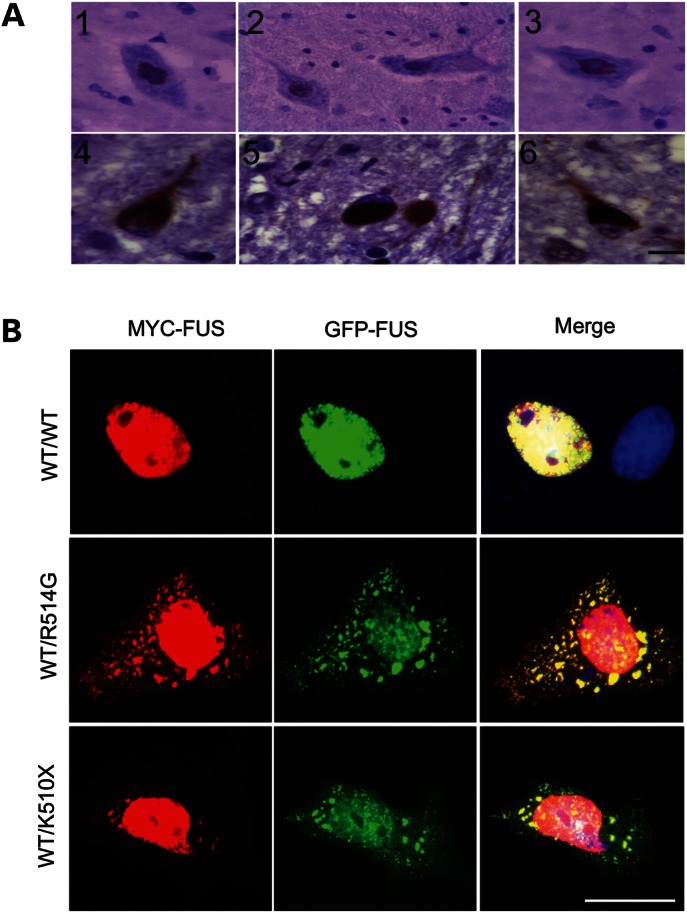
Mutant FUS sequesters wild-type FUS within cytoplasmic inclusions. (A) Anti-FUS immunostaining on sections from the anterior horn of the spinal cord of control individuals (1, 2, 3) and a patient with the FUSR521H mutation (4, 5, 6). Nuclear staining is present in control motor neurons and virtually absent in motor neurons in ALS with large cytoplasmic inclusions. (B) Confocal fluorescent microscopy of CV-1 cells with a double transfection of Myc-FUSWT and GFP-FUSWT or GFP-FUS mutants showing co-localization of the GFP signal from the mutant and Myc signal from wild-type FUS proteins within cytoplasmic inclusions. Scale bar: 15 μm (A), 25 μm (B).
FUS interactions with PABP are RNA dependent while FUS self-interaction is non-RNA dependent
In order to establish whether the recruitment of wild-type FUS into the cytoplasmic granules with the mutant FUS was due to the two proteins directly interacting, we performed a series of immunoprecipitation experiments. First, Myc-tagged wild-type FUS (Myc-FUSWT) was co-transfected with HA-FUSWT, HA-FUSR514G or HA-FUSK510X, and Myc-FUSWT protein immunoprecipitated using an anti-Myc antibody. For each co-transfection, a proportion of the HA-FUS was co-immunoprecipitated (Fig. 7A). To determine whether the FUS proteins also interacted with stress granule proteins, the blots were probed for the presence of PABP. This showed that an endogenous PABP protein was also co-immunoprecipitated, suggesting that all three proteins form a complex. Immunoprecipitation using an anti-HA antibody resulted in a similar pull down of the Myc-FUS protein and PABP (Supplementary Material, Fig. S5).
Figure 7.

FUS forms a complex with itself, PABP and RNA. (A) Co-immunoprecipitation of HA-FUS, Myc-FUS and PABP from HEK293T cells. Immunoprecipitation with a Myc (mouse anti-Myc, CST) antibody pulled down PABP (mouse anti-PABP, Sigma) along with anti-HA-FUS (rabbit HA, CST). RNasin was added to block all RNase activity. (B) The interaction between HA-FUS and Myc-FUS was not altered by the addition of RNase, while the co-immunoprecipitation of PABP was completely abolished. (C) Immunoprecipitation of FUS (mouse anti-FUS, Santa Cruz) from untransfected cells shows that endogenous FUS (rabbit anti-FUS, Novus Biologicals) also interacts with PABP (mouse anti-PABP, Sigma) and treatment with RNase shows that this interaction is also dependent on RNA. (D) Mock immunoprecipitation experiments with either untransfected cells or a single transfection of either HA-FUSWT or Myc-FUSWT with no antibody showed that neither endogenous nor tagged FUS binds to the beads. (E) Immunoprecipitation of single transfections with an antibody to the wrong tag showed that a Myc antibody does not pull down HA-FUS and a HA antibody does not precipitate Myc-FUS. FT indicates flow-through of proteins that did not bind to the beads.
Given that both FUS and PABP bind RNA, we next sought to determine whether the interaction between these proteins was RNA dependent. Treatment with RNase completely abolished co-immunoprecipitation of PABP by Myc-FUS, demonstrating that this interaction is RNA binding dependent. RNase treatment did not, however, alter the binding of wild-type FUS to itself or to mutant FUS. This was true for both HA-FUSR514G and HA-FUSK510X, indicating that FUS self-interaction is not dependent on the C-terminus (Fig. 7B). Finally, immunoprecipitation of endogenous FUS yielded endogenous PABP only in the absence of RNase, demonstrating that the interaction between these proteins is physiological and not due to the tagged epitopes (Fig. 7C). Collectively, these findings demonstrate that wild-type FUS interacts with itself and with mutant FUS and both can be recruited to PABP-positive stress granules via their binding to RNA.
FUS is recruited into stress granules from the cytoplasmic pool of FUS which is increased in ALS patients carrying FUS mutations
In order to determine whether mutant FUS is mislocalized in a more physiological context, we explored the effects of arsenite on cultured ALS-patient fibroblasts carrying the FUSR521C or FUSR514G mutations. These cells are derived from patients with a heterozygous mutation and therefore express both mutant and wild-type proteins, at physiological levels.
We found that FUS mutant fibroblasts have significantly greater levels of cytoplasmic FUS than control fibroblasts under basal culture conditions (Fig. 8A and C) (one-way ANOVA, P < 0.01, Dunnett's T3, both P < 0.05). Stress granules (as measured by PABP granule formation) were almost absent from mutant and control fibroblasts under these conditions. Following treatment with 1 mm arsenite, the mean number of stress granules per cell was significantly increased in both mutant and control fibroblasts compared with untreated cells (Fig. 8B and D, repeated measures ANOVA with a Greenhouse-Geisser correction, P < 0.001). A striking difference in the distribution of FUS was observed only in mutant fibroblasts treated with arsenite, as cytoplasmic FUS was recruited to stress granules (Fig. 8B and E). The mean number of FUS-positive cytoplasmic granules per cell was significantly increased following arsenite treatment only in mutant fibroblasts [ANOVA, with a Greenhouse-Geisser correction, P < 0.001, Dunnett's T3 post-hoc tests for FUSR521C (P < 0.01) and FUSR514G (P < 0.01)]. These experiments demonstrate that endogenous levels of cytoplasmic FUS are increased in mutant fibroblasts compared with controls, and that only the cytoplasmic pool of FUS is recruited into stress granules following oxidative stress.
Figure 8.

FUS co-localizes with stress granules in fibroblasts from patients with FUS mutations. (A) Immunofluorescent images of human fibroblasts stained with antibodies against PABP (left panels) and FUS (middle panels). (B) Addition of 1 mm arsenite to the cells resulted in the formation of distinct stress granules. Nuclei are stained with DAPI and merged images are shown on the right. Scale bar 25 μm. (C) There is a significant increase in the mean intensity of cytoplasmic FUS between mutant FUSR514G and FUSR521C fibroblast and control (*P < 0.05 for both). (D) Upon addition of arsenite, there is a significant increase in PABP-positive granules in control and mutant fibroblasts (***P < 0.001). (E) There is a significant increase in FUS-positive granules in FUSR521C and FUSR514G fibroblasts (**P < 0.01) compared with the control (n = 3 with eight images per condition as a minimum for analysis).
Discussion
The majority of FUS mutations linked to ALS result in single amino acid substitutions in the extreme C-terminus of the protein (12) (14). In this study, we observed that mutation and truncation of this region lead to FUS mislocalization, confirming data previously published in which the C-terminus has been shown to contain an NLS (14,32–37). Here, we demonstrate that this mislocalization of FUS is abolished by the addition of the wild-type C-terminus to the mutant protein, confirming that it is purely the disruption to the NLS and not a secondary gain of function effect of the amino acid change that causes the mislocalization. The C-terminus of FUS shows strong homology to another FET family member, EWS, which has an NLS in the same region (38,39). This type of domain is known as a PY (proline–tyrosine) NLS and is found in other RNA-binding proteins such as HNRPA1, which are known to be imported into the nucleus by transportins (40,41). Consistent with this observation, FUS has been shown to bind to transportins 1 and 2 (40,41) and knockdown of both inhibits the import of FUS into the nucleus (35). The interaction between FUS and transportin 1 has been shown to range from amino acid 508 to the very end of FUS indicating that the actual NLS extends further than previously thought (42).
These results help us to explain discrepancies in the literature about whether the presence of FUS in the cytoplasm is sufficient to cause stress granules or whether this is an artefact of transient transfection. Consistent with other work, the transient transfection of mutant FUS resulted in cytoplasmic stress granules (33,34), while we have also shown that cytoplasmic mutant FUS is incorporated into PABP-positive stress granules only following cellular stress when stably transfected (35). Of most importance, is that it has been possible to validate the findings from cell lines in fibroblasts from ALS patients who carry a single mutant and a single wild-type copy of the FUS gene. In particular, the data show that the stable cell lines are the best representation of the patients' cells. In these patients, there is a significantly higher incorporation of FUS into the stress granules compared with fibroblasts from patients with two wild-type copies.
Importantly, we have shown that FUS interacts with itself in a non-RNA dependent fashion. The clearing of nuclear FUS in motor neurons containing large cytoplasmic inclusions in FUS-ALS cases suggests that mutant FUS can act in a dominant-negative fashion to sequester the wild-type protein. Our data are consistent with histology data from ALS and FTLD cases with FUS pathology, where cytoplasmic inclusions of FUS are observed with a commensurate loss of FUS from the nucleus (5,17,43,44). A recent in vitro binding study has found that EWS interacts with itself via its RGG domains, so it is possible that a similar mechanism may mediate FUS self-interaction (45). While this also means that wild-type FUS might act on the mutant protein to import it into the nucleus, which might explain why a portion of the truncated FUS is found in the nucleus, it is unable to fully compensate for the effect of the NLS disruption.
We have shown that FUS and the stress granule protein PABP are in close proximity to each other in the cytoplasm under basal conditions. This interaction occurs in an RNA-dependent manner, which suggests that they are both involved in the same cellular processes, such as the shuttling of RNA between the nucleus and the cytoplasm. The presence of an increased level of cytoplasmic FUS due to C-terminal mutations, as seen in the patient fibroblasts, leads to an increased amount of FUS bound to PABP that is then sequestered into stress granules.
Our findings suggest that there are distinct parallels and important differences between FUS and another RNA processing protein, TDP-43, which is detected within cytoplasmic inclusions in a majority of frontotemporal lobar degeneration (FTLD-TDP) and ALS cases [for review see (46)]. Like FUS, TDP-43 normally resides in the nucleus but in FTLD and ALS it forms large aggregates in the cytoplasm with a corresponding loss of TDP-43 from the nucleus (31,47). TDP-43 has also been shown to exit the nucleus and co-localize with stress granule markers after axotomy of the mouse sciatic nerve (48) and in NSC34 cell lines under conditions of oxidative stress (49). However, the majority of ALS-linked TDP-43 mutations lie within the C-terminal glycine rich domain, which is distant from the dominant N-terminal NLS, and the mechanisms responsible for TDP-43 mislocalization are unknown (50–56). Recently, FTLD-related changes in the nuclear transport process have been implicated in the misaccumulation of TDP-43 (57). The two pathogenic processes appear to be independent, however, as TDP-43 is not present in FUS inclusions (58,59).
Our results confirm that ALS-linked mutations disrupt the NLS leading to increased cytoplasmic levels of FUS, and that cytoplasmic FUS coalesces with granules induced by oxidative stress. Moreover, we show that mutant FUS binds to wild-type FUS, which may result in the nuclear depletion and cytoplasmic sequestration of wild-type FUS, disrupt RNA processing and lead to neurodegeneration.
MATERIALS AND METHODS
Generation of constructs
The untagged or HA-tagged cDNA for the wild-type and mutant constructs was amplified by PCR from HA-tagged constructs generated previously (14), and sub-cloned into pDEST-30 (Invitrogen, Carlsbad, CA) to generate stable cell lines, or pEGFP-C1 (Clontech) to generate N-terminally tagged fusion proteins. Direct sequencing of the constructs confirmed successful cloning. Generation of the truncated construct was achieved by mutagenesis using the Quickchange II site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA).
Cell lines
CV-1 fibroblasts, HEK 293T and patient and control fibroblasts obtained from biopsies were maintained in Dulbecco's Modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 2 mm l-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin (all from Invitrogen). Stable inducible SH-SY5Y cell lines were maintained in DMEM:F12 (1:1), 10% tetracycline-free FBS, 2 mm l-glutamine, 1 mm sodium pyruvate, 5 µg/ml blasticidin and 650 µg/ml Geneticin (all from Invitrogen). Stable cell lines were generated by Lipofectamine 2000 (Invitrogen) transfection of the pcDNA6/TR Tet-repressor plasmid (Invitrogen) into early-passage SH-SY5Y human neuroblastoma cells, followed by selection using 5 µg/ml blasticidin and clonal isolation. The Tet-repressor stable cell line was then transfected with N-terminally HA-tagged FUS cDNAs subcloned into pDEST-30. FUS stable cell lines were selected using 650 µg/ml Geneticin and clonally isolated. FUS expression was induced by the addition of 0.5 µg/ml DOX (Sigma-Aldrich, Dorset, UK) 48 h prior to assay.
Transfection and imaging
CV-1 cells were transfected using FuGene HD (Roche, Burgess Hill, UK) according to the manufacturer's protocol. Rat cortical neurons were transfected at DIV5 using Lipofectamine 2000 according to the manufacturer's protocol. Cells were plated on 13 mm diameter glass coverslips in 24-well dishes and transfected with 0.25 µg (CV-1) or 1 µg (cortical neurons) plasmid DNA. Stably transfected SH-SY5Y cells were induced by the addition of 0.5 µg/ml DOX 24 h after plating. All cells were used for experiments 24–48 h after transfection or induction. 1 mm arsenite was added in the final 1 h of incubation as required. The cells were washed once with PBS and fixed in 4% paraformaldehyde in PBS (15 min; RT). Following washing with PBS, the cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min and blocked with PBS + 1% goat serum (Dako UK Ltd., Ely, UK) + 0.2% Triton X-100 for 30 min. The cells were incubated overnight at 4°C with primary antibody followed by washing with PBS and 1 h incubation with Alexa Fluor 568 anti-mouse IgG, Alexa Fluor 568 anti-rabbit IgG or Alexa Fluor 488 anti-mouse IgG as appropriate (all Invitrogen; 1:400 in PBS + 0.2% Triton X-100 + 1% goat serum). After washing with PBS, the cells were stained with DAPI, washed again and coverslips mounted using fluorescent mounting medium (Dako UK Ltd). Primary antibodies used were: mouse anti-HA (1:1000; Cell Signaling Technology, Beverley, MA, USA); mouse anti-PABP (1:1000; Sigma); rabbit anti-PABP (1:500; AbCam, Cambridge, UK); rabbit anti-TIA1 (1:100; AbCam); rabbit anti-XRN1 (1:100; Bethyl Laboratories, Montgomery, TX, USA); mouse anti-SC-35 (1:500; Sigma); rabbit anti-Staufen (1:250; AbCam); mouse anti-FUS (1:100; Santa Cruz Biotechnology, Inc., Heidelberg, Germany) and rabbit anti-FUS (1:500; Novus Biologicals, Littleton, CO, USA). The cells were observed using a Zeiss 510 meta confocal laser scanning microscope equipped with a 63×, 1.4NA Plan-Apochromat objective (Carl Zeiss Ltd., Hertfordshire, UK).
Quantification of cytoplasmic GFP-FUS in CV-1 cells (untreated) was performed blinded by counting of cells with cytoplasmic staining by eye on at least 200 cells on three coverslips, each of which was from a different transfection experiment, for each construct. Quantification of diffuse cytoplasmic FUS and cytoplasmic FUS and PABP granules in fibroblasts was performed blinded, using Metamorph software (v. 7.5, Molecular Devices, Sunnyvale, CA, USA) on a representative dataset (n = 3) comprising eight images per patient (or control), 3–19 cells per image. In order to examine the cytoplasmic compartment only, a binarized mask of the nuclear image was subtracted from FUS or PABP images. PABP granules were quantified using the Count Nuclei application, optimized for granule size and intensity above the local background using three randomly sampled images. FUS granules were also quantified using the Count Nuclei application, however, due to the strong fluorescent halo of nuclear FUS, this quantification was performed on images from which a dilated version of the binarized nuclear image was subtracted. The mean intensity of cytoplasmic FUS was quantified from non-arsenite-treated FUS images modified in the same manner, which had then been segmented by thresholding.
Co-immunoprecipitation
HEK 293T cells were grown in 10 cm dishes and then transfected using FuGene HD (Roche) according to the manufacturer's protocol. After 24 h, the cells were washed once with cold PBS and scraped into 500 µl ice cold Triton X-100 buffer (50 mm Tris pH 7.4, 150 mm NaCl, 1% Triton X-100, Roche Complete protease inhibitors). After 15 min on ice, the lysed cells were spun at 21 000g for 30 s at 4°C and the supernatant removed. 5 µl of an RNase A/T1 mix (Fermentas, Yorkshire, UK) or RNasin Plus RNase inhibitor (Promega, Southampton, UK) was added to the tubes and incubated at 37°C for 5 min, then placed back on ice. Total lysate samples were taken at this point. Mouse anti-Myc (Cell Signalling Technology, 1:100), rabbit anti-HA (Cell Signalling Technology, 1:100) or mouse anti-FUS (Santa Cruz Biotechnology, Inc., 1:100) was added to the lysates as appropriate followed by the addition of 20 µl of protein A beads (Invitrogen Dynabeads). After rotation at 4°C for 4 h, the beads were washed three times with 500 µl of Triton X-100 buffer and then resuspended in 2× SDS buffer (0.35 M Tris-Cl pH 6.8, 30% glycerol, 0.6 M DTT, 10% SDS, 0.0001% bromophenol blue) and boiled for 10 min.
Cellular fractionation
Stably transfected SH-SY5Y cells were grown to confluence in 3 cm dishes 48 h after induction of HA-FUS expression and were washed with ice-cold PBS and then collected in 150 µl ice-cold hypotonic lysis buffer (10 mm HEPES pH7.9, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, Roche Complete protease inhibitors). Cells were lysed by Dounce homogenization using 20 strokes of the tight pestle. Nuclei were pelleted by centrifugation at 800g for 5 min at 4°C. The supernatant was further clarified by centrifugation at 18 000g and extraction of the supernatant with 0.25 vol of 5× RIPA buffer (0.5% SDS, 0.75 m NaCl, 50 mm NaPO4 pH 7.2, 10 mm EDTA, 5% sodium deoxycholate, 5% NP-40) was used to generate the cytosolic fraction. Nuclei were then purified by resuspension in 100 µl isotonic sucrose buffer (10 mm HEPES pH 7.9, 0.25 m sucrose, 10 mm KCl, 10 mm MgCl2, 1 mm EDTA, 1 mm EGTA, Roche Complete protease inhibitors), layering over a cushion of 0.88 m sucrose, and centrifugation at 6000g for 10 min at 4°C. Nuclear proteins were extracted using 1× RIPA buffer. Samples were denatured and reduced by the addition of 6× Laemmli SDS buffer (0.35 M Tris-Cl pH 6.8, 30% glycerol, 0.6 M DTT, 10% SDS, 0.0001% bromophenol blue) and boiling for 10 min.
Western blotting
Following SDS–PAGE, proteins were transferred to a nitrocellulose membrane using the iBlot system, blocked using 5% non-fat milk for 30 min at room temperature and then incubated with primary antibodies as required; rabbit anti-Myc, (Cell Signalling Technology 1:1000), mouse anti-HA (Cell Signalling Technology 1:1000) and mouse anti-PABP (Sigma 1:2000); or mouse anti-Myc (Cell Signalling Technology 1:1000), rabbit anti-HA (Cell Signalling Technology 1:1000) and mouse anti-PABP (Sigma 1:2000); or rabbit anti-FUS (Novus Biologicals 1:5000) and mouse anti-PABP (Sigma 1:2000); mouse anti-GAPDH (Sigma 1:5000); mouse anti-HA (Cell Signalling Technology 1:1000); rabbit anti-Lamin B1 (AbCam 1:2000) overnight at 4°C. Following three washes with TBS-T (10 mm Tris, 150 mm NaCl, 0.1% Tween-20), membranes were either incubated in the dark in 0.1% Tween-20 in blocking solution with fluorescent secondary antibodies (IRDye 680 anti-mouse and 800 anti-rabbit, LI-COR, Biosciences, NE, USA, 1:10 000), or HRP-conjugated secondary antibodies (goat anti-mouse-HRP and goat anti-rabbit-HRP, both Chemicon, 1:5000) before three further washes in TBS-T. Images were either acquired with the Odyssey infrared imaging system (fluorescent, LI-COR) or detected using chemiluminescence (HRP, Immobilon Western, Millipore). Blot images in TIF format were quantified using the ImageJ gel analyzer tool (Image J 1.45e, NIH, Bethesda, USA, http://rsb.info.nih.gov/ij/, last accessed date on 14 March, 2013). Integrated band intensities were normalized to band intensities of loading controls, and also to relative input (lysate fraction).
Brain tissue collection and neuropathological assessment
Ten percent formalin-fixed, paraffin-embedded tissue blocks were available from the MRC London Neurodegenerative Diseases Brain Bank (Institute of Psychiatry, King's College London, UK). Consent for autopsy, neuropathological assessment and research was obtained from all subjects in accordance with local and national Research Ethics Committee-approved donation. Block-taking for histological and immunohistochemical studies and neuropathological assessment of motor neuron disease and control cases was performed in accordance with the published guidelines.
Histological staining and immunohistochemistry
Sections of 7 µm thickness were cut from the formalin-fixed, paraffin-embedded spinal cord blocks, and immunohistochemical reactions were performed. The tissue sections were processed for FUS immunohistochemistry as described previously (60). In brief, the sections were deparaffinized in xylene, endogenous peroxidase was blocked by H2O2 in methanol and immunohistochemistry performed with an overnight incubation of rabbit anti-FUS antibody at 4°C (Novus Biologicals NB100-2599, 1:50). To enhance antigen retrieval, the sections were kept in citrate buffer for 10 min following microwave treatment. The sections were counterstained with haematoxylin and immunostaining was analysed using an Olympus microscope (Olympus, Southend-on-Sea, UK).
Proximity ligation assay
The PLA was performed in the stably transfected SH-SY5Y cell lines 24 h after induction with 0.5 µg/ml doxycyline. Following fixing and blocking, the cells were incubated with rabbit anti-HA tag (Cell Signalling Technology 1:100) and mouse anti-PABP antibodies (Sigma 1:1000). The PLA assay was further performed using the Duolink™ kit (Olink) according to the manufacturer's protocol.
Quantification of the co-localization of FUS and PABP by the PLA was performed using Metamorph software on a representative data set (n = 3) comprising 8 images per condition per mutant (or control), 3–19 cells per image. A minimum of 70 nuclei per condition were counted. PLA-positive granules, representing areas of co-localization of FUS and PABP, were quantified using the Granularity application. Size and intensity thresholds defining both granules and nuclei were kept consistent for all conditions and mutants. PLA positivity is a function of both the number of granules and the average integrated intensity (average pixel area × average pixel intensity) of the granules, because individual PLA events cannot always be resolved to individual points of interaction and thus appear as brighter and/or larger granules. PLA was thus calculated as the number of PLA-positive granules per cell multiplied by the average integrated intensity of each granule.
Statistical analyses
Differences between multiple groups were analysed using one-way ANOVA. Where measurements were taken from multiple groups before and after cellular treatment, a two-way repeated measures ANOVA was used. Homogeneity of variance within groups was assessed using Levene's statistic. Where the assumption of homogeneity of variance was upheld (Levene's statistic not significant at the 0.05 threshold), Bonferroni post-hoc tests were used to assess which groups were significantly different (0.05 threshold). Where there was significant heterogeneity of variance, Dunnett's T3 post-hoc test was used. Sphericity of data within subjects was tested using Mauchly's test and where this assumption was not upheld, the Greenhouse-Geisser correction was applied. Data were analysed using SPSS v. 15.0 (SPSS Inc.).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from: the American ALS Association, the Middlemass family, Lady Edith Wolfson Trust, Motor Neurone Disease Association UK, The Wellcome Trust, European Union (APOPIS consortium, contract LSHM-CT-2003-503330; NeuroNE Consortium; and Marie Curie International Incoming Fellowship), NIHR Biomedical Research Centre for Mental Health, The South London and Maudsley NHS Foundation Trust, Medical Research Council UK, a Jack Cigman grant from King's College Hospital Charity and The Psychiatry Research Trust of the Institute of Psychiatry, Coker Charitable Trust, Health Research Council of New Zealand. Funding to pay the Open Access publication charges for this article was provided by The Wellcome Trust.
Supplementary Material
ACKNOWLEDGEMENTS
This publication is dedicated to the patients and families who have contributed to this project. Post-mortem tissues were provided by MRC London Neurodegenerative Diseases Brain Bank.
Conflict of interest statement. None declared.
REFERENCES
- 1.Shaw C.E., Enayat Z.E., Chioza B.A., Al-Chalabi A., Radunovic A., Powell J.F., Leigh P.N. Mutations in all five exons of SOD-1 may cause ALS. Ann. Neurol. 1998;43:390–394. doi: 10.1002/ana.410430319. [DOI] [PubMed] [Google Scholar]
- 2.Chio A., Calvo A., Moglia C., Ossola I., Brunetti M., Sbaiz L., Lai S.L., Abramzon Y., Traynor B.J., Restagno G. A de novo missense mutation of the FUS gene in a ‘true’ sporadic ALS case. Neurobiol. Aging. 2010;32:553 E23–553 E26. doi: 10.1016/j.neurobiolaging.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damme P.V., Goris A., Race V., Hersmus N., Dubois B., Bosch L.V., Matthijs G., Robberecht W. The occurrence of mutations in FUS in a Belgian cohort of patients with familial ALS. Eur. J. Neurol. 2009;17:754–756. doi: 10.1111/j.1468-1331.2009.02859.x. [DOI] [PubMed] [Google Scholar]
- 4.DeJesus-Hernandez M., Kocerha J., Finch N., Crook R., Baker M., Desaro P., Johnston A., Rutherford N., Wojtas A., Kennelly K., et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum. Mutat. 2010;31:E1377–E1389. doi: 10.1002/humu.21241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hewitt C., Kirby J., Highley J.R., Hartley J.A., Hibberd R., Hollinger H.C., Williams T.L., Ince P.G., McDermott C.J., Shaw P.J. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2010;67:455–461. doi: 10.1001/archneurol.2010.52. [DOI] [PubMed] [Google Scholar]
- 6.Lai S.L., Abramzon Y., Schymick J.C., Stephan D.A., Dunckley T., Dillman A., Cookson M., Calvo A., Battistini S., Giannini F., et al. FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging. 2010;32:550 E1–550 E4. doi: 10.1016/j.neurobiolaging.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rademakers R., Stewart H., Dejesus-Hernandez M., Krieger C., Graff-Radford N., Fabros M., Briemberg H., Cashman N., Eisen A., Mackenzie I.R. Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve. 2010;42:170–176. doi: 10.1002/mus.21665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai C.P., Soong B.W., Lin K.P., Tu P.H., Lin J.L., Lee Y.C. FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol. Aging. 2010;32:553 E13–553 E21. doi: 10.1016/j.neurobiolaging.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Waibel S., Neumann M., Rabe M., Meyer T., Ludolph A.C. Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology. 2010;75:815–817. doi: 10.1212/WNL.0b013e3181f07e26. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto-Watanabe Y., Watanabe M., Okamoto K., Fujita Y., Jackson M., Ikeda M., Nakazato Y., Ikeda Y., Matsubara E., Kawarabayashi T., et al. A Japanese ALS6 family with mutation R521C in the FUS/TLS gene: a clinical, pathological and genetic report. J. Neurol. Sci. 2010;296:59–63. doi: 10.1016/j.jns.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Yan J., Deng H.X., Siddique N., Fecto F., Chen W., Yang Y., Liu E., Donkervoort S., Zheng J.G., Shi Y., et al. Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology. 2010;75:807–814. doi: 10.1212/WNL.0b013e3181f07e0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwiatkowski T.J., Jr., Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 13.Ticozzi N., Silani V., Leclerc A.L., Keagle P., Gellera C., Ratti A., Taroni F., Kwiatkowski T.J., Jr., McKenna-Yasek D.M., Sapp P.C., et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology. 2009;73:1180–1185. doi: 10.1212/WNL.0b013e3181bbff05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corrado L., Del Bo R., Castellotti B., Ratti A., Cereda C., Penco S., Soraru G., Carlomagno Y., Ghezzi S., Pensato V., et al. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2009;47:190–194. doi: 10.1136/jmg.2009.071027. [DOI] [PubMed] [Google Scholar]
- 16.Belzil V.V., Valdmanis P.N., Dion P.A., Daoud H., Kabashi E., Noreau A., Gauthier J., Hince P., Desjarlais A., Bouchard J.P., et al. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology. 2009;73:1176–1179. doi: 10.1212/WNL.0b013e3181bbfeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urwin H., Josephs K.A., Rohrer J.D., Mackenzie I.R., Neumann M., Authier A., Seelaar H., Van Swieten J.C., Brown J.M., Johannsen P., et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120:33–41. doi: 10.1007/s00401-010-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Blitterswijk M., Landers J.E. RNA processing pathways in amyotrophic lateral sclerosis. Neurogenetics. 2010;11:275–290. doi: 10.1007/s10048-010-0239-4. [DOI] [PubMed] [Google Scholar]
- 19.Law W.J., Cann K.L., Hicks G.G. TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief. Funct. Genomic. Proteomic. 2006;5:8–14. doi: 10.1093/bfgp/ell015. [DOI] [PubMed] [Google Scholar]
- 20.Lagier-Tourenne C., Polymenidou M., Cleveland D.W. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010;19:R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwon M.J., Baek W., Ki C.S., Kim H.Y., Koh S.H., Kim J.W., Kim S.H. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobiol. Aging. 2012;33:1017 e1017–1017 e1023. doi: 10.1016/j.neurobiolaging.2011.12.003. –. [DOI] [PubMed] [Google Scholar]
- 22.Yamashita S., Mori A., Sakaguchi H., Suga T., Ishihara D., Ueda A., Yamashita T., Maeda Y., Uchino M., Hirano T. Sporadic juvenile amyotrophic lateral sclerosis caused by mutant FUS/TLS: possible association of mental retardation with this mutation. J. Neurol. 2011;259:1039–1044. doi: 10.1007/s00415-011-6292-6. [DOI] [PubMed] [Google Scholar]
- 23.Zou Z.Y., Peng Y., Feng X.H., Wang X.N., Sun Q., Liu M.S., Li X.G., Cui L.Y. Screening of the FUS gene in familial and sporadic amyotrophic lateral sclerosis patients of Chinese origin. Eur. J. Neurol. 2012;19:977–983. doi: 10.1111/j.1468-1331.2012.03662.x. [DOI] [PubMed] [Google Scholar]
- 24.Belzil V.V., Daoud H., St-Onge J., Desjarlais A., Bouchard J.P., Dupre N., Lacomblez L., Salachas F., Pradat P.F., Meininger V., et al. Identification of novel FUS mutations in sporadic cases of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011;12:113–117. doi: 10.3109/17482968.2010.536840. [DOI] [PubMed] [Google Scholar]
- 25.Belzil V.V., St-Onge J., Daoud H., Desjarlais A., Bouchard J.P., Dupre N., Camu W., Dion P.A., Rouleau G.A. Identification of a FUS splicing mutation in a large family with amyotrophic lateral sclerosis. J. Hum. Genet. 2011;56:247–249. doi: 10.1038/jhg.2010.162. [DOI] [PubMed] [Google Scholar]
- 26.Hara M., Minami M., Kamei S., Suzuki N., Kato M., Aoki M. Lower motor neuron disease caused by a novel FUS/TLS gene frameshift mutation. J. Neurol. 2012;259:2237–2239. doi: 10.1007/s00415-012-6542-2. [DOI] [PubMed] [Google Scholar]
- 27.Nagayama S., Minato-Hashiba N., Nakata M., Kaito M., Nakanishi M., Tanaka K., Arai M., Akiyama H., Matsui M. Novel FUS mutation in patients with sporadic amyotrophic lateral sclerosis and corticobasal degeneration. J. Clin. Neurosci. 2012;19:1738–1739. doi: 10.1016/j.jocn.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 28.van Blitterswijk M., van Es M.A., Hennekam E.A., Dooijes D., van Rheenen W., Medic J., Bourque P.R., Schelhaas H.J., van der Kooi A.J., de Visser M., et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012;21:3776–3784. doi: 10.1093/hmg/dds199. [DOI] [PubMed] [Google Scholar]
- 29.Hanson D.A., Ziegler S.F. Fusion of green fluorescent protein to the C-terminus of granulysin alters its intracellular localization in comparison to the native molecule. J. Negat. Results Biomed. 2004;3:2. doi: 10.1186/1477-5751-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Souquere S., Mollet S., Kress M., Dautry F., Pierron G., Weil D. Unravelling the ultrastructure of stress granules and associated P-bodies in human cells. J. Cell Sci. 2009;122:3619–3626. doi: 10.1242/jcs.054437. [DOI] [PubMed] [Google Scholar]
- 31.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 32.Kino Y., Washizu C., Aquilanti E., Okuno M., Kurosawa M., Yamada M., Doi H., Nukina N. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2010;39:2781–2798. doi: 10.1093/nar/gkq1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito D., Seki M., Tsunoda Y., Uchiyama H., Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 2011;69:152–162. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- 34.Gal J., Zhang J., Kwinter D.M., Zhai J., Jia H., Jia J., Zhu H. Nuclear localization sequence of FUS and induction of stress granu2323les by ALS mutants. Neurobiol. Aging. 2010;32:2323 E27–2323 E40. doi: 10.1016/j.neurobiolaging.2010.06.010. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dormann D., Rodde R., Edbauer D., Bentmann E., Fischer I., Hruscha A., Than M.E., Mackenzie I.R., Capell A., Schmid B., et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bosco D.A., Lemay N., Ko H.K., Zhou H., Burke C., Kwiatkowski T.J., Jr., Sapp P., McKenna-Yasek D., Brown R.H., Jr., Hayward L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bentmann E., Neumann M., Tahirovic S., Rodde R., Dormann D., Haass C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43) J. Biol. Chem. 2012;287:23079–23094. doi: 10.1074/jbc.M111.328757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zakaryan R.P., Gehring H. Identification and characterization of the nuclear localization/retention signal in the EWS proto-oncoprotein. J. Mol. Biol. 2006;363:27–38. doi: 10.1016/j.jmb.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 39.Shaw D.J., Morse R., Todd A.G., Eggleton P., Lorson C.L., Young P.J. Identification of a tripartite import signal in the Ewing Sarcoma protein (EWS) Biochem. Biophys. Res. Commun. 2009;390:1197–1201. doi: 10.1016/j.bbrc.2009.10.120. [DOI] [PubMed] [Google Scholar]
- 40.Guttinger S., Muhlhausser P., Koller-Eichhorn R., Brennecke J., Kutay U. Transportin2 functions as importin and mediates nuclear import of HuR. Proc. Natl Acad. Sci. USA. 2004;101:2918–2923. doi: 10.1073/pnas.0400342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee B.J., Cansizoglu A.E., Suel K.E., Louis T.H., Zhang Z., Chook Y.M. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell. 2006;126:543–558. doi: 10.1016/j.cell.2006.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niu C., Zhang J., Gao F., Yang L., Jia M., Zhu H., Gong W. FUS-NLS/transportin 1 complex structure provides insights into the nuclear targeting mechanism of FUS and the implications in ALS. PLoS ONE. 2012;7:e47056. doi: 10.1371/journal.pone.0047056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neumann M., Rademakers R., Roeber S., Baker M., Kretzschmar H.A., Mackenzie I.R. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng H.X., Zhai H., Bigio E.H., Yan J., Fecto F., Ajroud K., Mishra M., Ajroud-Driss S., Heller S., Sufit R., et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010;67:739–748. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw D.J., Morse R., Todd A.G., Eggleton P., Lorson C.L., Young P.J. Identification of a self-association domain in the Ewing's sarcoma protein: a novel function for arginine–glycine–glycine rich motifs? J. Biochem. 2010;147:885–893. doi: 10.1093/jb/mvq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lagier-Tourenne C., Cleveland D.W. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mackenzie I.R., Bigio E.H., Ince P.G., Geser F., Neumann M., Cairns N.J., Kwong L.K., Forman M.S., Ravits J., Stewart H., et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 48.Moisse K., Volkening K., Leystra-Lantz C., Welch I., Hill T., Strong M.J. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009;1249:202–211. doi: 10.1016/j.brainres.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 49.Colombrita C., Zennaro E., Fallini C., Weber M., Sommacal A., Buratti E., Silani V., Ratti A. Tdp-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009;111:1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- 50.Yokoseki A., Shiga A., Tan C.F., Tagawa A., Kaneko H., Koyama A., Eguchi H., Tsujino A., Ikeuchi T., Kakita A., et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 2008;63:538–542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- 51.Van Deerlin V.M., Leverenz J.B., Bekris L.M., Bird T.D., Yuan W., Elman L.B., Clay D., Wood E.M., Chen-Plotkin A.S., Martinez-Lage M., et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E., et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabashi E., Valdmanis P.N., Dion P., Spiegelman D., McConkey B.J., Vande Velde C., Bouchard J.P., Lacomblez L., Pochigaeva K., Salachas F., et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 54.Guerreiro R.J., Schymick J.C., Crews C., Singleton A., Hardy J., Traynor B.J. TDP-43 is not a common cause of sporadic amyotrophic lateral sclerosis. PLoS ONE. 2008;3:e2450. doi: 10.1371/journal.pone.0002450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gitcho M.A., Baloh R.H., Chakraverty S., Mayo K., Norton J.B., Levitch D., Hatanpaa K.J., White C.L., III, Bigio E.H., Caselli R., et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cairns N.J., Neumann M., Bigio E.H., Holm I.E., Troost D., Hatanpaa K.J., Foong C., White C.L., III, Schneider J.A., Kretzschmar H.A., et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishimura A.L., Zupunski V., Troakes C., Kathe C., Fratta P., Howell M., Gallo J.M., Hortobagyi T., Shaw C.E., Rogelj B. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133:1763–1771. doi: 10.1093/brain/awq111. [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi Z., Tsuchiya K., Arai T., Aoki M., Hasegawa M., Ishizu H., Akiyama H., Mizusawa H. Occurrence of basophilic inclusions and FUS-immunoreactive neuronal and glial inclusions in a case of familial amyotrophic lateral sclerosis. J. Neurol. Sci. 2010;293:6–11. doi: 10.1016/j.jns.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki N., Aoki M., Warita H., Kato M., Mizuno H., Shimakura N., Akiyama T., Furuya H., Hokonohara T., Iwaki A., et al. FALS with FUS mutation in Japan, with early onset, rapid progress and basophilic inclusion. J. Hum. Genet. 2010;55:252–254. doi: 10.1038/jhg.2010.16. [DOI] [PubMed] [Google Scholar]
- 60.Hortobagyi T., Wise S., Hunt N., Cary N., Djurovic V., Fegan-Earl A., Shorrock K., Rouse D., Al-Sarraj S. Traumatic axonal damage in the brain can be detected using beta-APP immunohistochemistry within 35 min after head injury to human adults. Neuropathol. Appl. Neurobiol. 2007;33:226–237. doi: 10.1111/j.1365-2990.2006.00794.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.