

Abstract
The Vesicle-associated membrane protein (VAMP)-Associated Protein B (VAPB) is the causative gene of amyotrophic lateral sclerosis 8 (ALS8) in humans. Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by selective death of motor neurons leading to spasticity, muscle atrophy and paralysis. VAP proteins have been implicated in various cellular processes, including intercellular signalling, synaptic remodelling, lipid transport and membrane trafficking and yet, the molecular mechanisms underlying ALS8 pathogenesis remain poorly understood. We identified the conserved phosphoinositide phosphatase Sac1 as a Drosophila VAP (DVAP)-binding partner and showed that DVAP is required to maintain normal levels of phosphoinositides. Downregulating either Sac1 or DVAP disrupts axonal transport, synaptic growth, synaptic microtubule integrity and the localization of several postsynaptic components. Expression of the disease-causing allele (DVAP-P58S) in a fly model for ALS8 induces neurodegeneration, elicits synaptic defects similar to those of DVAP or Sac1 downregulation and increases phosphoinositide levels. Consistent with a role for Sac1-mediated increase of phosphoinositide levels in ALS8 pathogenesis, we found that Sac1 downregulation induces neurodegeneration in a dosage-dependent manner. In addition, we report that Sac1 is sequestered into the DVAP-P58S-induced aggregates and that reducing phosphoinositide levels rescues the neurodegeneration and suppresses the synaptic phenotypes associated with DVAP-P58S transgenic expression. These data underscore the importance of DVAP–Sac1 interaction in controlling phosphoinositide metabolism and provide mechanistic evidence for a crucial role of phosphoinositide levels in VAP-induced ALS.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive, degenerative disorder characterized by the selective loss of motor neurons in the brain and spinal cord leading to paralysis, muscle atrophy and eventually, death (1). Two missense mutations in the gene encoding the human Vesicle-associated membrane protein (VAMP)-Associated Protein B (hVAPB) causes a range of dominantly inherited motor neuron diseases including ALS8 (2,3). VAP family proteins are characterized by an N-terminal major sperm protein (MSP) domain, a coiled-coil (CC) motif and a transmembrane (TM)-spanning region. They are implicated in several biological processes, including regulation of lipid transport, endoplasmic reticulum (ER) morphology and membrane trafficking (4). Drosophila Vap-33-1 (hereafter, DVAP) regulates synaptic structure, synaptic microtubule (MT) stability and the composition of postsynaptic glutamate receptors (5,6). MSP domains in DVAP are cleaved and secreted into the extracellular space where they bind Ephrin receptors (7). MSPs also bind postsynaptic Roundabout and Lar-like receptors to control muscle mitochondria morphology, localization and function (8). Transgenic expression of the disease-linked alleles (DVAP-P58S and DVAP-T48I) in the larval motor system recapitulates major hallmarks of the human disease, including aggregate formation, locomotion defects and chaperone upregulation (3,6,9). Several studies have also implicated the ALS mutant allele in abnormal unfolded protein response (UPR) (3,10–13) and in the disruption of the anterograde axonal transport of mitochondria (14). However, it is unclear how these diverse VAP functions are achieved and which mechanisms underlie the disease pathogenesis in humans. One way to address these questions is to search for DVAP-interacting proteins. We identified Sac1 (Suppressor of Actin 1), an evolutionarily conserved phosphoinositide phosphatase, as a DVAP-binding protein. Phosphoinositides are low-abundance lipids that localize to the membrane–cytoplasm interface and function by binding various effector proteins. The inositol group can be reversibly phosphorylated at the 3′, 4′ and 5′ positions to generate seven possible phosphoinositide derivatives, each with a specific intracellular dynamic distribution (15). Sac1 predominantly dephosphorylates PtdIns4P pools, although PtdIns3P and PtdIns(3,5)P2 can also function as substrates (16). In yeast, Sac1 has been linked to several processes, including actin organization, vacuole morphology and sphingomyelin synthesis (17,18). Drosophila Sac1 mutants die as embryos and exhibit defects in dorsal closure and axonal pathfinding (19,20). Mouse lines deficient for Sac1 are cell lethal, whereas Sac1 downregulation in mammalian cell cultures results in disorganization of Golgi membranes and mitotic spindles (21). Interestingly, SAC3 (also known as FIG4), another member of the Sac phosphatase family, is mutated in familial and sporadic cases of ALS (22). Inactivation of SAC3 in mice also results in extensive degeneration and neuronal vacuolization in the brain, most relevantly in the motor cortex (23). We identified Sac1 and DVAP as binding partners and show that DVAP is required to maintain normal levels of PtdIns4P. Loss of either Sac1 or DVAP function disrupts axonal transport, MT stability, synaptic growth and the localization of a number of postsynaptic markers. We also show that the disease-causing mutation (DVAP-P58S) induces neurodegeneration and displays synaptic phenotypes similar to those of either Sac1 or DVAP loss-of-function, including an increase in PtdIns4P levels. Importantly, reducing PtdIns4P levels rescues the neurodegeneration associated with DVAP-P58S and suppresses the synaptic phenotypes associated with DVAP-P58S and DVAP loss-of-function alleles. Consistent with these observations, Sac1 is sequestered into DVAP-P58S-mediated aggregates and downregulation of Sac1 in neurons induces increased PtdIns4P levels and degeneration. These data highlight the crucial role of DVAP and Sac1 in regulating phosphoinositides and support a causative role for PtdIns4P levels in ALS8 pathogenesis.
RESULTS
DVAP physically interacts with Sac1
A previous yeast two-hybrid screen for large-scale mapping of protein–protein interactions in Drosophila identified Sac1 as a DVAP-interacting protein (24). We confirmed this by yeast two-hybrid binary interaction and co-immunoprecipitation assays. A yeast two-hybrid analysis to detect a binary interaction between DVAP and Sac1 showed that these proteins can bind each other and that both the disease-causing alleles DVAP-P58S and DVAP-T48I retained their ability to bind Sac1 (Fig. 1A). For the co-immunoprecipitation assays, Myc-tagged Sac1 and Flag-tagged DVAP constructs were transfected in COS7 cells. In cell extracts expressing both Myc-tagged Sac1 and Flag-tagged DVAP, Flag-DVAP was clearly co-immunoprecipitated with Myc-Sac1 by the anti-Myc antibody. In agreement with the yeast two-hybrid data, Flag-DVAP-P58S was also significantly co-immunoprecipitated with Myc-Sac1. As a control, little co-immunoprecipitation was observed using cell extracts expressing either Flag-DVAP or Flag-DVAP-P58S but not Myc-Sac1 (Fig. 1B).
Figure 1.

Sac1 interacts with both DVAP and DVAP-P58S. (A) Sac1 interaction with DVAP and DVAP-P58S by yeast two-hybrid assay. p53/T-Ag interaction is the positive control and the lack of interaction between T-Ag and Lamin (LAM) is used as a negative control. Sac1 interacts with DVAP as well as with DVAP-P58S and DVAP-T48I. AD, Gal4 activation domain. DB, Gal4 DNA-binding domain. (B) Co-immunoprecipitation of Myc-Sac1 and Flag-DVAP or Flag-DVAP-P58S from cell lysates transfected with the indicated plasmids. Immunoprecipitates were collected on anti-Myc Sepharose beads. Input (I), immunoprecipitated complexes indicated as pellet (P) and the unbound fractions indicated as supernatant (S) were analysed by western blots with antibodies to DVAP or Myc. (C) DVAP interaction with Sac1-truncated proteins. LZ, leucine zipper; CD, catalytic domain; TM1,TM2, transmembrane domains 1, 2. On the right column, interactions (Int) are indicated by plus signs and lack of interaction by minus signs. In (D), Sac1 interaction with DVAP-truncated proteins is reported. MSP, major sperm protein; CC, coiled coil; TM, transmembrane domains. Interactions (Int) are indicated by plus signs and lack of interaction is indicated by minus signs. The CC domain (amino acids 184–227) and the TM domain (amino acids 250–266) were deleted from DVAP.
To further extend this analysis, we mapped the interaction domains on both proteins by yeast two-hybrid assay. Sac1 contains a conserved phosphoinositide–phosphatase module termed the Sac domain, comprising seven highly conserved motifs, including the catalytic domain. It also contains an N-terminal leucine zipper motif and two C-terminal putative TM domains, TM1 and TM2. We found that whereas TM2 was dispensable for the binding activity of Sac1, truncation of the first 18 amino acids encompassing the β1 helix effectively abolished Sac1 binding to DVAP (Fig. 1C). DVAP also exhibits conserved structural domains, including the MSP domain, a CC motif and a TM domain. Surprisingly, we found that the MSP domain was dispensable for the interaction, whereas removal of the C-terminal TM domain abolished the ability of DVAP to physically interact with Sac1 (Fig. 1D). Collectively, our data indicate that DVAP binds Sac1 via its TM and that a Sac1 fragment of 546 amino acids lacking the most C-terminal TM is necessary to bind DVAP. These data also show that ALS8-causing mutations do not affect the ability of DVAP to bind Sac1.
Presynaptic Sac1 is required for synaptic morphology, MT organization and axonal transport
Since DVAP and Sac1 physically interact, we asked whether mutations in Sac1 could affect the same biological processes as those affected by mutations in DVAP. DVAP loss-of-function mutations lead to a synaptic phenotype at the larval neuromuscular junction (NMJ) characterized by a reduction in the number of boutons and an increase in their size (5). Strong hypomorphic mutations in Sac1 cause embryonic lethality due to an early requirement of the gene during development (19). We therefore undertook a transgenic RNA interference (RNAi) approach to reduce Sac1 protein level during postembryonic development from the presynaptic side. We selectively expressed Sac1RNAi in neurons, using the UAS-Gal4 system and the neuronal-specific driver elav-Gal4. Staining with anti-HRP antibodies that label and allow for visualization of synaptic terminals showed that reducing Sac1 expression in presynaptic cells (elav;Sac1RNAi) led to a decrease in bouton number (181 ± 3.9 versus 280 ± 4.6 in controls) and a concomitant increase in their size (Fig. 2A and C and Supplementary Material, Fig. S1A and B). To dissect the cell-specific role of DVAP, we downregulated DVAP expression in presynaptic cells, using DVAPRNAi lines and the elav-Gal4 driver. Presynaptic knockdown of DVAP (elav;DVAPRNAiGD3990) induced a synaptic morphological phenotype similar to those of DVAP null mutations (5) and presynaptic Sac1RNAi expression (Fig. 2A–C and Supplementary Material, Fig. S1A and B).
Figure 2.

Presynaptic Sac1 and DVAP are required for synaptic morphology, MT integrity and axonal transport. (A–C) Representative confocal images of muscle 12 synapses stained for both HRP and Futsch in elav-Gal4/+ controls (A), elav;DVAPRNAiGD3990 larvae (B) and elav;Sac1RNAi transgenic mutants (C). elav;DVAPRNAiGD3990 and elav;Sac1RNAi NMJs exhibit a significant increase in the number of boutons with splayed (arrow) and lack or punctuate (arrowhead) Futsch staining. (D–F) Representative confocal images of nerves stained for both HRP and Bruchpilot (Brp) in controls (D), elav;DVAPRNAiGD3990 (E) and elav;Sac1RNAi transgenic larvae (F). Bar = 10 μm.
The presynaptic MT cytoskeleton, as visualized by staining with antibodies specific for the MAP1B-like protein Futsch, is severely perturbed in DVAP mutant NMJs (5). We tested whether Sac1 or DVAP downregulation in neurons affected MT stability at the NMJ. In control NMJs, MTs run mainly as a filament of Futsch staining across the nerve terminal branches (Fig. 2A). Occasionally, Futsch staining appears punctuate or splayed into a number of thinner filaments filling up the entire volume of the boutons. These two forms of staining, i.e. ‘punctuate and splayed’ have been described as two variant forms of disorganized MTs found in mutants affecting synaptic architecture (25,26). At both elav;Sac1RNAi and elav;DVAPRNAiGD3990 NMJs, there was an increase in the number of boutons showing MT disorganization. Although in controls the percentage of boutons exhibiting the ‘filled’ or ‘diffuse’ pattern of Futsch staining was only 35.7 ± 1.4, the number of boutons with the same pattern in elav;Sac1RNAi and elav;DVAPRNAiGD3990 NMJs was 58.3 ± 1.7 and 70.5 ± 2.4%, respectively (Fig. 2B and C; Supplementary Material, Fig. S1C).
Although the axonal MT cytoskeleton had no obvious morphological defects (data not shown), we observed abnormalities in the distribution of proteins and synaptic vesicles along the nerves. At control NMJs, Bruchpilot (Brp), an essential component of the synaptic active zones (27), was barely detectable along the nerves (Fig. 2D). Conversely, both elav;DVAPRNAiGD3990 and elav;Sac1RNAi mutants exhibited massive accumulation of Brp aggregates along the axons (Fig. 2E and F, Supplementary Material, Fig. S1D). Similar aggregates were observed using antibodies specific for the vesicle-associated cysteine string protein (data not shown). Hence, these results indicate that DVAP and Sac1 exhibit functional similarity and control several aspects of NMJ physiology, including synaptic structure, axonal transport and MT organization.
Postsynaptic downregulation of either Sac1 or DVAP affects synaptic morphology and the distribution of several synaptic markers
To assess the postsynaptic function of Sac1, we focused on two phosphoinositide-dependent proteins, adducin and β-spectrin, both with established NMJ function and involvement in ALS pathogenesis or neurodegeneration (28–31).
The muscle-specific driver BG57-Gal4 was used to express either Sac1RNAi or DVAPRNAi transgene postsynaptically. In controls, synaptic boutons had a rounded to oval appearance and were separated from each other by a well-defined neural process (Fig. 3A). Adducin, a predominantly postsynaptic marker, colocalized with and surrounded the presynaptic membrane defined by HRP staining (Fig. 3A). In both BG57/DVAPRNAiGD3990 and BG57;Sac1RNAi mutants, a complex phenotype affecting both the pre- and postsynaptic compartments was observed. The most striking effect was an aberrant synaptic morphology, although the number of boutons remained nearly normal. Boutons were larger and exhibited a highly irregular shape with a number of spike-like protrusions emerging from their contours. Remarkably, a mismatched apposition between the presynaptic membrane visualized by the HRP staining and the postsynaptic marker adducin was observed in mutants when compared with controls (Fig. 3B and C). At the postsynaptic compartment, adducin no longer tightly associated with the presynaptic boutons to entirely circumscribe the presynaptic membrane as in controls but it appeared confined to specific subregions of the membrane and absent or diffused in others (Fig. 3B and C). Similar phenotypes were observed for β-spectrin (Supplementary Material, Fig. S2A–C). Moreover, the postsynaptic localization of Discs-Large (DLG), the Drosophila homologue of the mammalian PSD-95, has been shown to depend on both adducin and β-spectrin (32,33). As expected, in both DVAPRNAiGD3990 and Sac1RNAi muscles, DLG was not surrounding the perimeter of the boutons but appeared rather disorganized and diffused (Supplementary Material, Fig. S2D–F). A high degree of heterogeneity among the different phenotypic classes was observed; however, the most abnormal bouton branches were those with the most aberrant organization in the distribution and localization of the adducin and β-spectrin postsynaptic markers. This is in agreement with previous data reporting that disruption of the actin–spectrin postsynaptic cytoskeleton can affect the shape and the appearance of presynaptic boutons (34). DVAP null alleles do not exhibit the morphological phenotype associated with postsynaptic downregulation of DVAP. A possible cause for this apparent discrepancy could be the non-cell autonomous effect of DVAP (7), as in this case phenotypic outcomes do not simply depend on the genotype of a mutant cell but may be also affected by the genotype of neighbouring cells.
Figure 3.

Postsynaptic downregulation of either DVAP or Sac1 induces changes in bouton morphology and adducin disorganization. (A–C) Representative confocal images of larval NMJs stained for HRP and adducin in BG57-Gal4/+controls (A), BG57/DVAPRNAi GD3990 transgenic mutants (B) and BG57;Sac1RNAi transgenic mutants (C). Asterisks indicate big and irregular boutons; arrowheads point to spikes; triangles show mismatched apposition of pre- and postsynaptic components and arrows indicate diffused and missing adducin staining. Bar = 10 μm.
It is well known that off-target activity can complicate the interpretation of phenotypic effects in gene silencing experiments by RNAi. To test the specificity of Sac1 knockdown by RNAi, we showed that expression of the Sac1RNAi construct in larval brains resulted in an almost complete depletion of Sac1 RNA (Supplementary Material, Fig. S3A). To exclude the possibility of off-target effects, we performed a phenotypic analysis of larvae expressing a Sac1 transgenic line (ΔSac1) in which the –RTNCIDCLDRTN– catalytic site has been removed. Expression of the ΔSac1 transgenic line in a wild-type background should have a dominant negative effect and should exhibit mutant phenotypes similar to those of a Sac1 loss-of-function allele. As expected, targeting the expression of ΔSac1 allele either pre- or postsynaptically led to a phenotype similar to those associated with Sac1 downregulation by Sac1RNAi (Supplementary Material, Fig. S3B–G).
For DVAP, two RNAi lines are available: DVAPRNAiGD3990 from the VDRC (Vienna Drosophila RNAi Center) collection and DVAPRNAiJF02621 from the TRiP collection. These lines were generated using constructs containing inverted repeats that target different, non-overlapping sequences within DVAP (see Materials and Methods for details). The DVAPRNAiGD3990 line effectively knocks down DVAP expression in salivary glands and brains from progeny of elav-Gal4 flies crossed to DVAPRNAiGD3990 flies (Supplementary Material, Fig. S4). A similar decrease in DVAP levels was associated with transgenic expression of DVAPRNAiJF02621 in the same tissues (data not shown). In addition, either pre- or postsynaptic expression of DVAPRNAiJF02621 induced mutant phenotypes reminiscent of those associated with DVAPRNAiGD3990 transgenic expression (Supplementary Material, Fig. S5). Together, these data indicate that for both DVAP and Sac1, gene knockdown by RNAi was specific and exclusively affected the intended genes.
Studies in Drosophila have suggested that DVAP-P58S could act as a dominant negative allele by sequestering wild-type DVAP into aggregates and depleting it from its normal localization (6,9). If this holds true for the DVAP–Sac1 interaction as well, then DVAP-P58S flies should exhibit mutant phenotypes similar to both DVAP and Sac1 downregulation. Indeed, the mutant phenotypes associated with both pre- and postsynaptic expression of DVAP-P58S (Supplementary Material, Figs S1, S6 and S7) were highly similar to those of DVAPRNAi and Sac1RNAi in the same tissues, thus confirming the proposed genetic effect of the DVAP-P58S allele.
The phenotypes associated with synaptic downregulation of Sac1 are highly reminiscent of those of DVAPRNAi mutants, suggesting that DVAP and Sac1 function in the same pathway. To assess additive effects, we focused on the easily quantifiable presynaptic morphology phenotype. Both elav;Sac1RNAi and elav;DVAPRNAiGD3990 exhibited a reduction in bouton number with a concomitant increase in their size. We noticed that elav;DVAPRNAiGD3990;Sac1RNAi double mutants did not show any further severity in the phenotype when compared with either single mutant alone (Supplementary Material, Fig S8). Taken together, these data indicate that Sac1 and DVAP do indeed function in a common pathway.
Sac1 colocalizes with DVAP in vivo
To assess the expression pattern of Sac1 in vivo, we generated antibodies against two subfragments of the protein. However, these antibodies performed rather poorly, so we generated transgenic flies expressing a functional Flag-Sac1 construct, which rescued the lethality of Sac12107 mutants and facilitated Sac1 localization using Flag-specific antibodies. We found that both Sac1 and DVAP proteins colocalized in larval brains, eye imaginal discs and salivary glands (Supplementary Material, Fig. S9A, C and D). DVAP and Sac1 closely associated in muscles where they colocalized in a perinuclear region (Supplementary Material, Fig. S9B) that has been previously shown to correspond to the ER/sarcoplasmic reticulum (3).
In yeast and mammalian cells, Sac1 associated with the ER and the Golgi (35,36). To define the subcellular localization of Drosophila Sac1, we cotransfected Flag-Sac1 and Myc-DVAP constructs in COS-7 cells. Staining with antibodies specific for DVAP and Flag revealed a considerable overlap of the two proteins in a reticular pattern that previous studies showed to be the region where DVAP overlapped with ER-specific markers (3). Sac1 also localized to a paranuclear region corresponding to the Golgi complex. DVAP staining appeared to be excluded from this region, confirming that DVAP was predominantly an ER-associated protein (Supplementary Material, Fig. S9E). Finally, to define the subcellular localization of Sac1 in vivo, we targeted expression of Flag-Sac1 in salivary glands. Consistent with the COS-7 data, Sac1 partially colocalized with both the ER marker Boca (37) and the cis-cisternal Golgi marker GM130 (Supplementary Material, Fig. S10). In summary, these data indicate that DVAP and Sac1 colocalize in all the tissues examined. Moreover, Sac1, like its yeast and mammalian homologues, associates with subcellular compartments corresponding to both the Golgi and the ER.
DVAP function is required to maintain normal PtdIns4P levels
In mammals, Sac1 controls PtdIns4P levels in the ER and in the Golgi, and in yeast, it can also modulate PtdIns4P associated with the plasma membrane (PM) (17,38). In yeast, VAP is required for Sac1 activity, and in VAP mutants PtdIns4P levels are increased (39). If such a mechanism were conserved in Drosophila, then one would also expect an increase in PtdIns4P levels in DVAPRNAiGD3990 mutants. We first stained larval salivary glands with a PtdIns4P antibody. In control cells, PtdIns4P was mainly detected at the PM, while under these staining conditions the intracellular pool was barely detectable. In contrast, both elav;Sac1RNAi and elav;DVAPRNAiGD3990 salivary glands exhibited increased PtdIns4P levels, particularly intracellularly (Supplementary Material, Fig. S11A). In control brains and muscles, PtdIns4P levels were basically below our detection limit (Supplementary Material, Fig. S11B and C). However, in Sac1RNAi and DVAPRNAiGD3990 brains, the expression levels of PtdIns4P were dramatically increased (Supplementary Material, Fig. S11B). In BG57;Sac1RNAi and BG57/DVAPRNAiGD3990 muscles, a similar increase in PtdIns4P levels was observed (Supplementary Material, Fig. S11C). These results support a role for DVAP in PtdIns4P metabolism, potentially by controlling Sac1 phosphatase function.
The mutant phenotypes associated with both pre- and postsynaptic expression of DVAP-P58S (Supplementary Material, Figs S1, S6 and S7) were highly similar to those of DVAPRNAi and Sac1RNAi in the same tissues. We should expect then to observe increased PtdIns4P levels in DVAP-P58S flies. Indeed, PtdIns4P levels were dramatically elevated in salivary glands, brains and muscles expressing the DVAP-P58S transgene (Supplementary Material, Fig. S11A–C). Finally, we showed that in every genotype the increase in PtdIns4P levels was not accompanied by a significant upregulation of PtdIns(4,5)P2, suggesting that the observed phenotypes are unlikely to be mediated by an upregulation of PtdIns(4,5)P2 (Supplementary Material, Fig. S12). Hence, these data indicate that DVAP function is required for PtdIns4P metabolism at the NMJ and that transgenic expression of ALS-causing mutations associates with an upregulation of PtdIns4P levels both in neurons and muscles.
Downregulation of Sac1 in neurons causes dosage-dependent neurodegeneration
Flies expressing DVAP-P58S in the adult nervous system fail to eclose, whereas targeted expression of DVAP-P58S in the eye induces extensive neurodegeneration, characterized by reduced eye size, with disorganized ommatidia showing missing, irregular or supernumerary bristles (3). As DVAP-P58S expression induces an increase in PtdIns4P levels and causes extensive neurodegeneration, one would expect the same outcome from Sac1 downregulation in neurons.
To test this hypothesis, we downregulated Sac1 in the nervous system, using the elav-Gal4 driver (elav;Sac1RNAi). Accordingly, elav;Sac1RNAi flies failed to eclose in 60% of individuals, whereas survivors exhibited marked neurodegeneration in the eye. The extent of neurodegeneration was, however, different in males and females. Females displayed extensive depigmentation, whereas males had the majority of the eye surface covered by patches of severe necrosis (Fig. 4A). The necrotic patch phenotype was fully penetrant as it was observed in 100% of males; however, differences in the degree and extent of the necrotic regions could be seen. The gender difference was consistent with higher levels of elav-Gal4 expression in males than in females owing to the X-chromosome dosage compensation effect in males (40). No difference in eye tissue survival between elav-Gal4 females and males was observed, indicating that the degenerative phenotype was not dependent on differences in gender or expression of Gal4 alone. Scanning electron micrographs of male eyes showed extensive regions of fused ommatidia and missing or misoriented interommatidial bristles (Fig. 4B). Eye sections showed a severe disruption in retinal organization, with thinning of the retina and loss of retinal neurons (Fig. 4C). This degenerative phenotype correlated with increased levels of PtdIns4P in elav;Sac1RNAi eye imaginal discs (data not shown).
Figure 4.
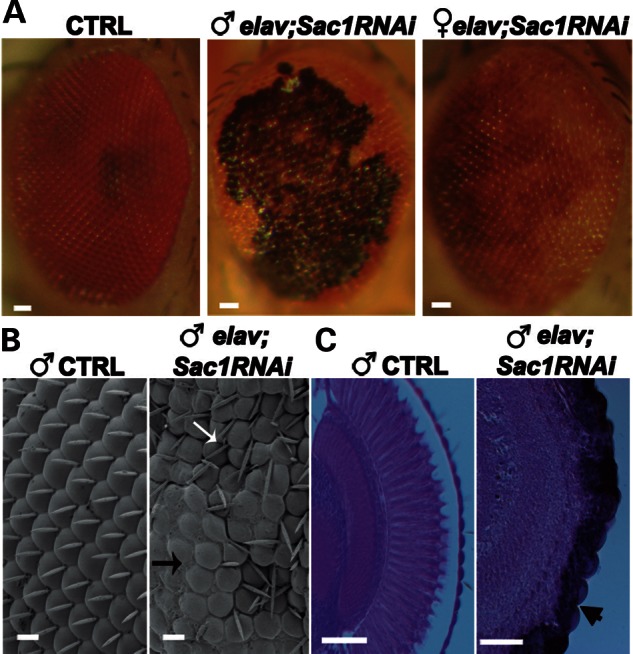
Downregulation of Sac1 in the eye induces neurodegeneration in a dosage-dependent manner. (A) Stereo microscope images of control (elav-Gal4/+), elav;Sac1RNAi male and elav;Sac1RNAi female adult eyes. (B) Scanning electron micrographs of control and elav;Sac1RNAi transgenic eyes of adult males. Although the adult Drosophila eye is composed of an ordered array of ommatidia with interspersed, regularly oriented bristles, transgenic eyes exhibit extensive regions of fused ommatidia (black arrow), with missing (black arrow) and misoriented bristles (white arrow). (C) Frontal sections of control and elav;Sac1RNAi transgenic eyes of adult males. The arrowhead points to tissue degeneration. Bar = 10 μm in (B) and 50 μm in (A) and (C).
In Sac1 mutant clones, activation of pro-apoptotic caspase 3 is suppressed by simultaneously overexpressing the Drosophila Inhibitor of Apoptosis 1 (DIAP1) gene (41). Likewise, DIAP1 overexpression strongly suppressed the reduction in eye size associated with DVAP-P58S expression in the eye. This suppression was not due to a dilution of Gal4, as coexpression of a GFP transgene had no effect (Supplementary Material, Fig. S13). These data indicate that in both cases neurodegeneration was, at least in part, due to death by apoptosis. In conclusion, these data demonstrate that Sac1 downregulation induces neurodegeneration in a dosage-dependent manner and that a similar phenotype is associated with transgenic expression of DVAP-P58S in neurons.
Upregulation in PtdIns4P levels induces DVAP-P58S-associated synaptic phenotypes and neurodegeneration
Our data so far indicate that upregulation of PtdIns4P could be a major cause of DVAP-P58S-mediated neurodegeneration. If this were true, then one would expect that blocking PtdIns4P production by downregulating either of the two PtdIns-kinases, PI4KIIIα and Four-wheel drive (Fwd), should suppress neurodegeneration. Coexpressing either FwdRNAi or PI4KIIIαRNAi transgenes in ey,DVAP-P58S recombinant flies markedly attenuated the neurodegenerative phenotype. Specifically, the structural phenotypes and ∼50% size reduction in ey,DVAP-P58S eyes were significantly suppressed in both ey,DVAP-P58S/PI4KIIIαRNAi and ey,DVAP-P58S;FwdRNAi flies (Fig. 5). This suppression was not due to a dilution of Gal4, as coexpression of a GFP transgene had no effect (Supplementary Material, Fig. S13).
Figure 5.
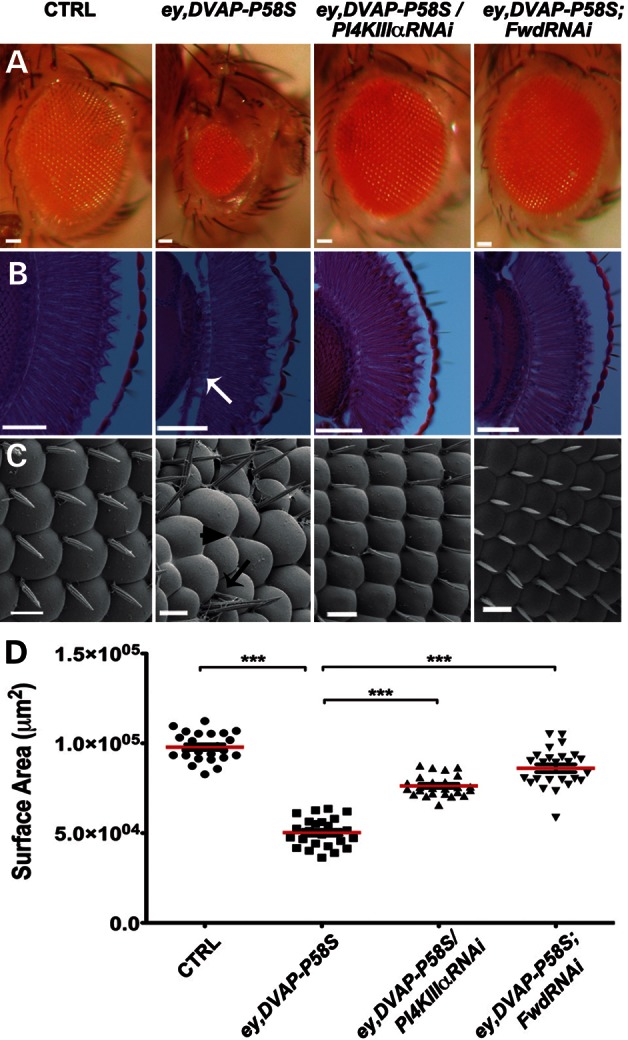
DVAP-P58S-mediated neurodegeneration is rescued by downregulating either PI4KIIIα or Fwd PI4-kinases. (A) Stereo microscope images, (B) frontal sections and (C) scanning electron micrographs of adult fly eyes of the following genotypes: controls (ey-Gal4/+), ey,DVAP-P58S, ey, DVAP-P58S/PI4KIIIαRNAi and ey,DVAP-P58S;FwdRNAi. Whereas control eyes are composed of ommatidia and regularly spaced mechanosensory bristles (C), ey,DVAP-P58S eyes are smaller (A) with missing (arrowhead in C), supernumerary (black arrow in C) bristles and occasionally fused ommatidia. In (B), white arrow points to tissue degeneration. The ey,DVAP-P58S-mediated neurodegeneration is rescued by downregulating either PI4KIIIα or Fwd. (D) Quantification of the eye surface area of every genotype. The reduced eye size of ey,DVAP-P58S flies is significantly suppressed by coexpressing either PI4KIIIα (ey,DVAP-P58S/PI4KIIIαRNAi) or FwdRNAi (ey,DVAP-P58S;FwdRNAi). Bar = 50 μm in (A) and (B) and 10 μm in (C). Error bars denote SEM. n = 25 eyes per genotype. ***P < 0.001.
To directly confirm the role of PtdIns4P in DVAP-P58S-induced neurodegeneration, we assessed PtdIns4P levels in eye imaginal discs of ey,DVAP-P58S; ey,DVAP-P58S/PI4KIIIαRNAi and ey,DVAP-P58S;FwdRNAi. A marked increase in PtdIns4P levels was observed in ey,DVAP-P58S eye imaginal discs, whereas PtdIns4P levels were reduced and similar to wild-type in ey,DVAP-P58S/PI4KIIIα RNAi and ey,DVAP-P58S;FwdRNAi animals (Fig. 6A, B, D and E). The decrease in the PtdIns4P levels was not a consequence of diluting the Gal4, as coexpression of a GFP transgene with DVAP-P58S had no effect (Fig. 6C).
Figure 6.

Suppression of DVAP-P58S-mediated neurodegeneration by FwdRNAi and PI4KIIIαRNAi correlates with a decrease in PtdIns4P levels. (A–E) Representative confocal images of eye imaginal discs stained with an antibody specific to PtdIns4P phosphoinositides from ey-Gal4/+ control larvae in (A), ey,DVAP-P58S larvae in (B), ey,DVAP-P58S/GFP larvae in (C), ey,DVAP-P58S/ PI4KIIIαRNAi larvae in (D) and ey,DVAP-P58S;FwdRNAi larvae in (E). The control levels of PtdIns4P are barely detectable in our experimental conditions, but there is a massive upregulation of PtdIns4P levels in ey,DVAP-P58S eye imaginal discs. A decrease in PtdIns4P levels is observed when either the PI4KIIIα or Fwd kinases are downregulated in eye imaginal discs expressing DVAP-P58S, whereas coexpression of a GFP transgene has no effect. Bar = 50 μm.
Synaptic phenotypes associated with the expression of DVAP-P58S in the motor system could also be due to an upregulation of PtdIns4P and therefore knocking down either Fwd or PI4KIIIα kinase should suppress DVAP-P58S-mediated phenotypes. In both cases, the motor neuron-specific driver OK6-Gal4 was used. We found that the NMJ phenotypes of OK6;DVAP-P58S were significantly rescued by coexpressing the FwdRNAi transgene (Fig. 7). A similar effect was observed when PI4KIIIαRNAi was expressed in motor neurons together with DVAP-P58S (data not shown). In addition, the effect of coexpressing FwdRNAi and DVAP-P58S transgenes in muscles (BG57/DVAP-P58S;FwdRNAi) was assessed. Interestingly, we found that BG57/DVAP-P58S;FwdRNAi NMJs did not resemble wild-type but rather NMJs expressing BG57;FwdRNAi alone. Specifically, NMJs exhibited an abnormal structure and were composed mainly of a few fused boutons lacking their characteristic smooth and round shape. These boutons appeared deformed, with elongated and misshapen profiles (Supplementary Material, Fig. S14A–C). This phenotypic similarity suggests that FwdRNAi expression in BG57/DVAP-P58S postsynaptic terminals decreased PtdIns4P below wild-type levels. An analogous effect was observed when PI4KIIIαRNAi transgene was expressed in BG57/DVAP-P58S muscles (data not shown). Interestingly, we found that presynaptic and postsynaptic coexpression of FwdRNAi transgene in DVAPRNAiGD3990 NMJs had a similar effect, further supporting the importance of PtdIns4P levels in DVAP-mediated function (Supplementary Material, Figs S15 and S14D, respectively). In conclusion, these data provide strong evidence for a direct role of PtdIns4P upregulation in DVAP-P58S-mediated neurodegeneration and synaptic phenotypes.
Figure 7.

Downregulation of Fwd PI4-kinase in neurons rescues the DVAP-P58S presynaptic phenotypes. (A–C) Representative confocal images of NMJs stained for HRP and Futsch in OK6-Gal4/+control (A), in OK6/DVAP-P58S larvae (B) and in OK6/DVAP-P58S;FwdRNAi larvae (C). (D–F) Representative confocal images of nerves stained for HRP and Brp antibodies in OK6-Gal4/+ control (D), in OK6/DVAP-P58S larvae (E) and in OK6/DVAP-P58S;FwdRNAi larvae (F). (G) Quantification of bouton size at muscle 12 (type I and type III boutons) of segment A3 in OK6-Gal4/+ control, OK6/DVAP-P58S and OK6/DVAP-P58S;FwdRNAi NMJs (n = 205, 149, 286, respectively). (H) Quantification of total number of boutons on muscle 12 and 13 of segment A3 in OK6-Gal4/+ controls (287.4 ± 3.2, n = 10), OK6/DVAP-P58S (160.9 ± 1.4, n = 10) and OK6/DVAP-P58S;FwdRNAi (265 ± 3, n = 10) NMJs. The decrease in number of boutons and the increase in their size in OK6/DVAP-P58S are significantly rescued by downregulating Fwd (P < 0.0001 in every case). Presynaptic downregulation of Fwd kinase rescues the morphological, the MT and the axonal transport phenotypes associated with both DVAP-P58S presynaptic downregulation. Bar = 10 μm. Error bars denote SEM. ***P < 0.0001.
Sac1 is sequestered in DVAP-P58S-induced aggregates
Our biochemical data and the analysis of mutant phenotypes indicate that ALS8 is due to DVAP loss-of-function, with Sac1 being downregulated and PtdIns4P levels subsequently increasing. However, our biochemical analysis also showed that both ALS8 mutations retain their ability to bind Sac1. How then is Sac1 function impaired in DVAP-P58S mutants? To address this question, we coexpressed a Flag-Sac1 transgene in ey-Gal4,DVAP-P58S recombinant flies and stained eye imaginal discs with antibodies specific for Flag and DVAP. We have previously shown that DVAP-P58S-mediated aggregates contain wild-type and mutant DVAP proteins that are both immunoreactive to DVAP-specific antibodies (3). We observed that, in larval eye imaginal discs, Sac1 was sequestered in the DVAP-P58S-mediated inclusions and was depleted from its normal localization (Fig. 8A). Similarly, in elav;DVAP-P58S; Flag-Sac1 larval brains, Sac1 appeared to be recruited into DVAP-P58S-mediated aggregates (Fig. 8B). This was in contrast to the diffuse and granular pattern of Sac1 staining observed in controls (Supplementary Material, Fig. S9A and C). In both instances, the association of Flag-Sac1 with the aggregates was specific since a coexpressed GFP was not recruited into the aggregates (data not shown). In conclusion, our data indicate that the DVAP–Sac1 interaction regulates PtdIns4P levels and support a model whereby sequestering Sac1 and DVAP into pathogenic aggregates compromises the function of both proteins, leading to an increase in PtdIns4P levels.
Figure 8.

Sac1 is sequestered into DVAP-P58S-mediated aggregates. (A) Eye imaginal discs and (B) brains stained for both DVAP and Flag in ey,DVAP-P58S/Flag-Sac1 and elav;DVAP-P58S/Flag-Sac1 larvae. Bar = 10 μm. (C) Graphical summary of the data. The left panel shows a chemical equilibrium between two reactions: one in which Sac1 interacts with DVAP to catalyse the conversion of PtdIns4P into PtdIns and another in which PI4K converts PtdIns into PtdIns4P. The right panel shows that depleting DVAP (i) or Sac1 (ii) or the aggregation of Sac1 and DVAP into DVAP-P58S-induced aggregates (iii) inhibits Sac1-mediated reaction leading to an increase in PtdIns4P levels.
DISCUSSION
Here we identified Sac1 as a DVAP-binding protein and uncovered a hitherto unknown function of Sac1 in postembryonic synaptic maturation and neurodegeneration. Presynaptic reduction of either DVAP or Sac1 levels induces structural changes, disruption of the synaptic MT cytoskeleton and accumulation of clusters of proteins and vesicles along the axons. In addition, muscle down-regulation of either Sac1 or DVAP leads to a strikingly aberrant synaptic morphology and abnormal localization and distribution of several postsynaptic markers, including adducin and β-spectrin. We also report that depletion of DVAP as well as Sac1 expression induces an increase in PtdIns4P levels.
We go on to show that Sac1 downregulation in the adult nervous system causes early death and neurodegeneration in a dosage-dependent manner, a phenotype similar to that of DVAP-P58S transgenic expression. Our analysis indicates that the DVAP-P58S allele has a dominant negative effect, as its transgenic expression leads to an upregulation of PtdIns4P and its mutant phenotypes are similar to those associated with either DVAP or Sac1 loss-of-function. In agreement with the hypothesis that neurodegeneration in the DVAP-P58S context is due to a loss-of-function of both DVAP and Sac1, we report that both wild-type DVAP and Sac1 are depleted from their normal localization and are sequestered into DVAP-P58S-mediated aggregates.
Altogether, these data are consistent with a model in which DVAP is required for Sac1 activity and for the regulation of intracellular PtdIns4P levels. Loss-of-function of DVAP and Sac1 by a DVAP-P58S-mediated dominant-negative mechanism induces cell degeneration by an upregulation of PtdIns4P, which is also responsible for the observed disruption of fundamental biological processes at the NMJs (Fig. 8C).
We and others have previously shown that transgenic expression of DVAP proteins carrying the equivalent ALS8 mutations in Drosophila mimic the human disease (3,6,9). Notably, expression of hVAPB in flies rescues the lethality and the phenotypes associated with DVAP mutants, indicating an evolutionarily conserved function for VAP proteins (6).
Collectively, these data indicate that DVAP-mediated molecular pathways are likely to be important for our understanding of the disease pathogenesis in humans.
DVAP and Sac1 interact to control PtdIns4P levels
There is evidence supporting that DVAP functions to maintain normal cellular levels of PtdIns4P by interacting with Sac1. First, Sac1 and DVAP bind to each other and colocalize in many different tissues. It has been reported that phosphoinositol transfer proteins/phosphoinositide-binding proteins associate directly with phosphatases and kinases to control their activities (39,42,43). Specifically, VAP has been shown to bind PtdIns4P in vitro (44) and to be required for Sac1 activity in yeast (39). Second, PtdIns4P levels are upregulated in DVAPRNAi mutants, suggesting that DVAP function is required for normal PtdIns4P levels. Similarly, in yeast, inactivation of Scs2/Scs22 VAP genes induces an increase in the levels of PtdIns4P (39,45). Third, the phenotypic similarity associated with either DVAP or Sac1 loss-of-function mutations supports the idea that the pool of PtdIns4P that is upregulated in DVAP mutants is the same as the one dephosphorylated by Sac1.
Previous studies attributed a prominent functional role to the N-terminal MSP domain of DVAP. The MSP domain is cleaved and secreted and binds to the extracellular domain of Ephrin receptors (7). Secreted MSP also binds to Robo and Lar-like receptors to control mitochondria morphology, localization and function in muscles (8). We identified a new DVAP-binding activity that is MSP-independent and involves a C-terminal fragment encompassing the TM domain. Interestingly, a new hVAPB mutation replacing valine at position 234 with an isoleucine in the conserved TM domain of hVAPB has been shown to cause ALS8 in humans (46). These data may provide direct evidence of a role of hVAPB-Sac1 interaction in the disease pathogenesis in humans.
Dynamic localization of Sac1 and ER/PM contact sites
In yeast and mammalian cells, Sac1 is an integral membrane protein localized to the ER and the Golgi (17,36,47). Here we report a similar localization for the Drosophila homologue of Sac1. In yeast and mammalian cells, Sac1 localization appears to be very dynamic, as this protein shuttles between ER and Golgi upon nutrient conditions. Specifically, glucose starvation in yeast or growth factor deprivation in mammalian cells causes relocalization of Sac1 from the ER to the Golgi complex, where it reduces PtdIns4P levels and slows protein trafficking (38). The ER–Golgi shuttling ability of Sac1 is reversed when nutrients or growth factors are added back to the growth medium (38). The growth factor-induced translocation of Sac1 from the Golgi to the ER requires p38 MAPK (mitogen-activated protein kinase) activity (48). These data suggest that Sac1 trafficking may be regulated by stressors that activate p38 MAPK. Some of these stressors such as oxidative damage and ER stress are triggers of neurodegeneration (49). This raises the intriguing possibility that a p38 MAPK-activated mechanism of PtdIns4P spatial regulation may be implicated in neurodegenerative processes.
A paper published after our paper was submitted revealed that Scs2/Scs22 VAP proteins in yeast play a pivotal role in tethering the ER to the PM to form ER/PM contact sites (45). Recent studies have highlighted the role of membrane junctions between organelles as important sites for lipid metabolism and intracellular signalling controlled by PtdIns4P (50). Depletion of Scs2/Scs22 VAP proteins located to the ER/PM contact sites leads to a retraction of the ER into internal structures, elevated levels of PtdIns4Ps at the PM and induction of the UPR (45). At the ER/PM contact sites, Sac1 dephosphorylates PtdIns4P on the PM in trans from the ER (39). This reaction requires the Scs2/Sc22p VAP proteins and the oxysterol-binding homology proteins that act as PtdIns4P sensors and activates Sac1 phosphatase activity (39). ER/PM junctions have been described in many organisms and cell types, including neurons and Drosophila photoreceptors (51,52). In addition, VAP proteins have been implicated in ER–Golgi, ER–endosomes and ER–mitochondria contacts in mammalian cells, suggesting that they may function as a tether for several organelle/membrane contact sites (53–55).
In conclusion, emerging evidence suggests that VAP proteins may be a crucial component of a hub controlling PtdIns4P metabolism in yeast and possibly, in higher eukaryotes as well.
The role of PtdIns4P in synaptic physiology and neurodegeneration
The ability of either PI4KIIIα or Fwd to suppress the synaptic and neurodegenerative phenotypes associated with transgenic expression of DVAP-P58S is somewhat surprising, as their yeast homologues (Stt4 and Pik1, respectively) are supposed to play non-redundant functions and to control spatially separate pools of PtdIns4P. This is based on previously published data showing that, in yeast, Stt4 and Pik1 are both essential for cell viability but control different cellular processes (56–58). Pik1 is essential for anterograde vesicular trafficking (59), whereas Stt4 plays a role in actin cytoskeleton organization and protein kinase C signalling (57). Both Pik1 and Stt4 play distinct roles in regulating MAPK signalling (60,61). Localization studies further suggest that Pik1p is primarily present in the nucleus and in the Golgi, whereas Stt4p is mainly cytoplasmic and is recruited to the PM for localized synthesis of PtdIns4P (56,57,59).
However, at present, the precise degree to which Stt4 and Pik1 functions have been conserved and apportioned among their homologues in flies remains unclear.
In Drosophila, the Stt4 homologue PI4KIIIα is required for oocyte polarization and its intracellular localization has not been determined (62). On the other hand, previous studies revealed that the fly Pik1 homologue Fwd is required for male germ-line cytokinesis (63). In spermatocytes, Fwd localizes to the Golgi and it is required for the accumulation of PtdIns4P on this organelle, implying that its function in providing PtdIns4P in the Golgi is evolutionarily conserved with yeast (64). However, whereas Pik1 is required for cell viability, Fwd appears to be dispensable for normal development, suggesting that it is redundant with similar genes in carrying out its function (56,64–66).
Another way to explain the rescue data would be to admit that upregulation of PtdIns(4,5P)2 and not PtdIns4P is responsible for DVAP-P58S mutant phenotypes.
PtdIns4P formed by the PM-associated STT4 can function as a substrate of PI4P 5-kinase to generate PtdIns(4,5)P2 at the cell cortex (15). It is also possible that PtdIns4P that is phosphorylated by plasmalemmal PI4P 5-kinase originates from intracellular sources. In the Golgi, PtdIns4P levels play a central role in the formation of vesicles delivered from the trans-Golgi network to the PM and their lipid cargo could be the substrate for the plasmalemmal PtdIns(4,5)P2 synthesis (67). As PtdIns4P in the Golgi is mainly produced by Pik1 is therefore possible that PtdIns(4,5)P2 associated with the PM and its effector proteins are downstream of both Stt4 and Pik1. Moreover, upregulation of PtdIns(4,5)P2 would explain the MT phenotypes, the mislocalization of post-synaptic markers and the axonal transport defects. Indeed, PtdIns(4,5)P2-enriched microdomains in the PM have been shown to participate in the regulation of MT plus-end capture and stabilization during polarized mobility (68). In Caenorhabditis elegans, the microtubular motor UNC-4 gene was shown to be anchored to synaptic vesicles, using a pleckstrin homology domain, thus implicating PtdIns(4,5)P2 in MT-based intracellular motility (69). Finally, spectrin proteins and adducin require PtdIns(4,5)P2 for their correct localization to the cell cortex (29,31).
However, if this were true, we should observe an increase in PtdIns(4,5)P2 levels wherever we observe an upregulation of PtdIns4P. By using an antibody specific for PtdIns(4,5)P2, we quantified PtdIns(4,5)P2 levels in tissues in which either Sac1 or DVAP were downregulated as well as in tissues expressing the DVAP-P58S transgene. Surprisingly, PtdIns(4,5)P2 levels were not affected by the dramatic upregulation of PtdIns4P in any of the genotypes described earlier. Consistent with these data, it was previously reported that, in Drosophila eye imaginal discs, depletion of Sac1 exhibits a dramatic increase in PtdIns4P levels, whereas PtdIns(4,5)P2 and PtdIns3P levels remain similar to wild-type (41). In addition, a paper published after our paper was submitted and another paper published a few months earlier showed that loss of PM PtdIns4P by downregulation of PI4KIIIα was not matched by a decrease in PtdIns(4,5)P2 levels (70,71). They showed that the major function of PtdIns4P is not to generate the pool of PtdIns(4,5)P2 on the PM but rather to contribute to the generation of a polyanionic lipid environment in the inner leaflet of the PM. PtdIns4P would then function in recruiting soluble proteins to the PM by electrostatic interaction with their polycationic surface. They also show that PtdIns4P contributes to processes such as modulation of ion channel activity that have been traditionally associated with changes in PtdIns(4,5)P2 (71). Finally, upregulation of PtdIns(4,5)P2 by inactivation of the Drosophila PI(4,5)P2 5-phosphatase synaptojanin leads to a distinct endocytotic phenotype due to defects in synaptic vesicle recycling. In synaptojanin mutants, synaptic vesicles are severely depleted and those remaining are clearly clathrin-coated. Intracellular recordings revealed enhanced synaptic depression during prolonged high-frequency stimulation (72). Ultrastructural and electrophysiological analysis of DVAP mutants do not exhibit a synaptojanin-like phenotype (6,9 and G. Pennetta, unpublished data), indicating that upregulation of PtdIns(4,5)P2 does not mimic the phenotype associated with increased levels of PtdIns4P.
Taken together, these considerations suggest that increased levels of PtdIns4P could be the main factor determining the observed synaptic and neurodegenerative phenotypes. Further studies using fluorescent phosphoinositide probes and genetic analyses will be needed to fully clarify the contribution of PtdIns4P versus PtdIns(4,5)P2 pools to NMJ physiology and neurodegeneration.
The role of DVAP–Sac1 interaction in lipid transport
Emerging evidence indicates that VAP and Sac1 may also play an important and specific role in membrane homeostasis (73). Biogenesis of sphingolipids, sterols and phosphoinositides that together determine the structural and functional properties of cell membranes must be closely coordinated. VAP interacts with both oxysterol-binding protein (OSBP) and ceramide transfer protein (CERT), recruiting them to contact sites between the ER and the Golgi complex (74). CERT has a FFAT (diphenylalanine in an acidic tract) motif that mediates its binding to ER-localized VAP and a PH domain that recognizes the PtdIns4P-enriched Golgi membrane. It has been proposed that CERT, because of its dual-binding ability, shuttles ceramide from the ER to the Golgi, where it is converted into sphingomyelin (75). Sphingomyelin continues to move through the secretory pathway to the PM, where it is most abundant. OSBP has an analogous function to CERT but instead mediates inter-membrane sterol transfer. This functional similarity is also reflected in OSBP's domain architecture: like CERT, it contains a PtdIns4P-binding PH domain and a VAP-binding FFAT motif (76). Ridgway and Perry (74) showed that sterols regulate sphingolipid metabolism by inducing a significant increase in SM synthesis that is dependent on OSBP, CERT and their shared binding partner VAP. The precise mechanism is not yet known but OSBP appears to activate CERT by promoting its recruitment to membranes and its binding to VAP. It is likely that disruption of the VAP–Sac1 interaction may have profound effects on the lipid composition of the PM, affecting its curvature and thickness and by consequence, vesicle budding and membrane remodelling (77). Interestingly, synaptic growth requires membrane remodelling and, at the Drosophila NMJs, occurs mainly by the budding of new boutons from pre-existing ones (78).
Putative roles of PtdIns4P in neurodegeneration
VAPB is involved in the IRE1/XBP1 signalling pathway of the UPR, an ER reaction inhibiting the accumulation of unfolded/misfolded proteins. In the disease-context, the hVAPB-P56S protein recruits its wild-type counterpart into the aggregates and it attenuates its ability to induce the UPR (10). This together with the observation that, in yeast, depletion of VAP proteins from the ER/PM contact sites induces a constitutive activation of the UPR suggests that motor neurons in ALS8 could be particularly vulnerable to cell death-induced ER stress. In addition, VAP proteins have been shown to be involved in lipid transfer and metabolism and accumulation of lipids and intermediates of lipid biosynthetic pathways are potent inducers of apoptosis (79). Finally, recent studies in yeast have shown that defects in the PtdIns4K Pik1 activity lead to a blockage of autophagy, a process controlling the degradation of long-lived proteins, damaged organelles and bulk cytoplasm in response to various types of stress (80).
Many questions remain to be explored concerning the precise molecular mechanism underlying neurodegeneration in ALS. However, over the last few years, an increasing number of experimental models have been generated and they represent an excellent tool for identifying molecular pathways in ALS and for evaluating their contribution to the disease pathogenesis.
MATERIALS AND METHODS
Drosophila stocks
Flies were raised on standard cornmeal food at 25°C. The following strains were used: elav-Gal4, ey-Gal4, OK6-Gal4, BG57-Gal4 and UAS-DIAP1 and the DVAPRNAiJF02621 (Bloomington Drosophila Stock Center) and Sac12107 (19). Sac1RNAi, PI4KIIIαRNAi, DVAPRNAiGD3990 and FwdRNAi stocks were obtained from the VDRC. For the DVAPRNAiJF02621 line, the P{TRiP.JF02621} construct was used and the target sequence is in the 3′ UTR of the cDNA, whereas the P{GD3990} construct used for the DVAPRNAiGD3990 line has a target sequence located within the ORF of the DVAP cDNA (http://flybase.org).
Immunohistochemistry
NMJs from wandering third-instar larvae were dissected in cold PBS before fixation and staining as previously described (5). The PtdIns4P staining and PtdIns(4,5)P2 staining were performed as previously reported (41) with tissue-specific modifications. For PtdIns4P and PtdIns(4,5)P2 staining, tissues were fixed with 4% paraformaldehyde in PBS for 30 min, then washed in PBS and quenched with 50 mm NH4Cl in PBS for 10 min, before washing. Salivary glands were permeabilized in TBS + 0.5% saponin (Sigma) for 1 h at room temperature before blocking overnight in TBS with 0.3% saponin and 10% NGS at 4°C. Primary antibodies were used at 1:100 in permeabilization buffer containing 5% NGS for 2 h at room temperature. Primary and secondary antibody washes were carried out in TBS containing 0.3% saponin and 1% NGS. Eye imaginal discs were processed similarly with a 3 h permeabilization and 4 h primary antibody incubation time. Larval brains and NMJs were processed similarly except that the permeabilization and the incubation with the primary antibody were performed overnight at 4°C.
Mouse monoclonal antibodies for Futsch, Adducin, dCSP, Brp and DLG were all used at a concentration of 1:50. Other antibodies were used at the following concentrations: anti-β-spectrin at 1:500, rabbit anti-DLG at 1:8000, anti-Boca at 1:1000, anti-DVAP at 1:1000, anti-GM130 (Abcam) at 1:100, anti-Flag (Sigma) at 1:100 and anti- PtdIns4P (Echelon) at 1:100. Goat anti-HRP (1:200), rabbit anti-HRP (1:500) and all secondary antibodies (1:500) were purchased from Jackson ImmunoResearch. Samples were mounted on microscope slides using Vectashield mounting medium (Vector Laboratories).
Eye histology and scanning electron microscopy
Frontal sections and scanning electron microscopy of adult fly eyes were processed as previously described (3).
Molecular biology
To generate the N-terminal Flag-tagged Sac1 (Flag-Sac1) construct, the full-length Sac1 cDNA was amplified from clone GH08349 with PCR primers containing BamHI and SalI sites and cloned in-frame into the pCMV-Tag2b vector (Clontech). The Flag-Sac1 fragment was then cloned into the pUAST vector using NotI and KpnI sites. For co-immunoprecipitation experiments, the Myc-Sac1 construct was generated by PCR amplification of the full-length Sac1 cDNA, using primers containing SalI and KpnI. The resulting fragment was cloned in-frame into pCMV-Myc vector (Clontech). The Flag-tagged DVAP (Flag-DVAP) construct was generated by amplifying the full-length DVAP cDNA with PCR primers containing EcoRI and SalI sites and by cloning the resulting DNA fragment in-frame into the pCMV-Tag2b vector. The DVAP-P58S construct was cloned into the pCMV-Tag2b vector, using the same strategy as for the cloning of DVAP cDNA in the same vector. For immunocytochemistry, a Myc-DVAP construct was generated by PCR amplification of the full-length DVAP cDNA, using EcoRI and NotI primers and cloned in-frame into pCMV-Myc vector (Clontech). For the same experiment, the Flag-Sac1 fragment was cloned into the pCMV-Tag2b vector as described earlier. For the semiquantitative PCR amplification, total RNA was purified from 20 dissected brains of the relevant genotypes and collected in TRIzol reagent (Sigma). cDNAs were produced using oligo-dT primers and following the manufacturer's instructions (SuperScript III First Strand Kit, Invitrogen). Amplification of Rp49 cDNA was made using the following primers: Rp49-L: CCGACCACGTTACAAGAACTCTC; Rp49-R: CGCTTCAAGGGACAGTATCTGA. For the amplification of Sac1 cDNA, the following primers were used: Sac1-L: TGCTGGGAACCATTCACCTA and Sac1-R: TGGCACTGACAGCG ATCATT. In both cases, PCR conditions were in the linear part of amplification. At least three different samples for each genotype were analysed. PCR products were visualized with ethidium bromide on a gel. For the Sac1Δ construct, the deletion of the RTNCIDCLDRTN catalytic motif was introduced into Sac1 cDNA (GH08349 clone) by site-directed mutagenesis using QuikChange IIXL Site-Directed Mutagenesis Kit (Agilent Technologies) and following the manufacturer's instructions. The Sac1cDNA carrying the RTNCIDCLDRTN motif deletion was isolated by PCR amplification and cloned into pUAST vector, using BamHI–KpnI linkers. Basic molecular biology techniques were performed following standard protocols.
Co-immunoprecipitation and immunocytochemistry
Cells were cultured in DMEM medium containing 10% FBS until 80% confluent and transfected with the relevant plasmids, using FuGENE Transfection Reagent (Roche) according to the manufacturer's instructions. Protein lysates for co-immunoprecipitation were prepared as previously described in Kawano et al. (81), with minor modifications. The protein complexes were immunoprecipitated using anti-Myc beads from Pierce Profound c-Myc Tag IP/Co-IP Application Set (Thermo Scientific). Inputs, precipitated protein complexes and supernatants were analysed by SDS–PAGE and western blots. Detection of Flag-DVAP proteins by western blot was performed by using a guinea pig anti-DVAP antibody at a concentration of 1:40 000 and a secondary HRP-conjugated anti-guinea pig antibody at 1: 60 000. Myc-Sac1 proteins were detected by western blot using a rabbit anti-Myc (Sigma) antibody at a concentration of 1:10 000 and an anti-rabbit HRP-conjugated secondary antibody at 1:30 000. For immunocytochemistry, cells were plated on BioCoat™ poly-l-lysine glass coverslips (BD Biosciences) and transfected as described earlier. Confluent cells were fixed in 4% paraformaldehyde for 20 min, blocked in PBS-T containing 10% NGS and incubated overnight with both rabbit anti-Myc (Sigma, 1:200) and mouse anti-Flag (Sigma, 1:200) antibodies. Fluorescent secondary antibodies were used at 1:500. Preparations were mounted in Vectashield medium.
Genetics and transgenics
Lethal transgenic insertions were maintained over GFP-containing balancers to select the relevant genotypes for larval phenotypic analysis. Transgenic lines were generated using standard protocols. To characterize the mutant phenotypes, Gal4 drivers were crossed with the transgenic lines. Embryos were collected for 20–24 h and then transferred to a water-bath at 30°C to maximize the expression of the transgene. The ey-Gal4 driver was recombined with DVAP-P58S on the same chromosome, following standard procedures.
Yeast two-hybrid assay
Binary interactions between DVAP and Sac1 were performed using the MATCHMAKER 3 GAL4 Yeast Two Hybrid System (Clontech). The full-length cDNA for DVAP was cloned in-frame into the pGADT7 vector, using PCR primers containing EcoRI and BamHI sites. Full-length cDNA for Sac1 was cloned in-frame into the pGBKT7 vector, using PCR primers containing NdeI and BamHI sites. All other fragments were cloned in-frame by PCR, using primers containing NdeI and BamHI sites. DVAP constructs lacking the CC and TMs were generated by using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) and following the manual's instructions. Protein interactions were analysed in media lacking histidine, adenine, leucine and tryptophan (−HALT medium) following transformation in AH109 yeast strain with the relevant plasmids.
Statistical analysis
Statistical analysis was performed and graphs were generated using GraphPad 5.0. The D'Agostino and Pearson omnibus test was applied to check for normality in the distributions of samples. For experiments with more than two samples which all passed the normality test, a one-way ANOVA test was applied to the samples. Tukey's multiple comparison test was then used as a post hoc-test when a significant difference was found in the ANOVA test. For experiments with more than two samples where at least one did not pass the normality test, statistical analysis was performed using the Kruskal–Wallis test and the Dunn's multiple comparison test as a post-test. For experiments with only two samples and where at least one of the two did not pass the normality test, statistical significance was evaluated using the Mann–Whitney U-test. Finally, for experiments with only two samples that both passed the normality test, a two-tailed unpaired Student's t-test was applied.
Imaging analysis and quantification
For imaging and quantification, NMJs, brains, eye imaginal discs and salivary glands from wandering third instar larvae were dissected and immunostained with the appropriate antibodies. Preparations were imaged on a Zeiss Axiovert LSM510 confocal microscope. The same confocal gain settings were applied to controls and samples. For all the images, the gain was chosen at the maximum gain that did not saturate the signal for each sample. Images were processed with the LSM software and Adobe Photoshop. Quantification of bouton number was performed by counting the total number of boutons on muscle 12 and 13 of the segment A3. The size of boutons was analysed by measuring the surface area of type I and type III boutons on muscle 12 of segment A3 and represented as ratio between the number of boutons with a given range of sizes and the total number of boutons (relative frequency). For the quantification of the axonal transport phenotype and PtdIns(4,5)P2 levels, a complete Z-stack was acquired and rendered as a maximum projection. Common appropriate intensity thresholds were selected to properly identify specific signals and ignore background intensities in the ImageJ package. The average number of detectable Brp puncta was quantified in 100 µm lengths of segmental nerves by using ImageJ. For the PtdIns(4,5)P2 levels, detectable puncta in 104 µm2 surface areas were quantified. Semi-quantitative analysis of the colocalization was performed and Pearson's overlap coeffcient was obtained with the JACoP plugin for ImageJ (82). Characterization of phenotypes in which elav-Gal4 was used as a driver was performed between gender-matched animals. However, besides the case of the Sac1 neurodegenerative phenotype, no major differences in phenotypes were observed between males and females in any other context in which elav-Gal4 was used as a driver.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Wellcome Trust (http://www.wellcome.ac.uk) (Pennetta8920), the Motor Neurone Disease Association (MNDA, http://www.mndassociation.org.uk) (Pennetta6231) and the Scottish Motor Neurone Disease Association (SMNDA; http://www.mndscotland.org.uk) (Pennetta R4107). Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
Supplementary Material
ACKNOWLEDGEMENTS
We are particularly grateful to Dr Trudy Gillespie for assistance with confocal imaging and to Steven Mitchell for help with scanning electron microscopy analysis. We thank Drs Maria Antonietta De Matteis, Jan Pielage, Richard Ribchester and James Withers for insightful discussions. We are grateful to Luigi Zechini for his technical assistance in the last part of this project. We also thank Dr Jan Pielage, Dr Joaquim Culi and the Developmental Studies Hybridoma Bank for antibodies, the Bloomington Drosophila Stock Center and the Vienna Drosophila RNAi Center for fly stocks.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. doi:10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 2.Nishimura A.L., Mitne-Neto M., Silva H.C.A., Richieri-Costa A., Middleton S., Cascio D., Kok F., Oliveira J.R.M., Gillingwater T., Webb J., et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004;75:822–831. doi: 10.1086/425287. doi:10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H.-J., Anagnostou G., Chai A., Withers J., Morris A., Adhikaree J., Pennetta G., De Belleroche J.S. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2010;285:40266–40281. doi: 10.1074/jbc.M110.161398. doi:10.1074/jbc.M110.161398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lev S., Ben Halevy D., Peretti D., Dahan N. The VAP protein family: from cellular functions to motor neuron disease. Trends Cell Biol. 2008;18:282–290. doi: 10.1016/j.tcb.2008.03.006. doi:10.1016/j.tcb.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Pennetta G., Hiesinger P.R., Fabian-Fine R., Meinertzhagen I.A., Bellen H.J. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron. 2002;35:291–306. doi: 10.1016/s0896-6273(02)00769-9. doi:10.1016/S0896-6273(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 6.Chai A., Withers J., Koh Y.H., Parry K., Bao H., Zhang B., Budnik V., Pennetta G. hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum. Mol. Genet. 2008;17:266–280. doi: 10.1093/hmg/ddm303. doi:10.1093/hmg/ddm303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsuda H., Han S.M., Yang Y., Tong C., Lin Y.Q., Mohan K., Haueter C., Zoghbi A., Harati Y., Kwan J., et al. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell. 2008;133:963–977. doi: 10.1016/j.cell.2008.04.039. doi:10.1016/j.cell.2008.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han S.M., Tsuda H., Yang Y., Vibbert J., Cottee P., Lee S.-J., Winek J., Haueter C., Bellen H.J., Miller M.A. Secreted VAPB/ALS8 major sperm protein domains modulate mitochondrial localization and morphology via growth cone guidance receptors. Dev. Cell. 2012;22:348–362. doi: 10.1016/j.devcel.2011.12.009. doi:10.1016/j.devcel.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ratnaparkhi A., Lawless G.M., Schweizer F.E., Golshani P., Jackson G.R. A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE. 2008;3:e2334. doi: 10.1371/journal.pone.0002334. doi:10.1371/journal.pone.0002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanekura K., Nishimoto I., Aiso S., Matsuoka M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8) J. Biol. Chem. 2006;281:30223–30233. doi: 10.1074/jbc.M605049200. doi:10.1074/jbc.M605049200. [DOI] [PubMed] [Google Scholar]
- 11.Langou K., Moumen A., Pellegrino C., Aebischer J., Medina I., Aebischer P., Raoul C. AAV-mediated expression of wild-type and ALS-linked mutant VAPB selectively triggers death of motoneurons through a Ca2+-dependent ER-associated pathway. J. Neurochem. 2010;114:795–809. doi: 10.1111/j.1471-4159.2010.06806.x. doi:10.1111/j.1471-4159.2010.06806.x. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki H., Kanekura K., Levine T.P., Kohno K., Olkkonen V.M., Aiso S., Matsuoka M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 2009;108:973–985. doi: 10.1111/j.1471-4159.2008.05857.x. doi:10.1111/j.1471-4159.2008.05857.x. [DOI] [PubMed] [Google Scholar]
- 13.Gkogkas C., Middleton S., Kremer A.M., Wardrope C., Hannah M., Gillingwater T.H., Skehel P. VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 2008;17:1517–1526. doi: 10.1093/hmg/ddn040. doi:10.1093/hmg/ddn040. [DOI] [PubMed] [Google Scholar]
- 14.Mórotz G.M., De Vos K.J., Vagnoni A., Ackerley S., Shaw C.E., Miller C.C.J. Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 2012;21:1979–1988. doi: 10.1093/hmg/dds011. doi:10.1093/hmg/dds011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Paolo G., De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. doi:10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 16.Guo S., Stolz L.E., Lemrow S.M., York J.D. SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J. Biol. Chem. 1999;274:12990–12995. doi: 10.1074/jbc.274.19.12990. doi:10.1074/jbc.274.19.12990. [DOI] [PubMed] [Google Scholar]
- 17.Foti M., Audhya A., Emr S.D. Sac1 lipid phosphatase and Stt4 phosphatidylinositol 4-kinase regulate a pool of phosphatidylinositol 4-phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol. Biol. Cell. 2001;12:2396–2411. doi: 10.1091/mbc.12.8.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brice S.E., Alford C.W., Cowart L.A. Modulation of sphingolipid metabolism by the phosphatidylinositol-4-phosphate phosphatase Sac1p through regulation of phosphatidylinositol in Saccharomyces cerevisiae. J. Biol. Chem. 2009;284:7588–7596. doi: 10.1074/jbc.M808325200. doi:10.1074/jbc.M808325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei H.-C., Sanny J., Shu H., Baillie D.L., Brill J.A., Price J.V., Harden N. The Sac1 lipid phosphatase regulates cell shape change and the JNK cascade during dorsal closure in Drosophila. Curr. Biol. 2003;13:1882–1887. doi: 10.1016/j.cub.2003.09.056. doi:10.1016/j.cub.2003.09.056. [DOI] [PubMed] [Google Scholar]
- 20.Lee S., Kim S., Nahm M., Kim E., Kim T.-I., Yoon J.H., Lee S. The phosphoinositide phosphatase Sac1 is required for midline axon guidance. Mol. Cell. 2011;32:477–482. doi: 10.1007/s10059-011-0168-6. doi:10.1007/s10059-011-0168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y., Boukhelifa M., Tribble E., Morin-Kensicki E., Uetrecht A., Bear J.E., Bankaitis V.A. The Sac1 phosphoinositide phosphatase regulates Golgi membrane morphology and mitotic spindle organization in mammals. Mol. Biol. Cell. 2008;19:3080–3096. doi: 10.1091/mbc.E07-12-1290. doi:10.1091/mbc.E07-12-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chow C.Y., Landers J.E., Bergren S.K., Sapp P.C., Grant A.E., Jones J.M., Everett L., Lenk G.M., McKenna-Yasek D.M., Weisman L.S., et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009;84:85–88. doi: 10.1016/j.ajhg.2008.12.010. doi:10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow C.Y., Zhang Y., Dowling J.J., Jin N., Adamska M., Shiga K., Szigeti K., Shy M.E., Li J., Zhang X., et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. doi:10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giot L., Bader J.S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y.L., Ooi C.E., Godwin B., Vitols E., et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. doi:10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 25.Roos J., Hummel T., Ng N., Klämbt C., Davis G.W. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/s0896-6273(00)81170-8. doi:10.1016/S0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- 26.Sherwood N.T., Sun Q., Xue M., Zhang B., Zinn K. Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2004;2:e429. doi: 10.1371/journal.pbio.0020429. doi:10.1371/journal.pbio.0020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagh D.A., Rasse T.M., Asan E., Hofbauer A., Schwenkert I., Dürrbeck H., Buchner S., Dabauvalle M.-C., Schmidt M., Qin G., et al. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. doi:10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Pielage J., Bulat V., Zuchero J.B., Fetter R.D., Davis G.W. Hts/Adducin controls synaptic elaboration and elimination. Neuron. 2011;69:1114–1131. doi: 10.1016/j.neuron.2011.02.007. doi:10.1016/j.neuron.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J., Gambhir A., Hangyás-Mihályné G., Murray D., Golebiewska U., McLaughlin S. Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J. Biol. Chem. 2002;277:34401–34412. doi: 10.1074/jbc.M203954200. doi:10.1074/jbc.M203954200. [DOI] [PubMed] [Google Scholar]
- 30.Pielage J., Fetter R.D., Davis G.W. A postsynaptic spectrin scaffold defines active zone size, spacing, and efficacy at the Drosophila neuromuscular junction. J. Cell Biol. 2006;175:491–503. doi: 10.1083/jcb.200607036. doi:10.1083/jcb.200607036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikeda Y., Dick K.A., Weatherspoon M.R., Gincel D., Armbrust K.R., Dalton J.C., Stevanin G., Dürr A., Zühlke C., Bürk K., et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 2006;38:184–190. doi: 10.1038/ng1728. doi:10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- 32.Wang S., Yang J., Tsai A., Kuca T., Sanny J., Lee J., Dong K., Harden N., Krieger C. Drosophila adducin regulates Dlg phosphorylation and targeting of Dlg to the synapse and epithelial membrane. Dev. Biol. 2011;357:392–403. doi: 10.1016/j.ydbio.2011.07.010. doi:10.1016/j.ydbio.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 33.Featherstone D.E., Davis W.S., Dubreuil R.R., Broadie K. Drosophila alpha- and beta-spectrin mutations disrupt presynaptic neurotransmitter release. J. Neurosci. 2001;21:4215–4224. doi: 10.1523/JNEUROSCI.21-12-04215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz-Canada C., Ashley J., Moeckel-Cole S., Drier E., Yin J., Budnik V. New synaptic bouton formation is disrupted by misregulation of microtubule stability in aPKC mutants. Neuron. 2004;42:567–580. doi: 10.1016/s0896-6273(04)00255-7. doi:10.1016/S0896-6273(04)00255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitters E.A., Cleves A.E., McGee T.P., Skinner H.B., Bankaitis V.A. SAC1p is an integral membrane protein that influences the cellular requirement for phospholipid transfer protein function and inositol in yeast. J. Cell Biol. 1993;122:79–94. doi: 10.1083/jcb.122.1.79. doi:10.1083/jcb.122.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nemoto Y., Kearns B.G., Wenk M.R., Chen H., Mori K., Alb J.G., Jr, De Camilli P., Bankaitis V.A. Functional characterization of a mammalian Sac1 and mutants exhibiting substrate-specific defects in phosphoinositide phosphatase activity. J. Biol. Chem. 2000;275:34293–34305. doi: 10.1074/jbc.M003923200. doi:10.1074/jbc.M003923200. [DOI] [PubMed] [Google Scholar]
- 37.Culi J., Mann R.S. Boca, an endoplasmic reticulum protein required for wingless signaling and trafficking of LDL receptor family members in Drosophila. Cell. 2003;112:343–354. doi: 10.1016/s0092-8674(02)01279-5. doi:10.1016/S0092-8674(02)01279-5. [DOI] [PubMed] [Google Scholar]
- 38.Piao H., Mayinger P. Growth and metabolic control of lipid signalling at the Golgi. Biochem. Soc. Trans. 2012;40:205–209. doi: 10.1042/BST20110637. doi:10.1042/BST20110637. [DOI] [PubMed] [Google Scholar]
- 39.Stefan C.J., Manford A.G., Baird D., Yamada-Hanff J., Mao Y., Emr S.D. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell. 2011;144:389–401. doi: 10.1016/j.cell.2010.12.034. doi:10.1016/j.cell.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 40.Warrick J.M., Chan H.Y., Gray-Board G.L., Chai Y., Paulson H.L., Bonini N.M. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 1999;23:425–428. doi: 10.1038/70532. doi:10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 41.Yavari A., Nagaraj R., Owusu-Ansah E., Folick A., Ngo K., Hillman T., Call G., Rohatgi R., Scott M.P., Banerjee U. Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev. Cell. 2010;19:54–65. doi: 10.1016/j.devcel.2010.06.007. doi:10.1016/j.devcel.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panaretou C., Domin J., Cockcroft S., Waterfield M.D. Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 3-kinase. Substrate presentation by phosphatidylinositol transfer protein to the p150.Ptdins 3-kinase complex. J. Biol. Chem. 1997;272:2477–2485. doi: 10.1074/jbc.272.4.2477. doi:10.1074/jbc.272.4.2477. [DOI] [PubMed] [Google Scholar]
- 43.Jones S.M., Alb J.G., Jr, Phillips S.E., Bankaitis V.A., Howell K.E. A phosphatidylinositol 3-kinase and phosphatidylinositol transfer protein act synergistically in formation of constitutive transport vesicles from the trans-Golgi network. J. Biol. Chem. 1998;273:10349–10354. doi: 10.1074/jbc.273.17.10349. doi:10.1074/jbc.273.17.10349. [DOI] [PubMed] [Google Scholar]
- 44.Kagiwada S., Hashimoto M. The yeast VAP homolog Scs2p has a phosphoinositide-binding ability that is correlated with its activity. Biochem. Biophys. Res. Commun. 2007;364:870–876. doi: 10.1016/j.bbrc.2007.10.079. doi:10.1016/j.bbrc.2007.10.079. [DOI] [PubMed] [Google Scholar]
- 45.Manford A.G., Stefan C.J., Yuan H.L., Macgurn J.A., Emr S.D. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell. 2012;23:1129–1140. doi: 10.1016/j.devcel.2012.11.004. doi:10.1016/j.devcel.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 46.Van Blitterswijk M., Van Es M.A., Koppers M., Van Rheenen W., Medic J., Schelhaas H.J., Van der Kooi A.J., De Visser M., Veldink J.H., Van den Berg L.H. VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol. Aging. 2012;33:2950.e1–2950.e4. doi: 10.1016/j.neurobiolaging.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 47.Faulhammer F., Kanjilal-Kolar S., Knödler A., Lo J., Lee Y., Konrad G., Mayinger P. Growth control of Golgi phosphoinositides by reciprocal localization of sac1 lipid phosphatase and pik1 4-kinase. Traffic. 2007;8:1554–1567. doi: 10.1111/j.1600-0854.2007.00632.x. doi:10.1111/j.1600-0854.2007.00632.x. [DOI] [PubMed] [Google Scholar]
- 48.Blagoveshchenskaya A., Cheong F.Y., Rohde H.M., Glover G., Knödler A., Nicolson T., Boehmelt G., Mayinger P. Integration of Golgi trafficking and growth factor signaling by the lipid phosphatase SAC1. J. Cell Biol. 2008;180:803–812. doi: 10.1083/jcb.200708109. doi:10.1083/jcb.200708109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim E.K., Choi E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. doi:10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 50.Toulmay A., Prinz W.A. Lipid transfer and signaling at organelle contact sites: the tip of the iceberg. Curr. Opin. Cell Biol. 2011;23:458–463. doi: 10.1016/j.ceb.2011.04.006. doi:10.1016/j.ceb.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayashi M., Raimondi A., O'Toole E., Paradise S., Collesi C., Cremona O., Ferguson S.M., De Camilli P. Cell- and stimulus-dependent heterogeneity of synaptic vesicle endocytic recycling mechanisms revealed by studies of dynamin 1-null neurons. Proc. Natl Acad. Sci. USA. 2008;105:2175–2180. doi: 10.1073/pnas.0712171105. doi:10.1073/pnas.0712171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suzuki E., Hirosawa K. Immunolocalization of a Drosophila phosphatidylinositol transfer protein (rdgB) in normal and rdgA mutant photoreceptor cells with special reference to the subrhabdomeric cisternae. J. Electron Microsc. (Tokyo) 1994;43:183–189. [PubMed] [Google Scholar]
- 53.Peretti D., Dahan N., Shimoni E., Hirschberg K., Lev S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell. 2008;19:3871–3884. doi: 10.1091/mbc.E08-05-0498. doi:10.1091/mbc.E08-05-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rocha N., Kuijl C., Van der Kant R., Janssen L., Houben D., Janssen H., Zwart W., Neefjes J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol. 2009;185:1209–1225. doi: 10.1083/jcb.200811005. doi:10.1083/jcb.200811005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Vos K.J., Mórotz G.M., Stoica R., Tudor E.L., Lau K.-F., Ackerley S., Warley A., Shaw C.E., Miller C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012;21:1299–1311. doi: 10.1093/hmg/ddr559. doi:10.1093/hmg/ddr559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Audhya A., Foti M., Emr S.D. Distinct roles for the yeast phosphatidylinositol 4-kinases, Stt4p and Pik1p, in secretion, cell growth, and organelle membrane dynamics. Mol. Biol. Cell. 2000;11:2673–2689. doi: 10.1091/mbc.11.8.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Audhya A., Emr S.D. Stt4 PI 4-kinase localizes to the plasma membrane and functions in the Pkc1-mediated MAP kinase cascade. Dev. Cell. 2002;2:593–605. doi: 10.1016/s1534-5807(02)00168-5. doi:10.1016/S1534-5807(02)00168-5. [DOI] [PubMed] [Google Scholar]
- 58.Strahl T., Hama H., DeWald D.B., Thorner J. Yeast phosphatidylinositol 4-kinase, Pik1, has essential roles at the Golgi and in the nucleus. J. Cell Biol. 2005;171:967–979. doi: 10.1083/jcb.200504104. doi:10.1083/jcb.200504104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walch-Solimena C., Novick P. The yeast phosphatidylinositol-4-OH kinase pik1 regulates secretion at the Golgi. Nat. Cell Biol. 1999;1:523–525. doi: 10.1038/70319. doi:10.1038/70319. [DOI] [PubMed] [Google Scholar]
- 60.Garrenton L.S., Stefan C.J., McMurray M.A., Emr S.D., Thorner J. Pheromone-induced anisotropy in yeast plasma membrane phosphatidylinositol-4,5-bisphosphate distribution is required for MAPK signaling. Proc. Natl Acad. Sci. USA. 2010;107:11805–11810. doi: 10.1073/pnas.1005817107. doi:10.1073/pnas.1005817107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cappell S.D., Dohlman H.G. Selective regulation of MAP kinase signaling by an endomembrane phosphatidylinositol 4-kinase. J. Biol. Chem. 2011;286:14852–14860. doi: 10.1074/jbc.M110.195073. doi:10.1074/jbc.M110.195073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yan Y., Denef N., Tang C., Schüpbach T. Drosophila PI4KIIIalpha is required in follicle cells for oocyte polarization and Hippo signaling. Development. 2011;138:1697–1703. doi: 10.1242/dev.059279. doi:10.1242/dev.059279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brill J.A., Hime G.R., Scharer-Schuksz M., Fuller M.T. A phospholipid kinase regulates actin organization and intercellular bridge formation during germline cytokinesis. Development. 2000;127:3855–3864. doi: 10.1242/dev.127.17.3855. [DOI] [PubMed] [Google Scholar]
- 64.Polevoy G., Wei H.-C., Wong R., Szentpetery Z., Kim Y.J., Goldbach P., Steinbach S.K., Balla T., Brill J.A. Dual roles for the Drosophila PI 4-kinase four wheel drive in localizing Rab11 during cytokinesis. J. Cell Biol. 2009;187:847–858. doi: 10.1083/jcb.200908107. doi:10.1083/jcb.200908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flanagan C.A., Schnieders E.A., Emerick A.W., Kunisawa R., Admon A., Thorner J. Phosphatidylinositol 4-kinase: gene structure and requirement for yeast cell viability. Science. 1993;262:1444–1448. doi: 10.1126/science.8248783. doi:10.1126/science.8248783. [DOI] [PubMed] [Google Scholar]
- 66.Yoshida S., Ohya Y., Goebl M., Nakano A., Anraku Y. A novel gene, STT4, encodes a phosphatidylinositol 4-kinase in the PKC1 protein kinase pathway of Saccharomyces cerevisiae. J. Biol. Chem. 1994;269:1166–1172. [PubMed] [Google Scholar]
- 67.Graham T.R., Burd C.G. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 2011;21:113–121. doi: 10.1016/j.tcb.2010.10.002. doi:10.1016/j.tcb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Golub T., Caroni P. PI(4,5)P2-dependent microdomain assemblies capture microtubules to promote and control leading edge motility. J. Cell Biol. 2005;169:151–165. doi: 10.1083/jcb.200407058. doi:10.1083/jcb.200407058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klopfenstein D.R., Vale R.D. The lipid binding pleckstrin homology domain in UNC-104 kinesin is necessary for synaptic vesicle transport in Caenorhabditis elegans. Mol. Biol. Cell. 2004;15:3729–3739. doi: 10.1091/mbc.E04-04-0326. doi:10.1091/mbc.E04-04-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakatsu F., Baskin J.M., Chung J., Tanner L.B., Shui G., Lee S.Y., Pirruccello M., Hao M., Ingolia N.T., Wenk M.R., et al. PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol. 2012;199:1003–1016. doi: 10.1083/jcb.201206095. doi:10.1083/jcb.201206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hammond G.R.V., Fischer M.J., Anderson K.E., Holdich J., Koteci A., Balla T., Irvine R.F. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science. 2012;337:727–730. doi: 10.1126/science.1222483. doi:10.1126/science.1222483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verstreken P., Koh T.-W., Schulze K.L., Zhai R.G., Hiesinger P.R., Zhou Y., Mehta S.Q., Cao Y., Roos J., Bellen H.J. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron. 2003;40:733–748. doi: 10.1016/s0896-6273(03)00644-5. doi:10.1016/S0896-6273(03)00644-5. [DOI] [PubMed] [Google Scholar]
- 73.Breslow D.K., Weissman J.S. Membranes in balance: mechanisms of sphingolipid homeostasis. Mol. Cell. 2010;40:267–279. doi: 10.1016/j.molcel.2010.10.005. doi:10.1016/j.molcel.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perry R.J., Ridgway N.D. Oxysterol-binding protein and vesicle-associated membrane protein-associated protein are required for sterol-dependent activation of the ceramide transport protein. Mol. Biol. Cell. 2006;17:2604–2616. doi: 10.1091/mbc.E06-01-0060. doi:10.1091/mbc.E06-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hanada K. Discovery of the molecular machinery CERT for endoplasmic reticulum-to-Golgi trafficking of ceramide. Mol. Cell. Biochem. 2006;286:23–31. doi: 10.1007/s11010-005-9044-z. doi:10.1007/s11010-005-9044-z. [DOI] [PubMed] [Google Scholar]
- 76.Raychaudhuri S., Prinz W.A. The diverse functions of oxysterol-binding proteins. Annu. Rev. Cell Dev. Biol. 2010;26:157–177. doi: 10.1146/annurev.cellbio.042308.113334. doi:10.1146/annurev.cellbio.042308.113334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Trajkovic K., Hsu C., Chiantia S., Rajendran L., Wenzel D., Wieland F., Schwille P., Brügger B., Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. doi:10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 78.Zito K., Parnas D., Fetter R.D., Isacoff E.Y., Goodman C.S. Watching a synapse grow: noninvasive confocal imaging of synaptic growth in Drosophila. Neuron. 1999;22:719–729. doi: 10.1016/s0896-6273(00)80731-x. doi:10.1016/S0896-6273(00)80731-X. [DOI] [PubMed] [Google Scholar]
- 79.Taha T.A., Mullen T.D., Obeid L.M. A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta. 2006;1758:2027–2036. doi: 10.1016/j.bbamem.2006.10.018. doi:10.1016/j.bbamem.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang K., Yang Z., Liu X., Mao K., Nair U., Klionsky D.J. Phosphatidylinositol 4-kinases are required for autophagic membrane trafficking. J. Biol. Chem. 2012;287:37964–37972. doi: 10.1074/jbc.M112.371591. doi:10.1074/jbc.M112.371591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kawano M., Kumagai K., Nishijima M., Hanada K. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 2006;281:30279–30288. doi: 10.1074/jbc.M605032200. doi:10.1074/jbc.M605032200. [DOI] [PubMed] [Google Scholar]
- 82.Bolte S., Cordelières F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. doi:10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.