

Abstract
The Duchenne and Becker forms of muscular dystrophy are associated with dilated cardiomyopathy and are diseases in which pulmonary function peaks, then progressively declines. In this report, we quantify cardiopulmonary function variability among brothers. Brothers in 3 of 7 eligible sibships had discordant pulmonary function, with significant differences between the brothers' peak forced vital capacities and their vital capacities at last comparable age. There was no relationship between pulmonary and cardiac function among the siblings. We concluded that despite identical genetic mutations, cardiac and pulmonary function variability was common among brothers in our clinic with Duchenne or Becker muscular dystrophy. If confirmed by larger studies, these results have negative implications for use of genetic testing to predict cardiopulmonary course and response to therapies in Duchenne or Becker muscular dystrophy.
Keywords: Becker muscular dystrophy, cardiomyopathy, Duchenne muscular dystrophy, genetics, genotype, muscular dystrophy, phenotype, pulmonary function
Introduction
The Duchenne and Becker forms of muscular dystrophy are X-linked dystrophinopathies caused by mutations in the gene encoding dystrophin, a protein essential for normal muscle function. These diseases cause pulmonary function to plateau, then predictably decline. Moreover, the association between the Duchenne and Becker forms of muscular dystrophy and dilated cardiomyopathy is well-described.1 We have observed variability of cardiac and pulmonary function among brothers with these muscular dystrophies and identical genetic mutations. In this pilot study, we assessed the prevalence of variability in cardiopulmonary function among brothers with the Duchenne or Becker forms of muscular dystrophy in eligible sibships in our clinic. The clinical implications of such variability would be significant, including the potential inability of genetic test results to predict respiratory and cardiac course, prognosis, and response to therapies in patients with Duchenne or Becker muscular dystrophy. Our study was approved by the hospital's institutional review board.
Patients and Methods
A retrospective chart review was done, covering the time period January 1992 through March 2009. The diagnosis of Duchenne or Becker muscular dystrophy was based on clinical presentation, elevated serum creatine phosphokinase levels, and genetic testing result. We identified sibships consisting of biological brothers with Duchenne or Becker muscular dystrophy and reviewed their echocardiograms and pulmonary function test results. At least one member of each sibship had genetic testing results that showed a known mutation of the gene coding for dystrophin (Table 1). Patients were excluded if they had received glucocorticoid therapy, if they had non-neuromuscular pulmonary diagnoses such as asthma, if cardiac or pulmonary function data was inadequate due to lack of follow-up in our clinic, or if the brothers were raised in separate households.
Table 1.
Peak FVC and Genetic Data
| Sibship Number | Patient | Exons Involved | Age at Peak FVC (y) | Peak FVC Level (L) |
|---|---|---|---|---|
| Discordant Sibships | ||||
| 1 | 1 | 51 | 13.42 | 1.97 |
| 2 | 14.17 | 2.88 | ||
| 3 | 14.5 | 2.71 | ||
| 2 | 1 | 44 | 17 | 3.62 |
| 2 | 11.33 | 2.67 | ||
| 3 | 1 | 31 | 18.33 | 3.38 |
| 2 | 18.17 | 4.6 | ||
| Concordant Sibships | ||||
| 4 | 1 | 45–50 | 14.67 | 2.48 |
| 2 | 15.08 | 2.81 | ||
| 5 | 1 | 52 | 11.08 | 1.5 |
| 2 | 13.08 | 2.04 | ||
| 6 | 1 | 44 | 12.42 | 1.4 |
| 2 | 12.83 | 1.44 | ||
| 7 | 1 | 56 | 14 | 2.02 |
| 2 | 14.5 | 1.67 | ||
Mean age at peak FVC +/− S.D. = 14.31 +/− 2.18 years
Abbreviation: FVC, forced vital capacity.
To characterize the patients' pulmonary courses, we identified each one's peak forced vital capacity level and the percent predicted forced vital capacity levels at the last comparable age of each fraternal pair (see Figure 1). (At last comparable age, we used percent predicted forced vital capacity rather than absolute value of forced vital capacity to minimize variability attributable to patient height. Arm span estimates of height were used to determine predicted forced vital capacity in all non-ambulatory patients and all tests were done by the same technician using standard validation criteria.) Visual comparison of the peak forced vital capacity levels and subsequent pulmonary course allowed us to classify the sibships into 2 groups: brothers who had similar or concordant pulmonary function and brothers who had dissimilar or discordant pulmonary function (see Figure 1 for an example of brothers with discordant pulmonary function). To test the validity of our categorizations, we assessed if the “concordant” and “discordant” groups really had significantly different pulmonary function by comparing the mean difference in peak forced vital capacity levels and the mean difference in percent predicted forced vital capacity levels at last comparable age, between the fraternal pairs in the concordant group versus those in the discordant group. The mean forced vital capacity differences between the 2 groups were assessed for statistical significance via 2-tailed t test.
Figure 1.
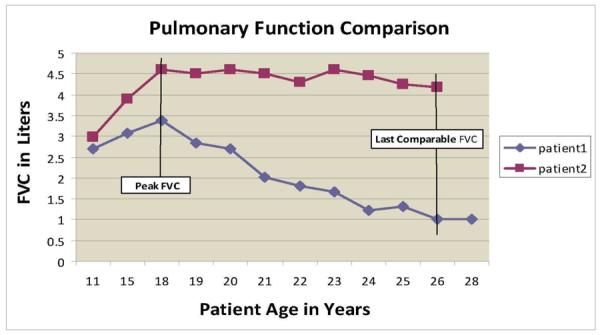
Forced vital capacity with aging in 2 brothers with discordant pulmonary function.
To assess cardiac function, the echocardiographic results for the patients were reviewed retrospectively. All the echocardiograms of each patient (most were performed each year) were reassessed by one of the co-authors (RCB), who was blinded to the patient's identifying information and to the pulmonary function data. The left ventricular internal dimension at end-diastole, or LVIDd, and the left ventricular internal diameter at end-systole, or LVIDs, were measured from the parasternal long axis view. Three cardiac cycles were measured for each study and the mean values were used to calculate the left ventricular shortening fraction, expressed as a percent: {[LVIDd − LVIDs/ LVIDd] × 100}.
Normal shortening fraction was defined as > 28%. Shortening fraction rather than ejection fraction was chosen for the serial observations because the quality of apical images in Duchenne or Becker muscular dystrophy patients markedly deteriorates with increasing age due to loss of intercostal windows. Furthermore, shortening fraction is not altered by age or body size. Shortening fraction at the brothers' oldest comparable age was compared with classify cardiac function as concordant or discordant within each sibship.
Results
Among the 97 patients with Duchenne or Becker muscular dystrophy in our clinic, we actively followed 9 sibships in our clinic. One sibship had to be excluded from both the pulmonary and cardiac function comparison because of steroid therapy, leaving 8 sibships. Two of the 8 sibships had to be excluded only from the cardiac function comparison because of inadequate echocardiographic data, leaving 6 eligible sibships for the cardiac portion of the study. One of the 8 sibships had to be excluded from the pulmonary function comparison because of inadequate forced vital capacity data, leaving 7 eligible sibships for the pulmonary portion of the study. No patients needed to be excluded because of non-neuromuscular respiratory illnesses or residence in separate households.
Pulmonary Function Comparisons
Of the 7 sibships eligible for pulmonary function comparison, 6 consisted of 2 brothers and one sibship had 3 brothers, 2 of whom had nearly identical pulmonary function. The mean values of peak and last comparable forced vital capacity were used to represent the 2 brothers with similar pulmonary function in the latter sibship, thereby creating 7 fraternal pairs from a total of 15 brothers. Each patient reached his peak forced vital capacity level before major respiratory illness, the onset of clinically significant cardiomyopathy, or the onset of noninvasive ventilation.
Peak pulmonary function occurred at mean age (+/− SD) 14.31 +/− 2.18 years (n = 15) (Table 1). The brothers in 4 sibships were found to have similar peak and last comparable forced vital capacity values, and these brothers were identified as having “concordant pulmonary function.” The brothers in 3 sibships were found to have disparate forced vital capacity values and these brothers were identified as having “discordant pulmonary function.” In the 4 sibships with concordant pulmonary function, the mean peak forced vital capacity difference between brothers was 0.32 +/− 0.21 L. In the 3 sibships with discordant pulmonary function, the mean peak forced vital capacity difference between brothers was 1.00 +/− 0.20 L (P < .01) (Table 2).
Table 2.
Comparison of Mean Peak FVC Differences Between Brothers by Group
| Sibship Number | FVC Difference (L) |
|---|---|
| Discordant Sibships | |
| 1 | 0.83 |
| 2 | 0.95 |
| 3 | 1.22 |
| Mean= 1.00+/−0.20 L | |
| Concordant Sibships | |
| 4 | 0.33 |
| 5 | 0.54 |
| 6 | 0.04 |
| 7 | 0.35 |
| Mean = 0.32+/−0.21 L* |
P < .01 concordant vs. discordant sibships
Abbreviation: FVC, forced vital capacity.
In the group with discordant pulmonary function, the brother with the highest peak forced vital capacity in each fraternal pair always maintained a superior forced vital capacity with aging. Mean difference in the percent predicted forced vital capacity between the brothers at their oldest comparable age was 28.5 +/− 16% in the discordant group and 5.3 +/− 4.4% in the concordant group (P < .04), attained at mean age 19.2 +/− 3.8 years (Table 3). Illustrative results for 2 brothers with pulmonary function discordance that is maintained over time are shown in Figure 1.
Table 3.
Comparision of Difference in % Predicted FVC Between Brother at Oldest Comparable Age by Group
| Sibship Number | Difference in % Predicted FVC | Age at Comparison |
|---|---|---|
| Discordant Sibships | ||
| 1 | 18.5 | 18.5 |
| 2 | 20 | 17.0 |
| 3 | 47 | 26.2 |
| Mean +/− SD = 28.5 +/−16% | ||
| Concordant Sibships | ||
| 4 | 10 | 16.2 |
| 5 | 1 | 21.7 |
| 6 | 2 | 14.9 |
| 7 | 7 | 19.7 |
| Mean +/− SD = 5.3 +/− 4.4%* |
P < .04 concordant vs. discordant sibships.
Abbreviations: FVC, forced vital capacity; SD, standard deviation.
Cardiac Function Comparisons
Of the 6 sibships eligible for cardiac function comparisons, 5 consisted of 2 brothers and one sibship had 3 brothers. Cardiac function was compared at a mean age of 20.6 years (range, 15–27 years). In Tables 4 and 5, which contain the cardiac function results, the numbering of the sibships and patients is identical to that in Tables 1 through 3, in which the pulmonary function results are presented. Both brothers in 3 of the 6 eligible sibships were found to have similarly impaired shortening fraction and these brothers were identified as having concordant cardiac function (Table 4). The brothers in the other 3 sibships were found to have disparate cardiac impairment; ie, only one of the brothers had an impaired shortening fraction, and these brothers were identified as having discordant cardiac function (Table 4).
Table 4.
Cardiomyopathy and Genetic Data
Table 5.
Cardiac Function versus Pulmonary Function
| Sibship Number | Patient | Cardiac Function | Pulmonary Function | Shortening Fraction (%) |
|---|---|---|---|---|
| Discordant Sibships by Cardiac Function | ||||
| 1 ** | 1 | Best | Worst | 30 |
| 2 | Best | Best | 36 | |
| 3 | Worst | Best | 21* | |
| 2 | 1 | Worst | Best | 16* |
| 2 | Best | Worst | 36 | |
| 7 | 1 | Best | Poor | 31 |
| 2 | Worst | Poor | 8* | |
| Concordant Sibships by Cardiac Function | ||||
| 4 | 1 | Poor | Better | 16* |
| 2 | Poor | Better | 22* | |
| 5 | 1 | Poor | Poor | 11* |
| 2 | Poor | Poor | 15* | |
| 8 | 1 | Poor | Poor | 28* |
| 2 | Poor | Poor | 25* | |
Therapy with an angiotensin-converting enzyme inhibitor was started only after the initial evidence of left ventricular dysfunction. Thus, this therapy was given only to those brothers who had dilated cardiomyopathy in the discordant group and to all patients in the concordant group. Differences in shortening fraction between brothers in the discordant sibships at oldest comparable age ranged from 9% to 23%, while the differences in shortening fraction between brothers in the concordant sibships ranged from only 3% to 6%. There was no apparent relationship between the particular exon(s) involved in a sibship's dystrophin mutation and the classification of that sibship as concordant or discordant for cardiac function.
Cardiopulmonary Correlations
There was little relationship between the cardiac and pulmonary function demonstrated by the brothers within the sibships (Table 5). Among the sibships concordant for cardiac function, only the brothers in sibships 5 and 8 showed parallel alterations in their cardiac and pulmonary function (ie, both brothers in each sibship had poor pulmonary function and poor cardiac function). In contrast, in 2 of the sibships discordant for cardiac function, the brother with best cardiac function had the worst pulmonary function and the brother with best pulmonary function had the worst cardiac function; but in the third sibship discordant for cardiac function, both brothers had similarly poor pulmonary function.
Discussion
Our report has numerous implications relevant to the care of patients with dystrophinopathy. At a basic level, the report illustrates how forced vital capacity can be used as a marker of respiratory muscle strength, and how serial forced vital capacity measurements can give important information about disease course.2 In contrast to formal muscle strength testing,3 forced vital capacity is easily measured and is widely available, making it an important part of the routine assessment of patients with neuromuscular diseases.4 Our report also illustrates how pulmonary and cardiac function can be highly discordant in any given patient, meaning, for example, that regular echocardiography is an important aspect of the clinical evaluation, even in patients whose pulmonary function is well-preserved.
The lack of consistent genotype-cardiopulmonary phenotype correlations manifested by the patients in our report shows that prediction of a Duchenne or Becker muscular dystrophy patient's cardiac or respiratory course and prognosis from the genotype might not be possible (illustrated in Figure 1 and Tables 1 and 4). In addition to prognostic uncertainty, the absence of consistent genotype-phenotype correlations has other interesting implications. Pulmonary function test results may be used as a primary outcome measure when evaluating therapies for patients with neuromuscular diseases (for example, assisted ventilation or enzyme replacement therapy for Pompe disease)5,6 and small improvements in pulmonary function may be viewed as evidence of therapeutic efficacy. Our report illustrates that large differences in both pulmonary and cardiac function can occur among patients with dystrophinopathy even when they have identical genetic mutations. Thus, differences in pulmonary or cardiac function that might be attributed to therapeutic interventions may instead be due to phenotypic diversity, and the evaluation of therapies begun in early childhood, such as glucocorticoids or genetic replacement therapies,7 may be confounded by variability of pulmonary and cardiac function affecting the natural history of individual patients.
Similarly, our results suggest the benefit of genetic and/or specific cardiac therapies with respect to protecting patients from dilated cardiomyopathy must be viewed in the context of phenotypic variability in the development of the condition among patients with various dystrophin genotypes. A prior report suggested that dilated cardiomyopathy may be correlated with mutations in certain exons of the dystrophin gene.8 In contrast, our results showed variability of cardiac function among patients with identical mutations of the dystrophin gene (Table 4), suggesting that identification of the exon(s) involved in a specific patient's dystrophin mutation may be a poor predictor of the presence of dilated cardiomyopathy. Not only does our report suggest that variability of cardiac function among patients with a particular dystrophin genotype may be common, it also suggests that in patients with identical mutations, correlations between pulmonary and cardiac function may be unpredictable (Table 5).
It has long been recognized that clinical heterogeneity complicates the evaluation of patients with dystrophinopathy and that evaluation of fraternal sibships can be used to maximize genetic homogeneity when assessing disease severity and prognosis.9 Brothers who manifest substantial intrafamilial clinical diversity have been studied in an attempt to identify the genes that modify the phenotypic expression of the gene encoding dystrophin production.10 Recognizing that phenotypic variability can confound evaluation of therapies for Duchenne muscular dystrophy, a recent report focused on identification of clinical groups, or sub-phenotypes, that could predict clinical course and disease severity with greater accuracy than a patient's genotype.11 Our report supplements this literature by suggesting that variability in cardiac and pulmonary function may be common in patients with dystrophinopathy who share identical genetic mutations.
Our report has significant limitations, chiefly that our sample size is small and the prevalence of genotype-cardiopulmonary phenotype discordance among larger populations of Duchenne or Becker muscular dystrophy patients is unknown. Thus, our results must be viewed as a pilot study or initial report. For example, our observations could be unique to these particular sibships; cardiopulmonary function variability might be characteristic of the particular genetic mutations found in our patients, making generalization of our findings to other Duchenne or Becker muscular dystrophy patients inaccurate. Because of the limitations of our study's retrospective design, it is possible that the variability in pulmonary or cardiac function seen in one or more of our discordant sibships was due to unrecognized causes that had a preferential effect on one sibling, such as illnesses (like pneumonia or cardiac dysfunction due to viral illness) or therapies (like assisted ventilation or inconsistencies of non-cardiac medication use among the siblings).
However, no major illnesses were retrospectively identified that affected just one member of the sibships. Moreover, our methodology attempted to control for potential confounders by including in our core definition of pulmonary function concordance and discordance the differences in the siblings' peak forced vital capacity values, which occurred before the onset of clinically significant cardiomyopathy or major respiratory illness, or the initiation of noninvasive ventilation. The fact that observed differences were preserved over time — ie, among discordant sibships, the sibling with the superior peak forced vital capacity always had the superior last comparable forced vital capacity — and given the absence of any retrospectively identifiable environmental factors preferentially affecting the cardiopulmonary function of any individual patient, supports the premise that the cardiopulmonary function variability we observed was indeed due to phenotypic variability. However, these important limitations illustrate that validation of our findings will require a prospective study carried out with a larger number of patients with the Duchenne or Becker forms of muscular dystrophy and identical genetic mutations.
An incidental finding of our study is that among patients with the Duchenne or Becker form of muscular dystrophy who had discordant pulmonary function and identical genetic mutations, patients with superior peak forced vital capacity retained their higher level of pulmonary function with aging (an example of which is shown in Figure 1). This finding indicates that peak forced vital capacity may be a useful predictor of future pulmonary function for some patients with the Duchenne or Becker form of muscular dystrophy. If the predictive value of peak forced vital capacity is confirmed in a larger number of patients with identical genetic mutations, then peak forced vital capacity level could be used to predict respiratory course and to assess response to therapies for some patients with the Duchenne or Becker form of muscular dystrophy.
In summary, cardiac and pulmonary function variability was common among brothers with the Duchenne or Becker form of muscular dystrophy and identical genetic mutations in our clinic, suggesting that genotype-pulmonary function and genotype-left ventricular cardiac function correlations may be unpredictable in these patients, with potentially negative implications for the use of genetic test results to predict cardiac and respiratory course, prognosis, and response to therapies. Peak forced vital capacity level may be a useful predictor of future respiratory course in some patients with the Duchenne or Becker form of muscular dystrophy. A prospective study involving a larger group of these patients with identical genetic mutations will be needed to validate our observations.
Acknowledgments
This study was presented in abstract form at the American College of Chest Physicians annual meeting in San Diego, California, on November 3, 2009, and at the Neurobiology of Disease in Children Symposium: Muscular Dystrophy, in conjunction with the 38th Annual Meeting of the Child Neurology Society, Louisville, Kentucky, October 14, 2009. Supported by grants from the National Institutes of Health (5R13NS040925-09), the National Institutes of Health Office of Rare Diseases Research, the Muscular Dystrophy Association, and the Child Neurology Society. The authors thank Melanie Fridl Ross, MSJ, ELS, for editing this manuscript.
Footnotes
Conflicts of interest: Ms Merrill and Drs Ashwath, Bahler, Birnkrant, Crowe, Noritz, and Shah have no real or potential conflicts of interest to disclose.
References
- 1.Cardamone M, Darras BT, Ryan MM. Inherited myopathies and muscular dystrophies. Semin Neurol. 2008;28(2):250–259. doi: 10.1055/s-2008-1062269. [DOI] [PubMed] [Google Scholar]
- 2.Phillips MF, Quinlivan RC, Edwards RH, Calverley PM. Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am J Repir Crit Care Med. 2001;164:2191–2194. doi: 10.1164/ajrccm.164.12.2103052. [DOI] [PubMed] [Google Scholar]
- 3.Kilmer DD, Abresch RT, Fowler WM., Jr Serial manual muscle testing in Duchenne muscular dystrophy. Arch Phys Med Rehabil. 1993;74:1168–1171. [PubMed] [Google Scholar]
- 4.Finder JD, Birnkrant D, Carl J, et al. Respiratory care of the patient with Duchenne muscular dystrophy: an ATS consensus statement. Am J Respir Crit Care Med. 2004;170:456–465. doi: 10.1164/rccm.200307-885ST. [DOI] [PubMed] [Google Scholar]
- 5.Pellegrini N, Laforet P, Orlikowski D, et al. Respiratory insufficiency and limb muscle weakness in adults with Pompe's disease. Eur Respir J. 2005;26:1024–1031. doi: 10.1183/09031936.05.00020005. [DOI] [PubMed] [Google Scholar]
- 6.van der Ploeg AT. Monitoring of pulmonary function in Pompe disease: a muscle disease with new therapeutic perspectives. Eur Respir J. 2005;26:984–985. doi: 10.1183/09031936.05.00112005. [DOI] [PubMed] [Google Scholar]
- 7.Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 8.Jefferies JL, Eidem BW, Belmont JW, et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112(18):2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- 9.Hyser CL, Province M, Griggs RC, et al. Genetic heterogeneity in Duchenne dystrophy. Ann Neurol. 1987;22:553–555. doi: 10.1002/ana.410220420. [DOI] [PubMed] [Google Scholar]
- 10.Sifringer M, Uhlenberg B, Lammel S, et al. Identification of transcripts from a subtraction library which might be responsible for the mild phenotype in an intrafamilially variable course of Duchenne muscular dystrophy. Hum Genet. 2004;114:149–156. doi: 10.1007/s00439-003-1041-2. [DOI] [PubMed] [Google Scholar]
- 11.Desguerre K, Christov C, Mayer M, et al. Clinical heterogeneity of Duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE. 2009;4:e4347. doi: 10.1371/journal.pone.0004347. [DOI] [PMC free article] [PubMed] [Google Scholar]