

Abstract
Stickler syndrome is a heterogeneous condition due to mutations in COL2A1, COL11A1, COL11A2, and COL9A1. To our knowledge, neither non-penetrance nor mosaicism for COL2A1 mutations has been reported for Stickler syndrome. We report on a family with two clinically affected sibs with Stickler syndrome who have clinically unaffected parents. Both sibs have a novel heterozygous mutation in exon 26 of COL2A1 (c.1525delT); this results in a premature termination codon downstream of the mutation site. One parent was found to have low level mosaicism in DNA extracted from whole blood. This scenario encourages consideration of molecular testing in seemingly unaffected parents for recurrence risks and potential screening for mild age-related manifestations.
Keywords: Stickler syndrome, Mosaicism, COL2A1, Recurrence risk
1. Introduction
Stickler syndrome is a connective tissue disorder with clinical variability. The phenotype can include micrognathia, cleft palate, midface hypoplasia, myopia, cataracts, retinal detachment, hearing loss, hypermobility, and arthropathy [1–8]. Both intra- and inter-familial variable expressivity has been reported [3], but the condition is thought to be fully penetrant.
Stickler syndrome is heterogeneous with mutations reported in COL2A1, COL11A1, COL11A2, COL9A1, and COL9A2 [9–13]. The phenotype can vary based on the causative gene. Autosomal dominant inheritance is observed for COL2A1, COL11A1, and COL11A2, while autosomal recessive inheritance is observed for COL9A1 and COL9A2. Although Stickler syndrome is heterogeneous, the majority of individuals with Stickler syndrome have mutations in COL2A1.
As far as we know there have been no reports of mosaicism for Stickler syndrome published to date. We report on a family with affected sibs who have seemingly unaffected parents; one parent displays mosaicism for the familial mutation.
2. Materials and methods
After voluntary informed consent, a comprehensive physical examination and medical history was obtained on the participants of this family (Fig. 1).
Fig. 1.

Pedigree of family. Black circles/squares indicate clinically affected status for Stickler syndrome.
DNA was extracted from lymphoctyes from peripheral blood in the parents and oldest affected child (I-1, I-2, II-1), and from buccal cheek swabs in the other children (II-2, II-3, II-4). Primers were designed to amplify exon 26 of the COL2A1 gene (GenBank Accession number NM_001844.4). BigDye sequencing chemistry (ABI) was used to sequence the PCR product in both directions using the ABI 3730x1 DNA analyzer (Applied Biosystems). The Mutation Surveyor program (Softgenetics, State College, PA) was used to analyze the sequences.
3. Results
3.1. Clinical reports
The proband (II-1) is a 10-year-old boy initially evaluated at four years of age with concern for Stickler syndrome. Presenting features were a cleft of the soft palate and myopia. Physical examination at four and a half years of age (Fig. 2A) showed mid-face hypoplasia, low nasal bridge, anteverted nares, epicanthal folds, bifid uvula, mild retrognathia, pectus excavatum, and joint hypermobility. His height was 107 cm (≈75th centile) and weight 15.6 kg (≈25th centile). His cleft palate was previously repaired, and he had multiple surgical procedures to correct esotropia. His ophthalmologic evaluation, including an examination under anesthesia, showed significant myopia requiring corrective lenses, but there were no retinal or vitreous abnormalities. He had frequent episodes of otitis media and middle ear effusions requiring pressure equalizing tubes. A skeletal survey performed at ≈3 years of age was reported as showing mild shortening of the ilia, and mild flattening of the epiphyses of the lower extremities, but otherwise the tubular bones were reported as normal. Mutation analysis of the COL2A1 gene was performed clinically and a mutation was identified (c.1525delT). He was re-evaluated at 10 years of age with similar physical manifestations (Fig. 2B). His audiologic findings included a persistent low frequency conductive hearing loss after pressure equalizing tube insertion. He also had a history of obstructive sleep apnea, and bony prominences of the lateral aspect of the knees. His cycloplegic refractions were −6.50 OD and −6.25 OS.
Fig. 2.
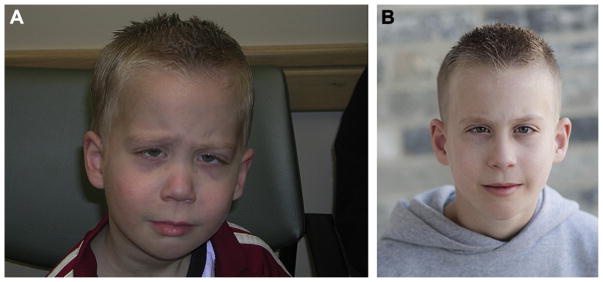
Photograph of face of boy at four years of age (A) and at 10 years of age (B) with physical features of Stickler syndrome with mutation in COL2A1 (c.1525delT).
The proband’s now three-year-old sister (II-4) was initially evaluated at seven months of age (Fig. 3A). She presented with facial features similar to her brother and significant myopia. On physical examination she had midface hypoplasia, low nasal bridge, epicanthal folds, bifid uvula with mild submucous cleft palate, high arched palate, mild retrognathia, and joint hypermobility. Her height was 69 cm (≈75th centile) and weight 7.3 kg (≈25th centile). At three years of age she was re-evaluated (Fig. 3B). She had developed chronic middle ear effusions and a slight high frequency conductive hearing loss. Her ophthalmologic evaluation showed myopia requiring corrective lenses, but there were no retinal or vitreous abnormalities. Her cycloplegic refractions were −3.50 OD and −2.25 OS. Targeted mutation analysis for the same mutation reported in her brother was performed clinically, and the same mutation in COL2A1 (c.1525delT) was identified.
Fig. 3.
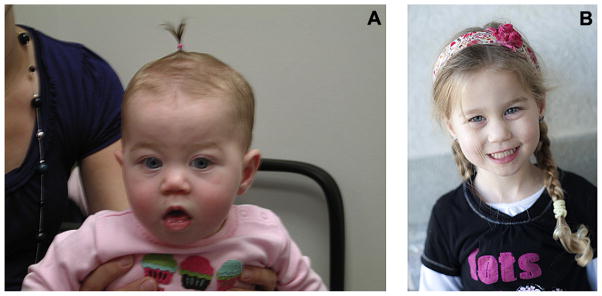
Photograph of face of a girl as an (A) infant, and at (B) three years with physical features of Stickler syndrome with mutation in COL2A1 (c.1525delT).
Both parents (I-1, I-2) (Fig. 4) and two additional sibs (II-2, II-3) were clinically unaffected. Notably, they did not have facial features consistent with Stickler syndrome. Parents report that their own previous ophthalmologic evaluations were normal and neither of them required corrective lenses. Additionally, neither parent reported a history of cleft palate, hearing aids, hypermobility, mitral valve prolapse, or arthritis. The 34-year-old father had a height of 183 cm (≈75th centile) and a weight of 78 kg (≈75th centile). Our subjective impression was that the father’s facial appearance did not show significant micrognathia, retrognathia, or low nasal bridge (Fig. 4).
Fig. 4.

(A) Photograph of clinically unaffected parents of 2 children with Stickler syndrome. (B) Photograph of face of the father who was found to have low level mosaicism for mutation in COL2A1 (c.1525delT) in peripheral blood.
Using the proposed scoring system developed by Hoonaert et al. [5] scores were calculated for each individual based on known clinical features. Hoonaert et al. [5] previously showed that 75% of patients with a COL2A1 mutation had a total score ≥9. Both parents (I-1, I-2) and unaffected sib (II-2, II-3) had scores of 5 based on positive family history. Both the proband (II-1) and affected sib (II-4) had scores of 11.
3.2. COL2A1 sequencing
In our research laboratory the novel heterozygous deletion in exon 26 of the COL2A1 gene, c.1525delT, resulting in a frame-shift and premature termination codon downstream of the mutation site (p.Ser509fs), was confirmed in both affected sibs (II-1, II-4). This is predicted to result in a null allele consistent with Stickler syndrome. No mutation was identified in the mother or two unaffected sibs (II-2, II-3). The same c.1525delT mutation was detected in the father at a low level in both forward and reverse sequencing directions (Fig. 5). Although this result is not quantitative, it is suggestive of low level mosaicism when the sequence data are compared to the affected individuals.
Fig. 5.

Sanger sequencing results of the family with individuals affected with Stickler syndrome. A. Affected proband (II-1). B. Affected sib (II-4). C. Unaffected sib (II-3). D. Mother (I-2). E. Father (I-1); forward sequence. F. Father (I-1); reverse sequence. The arrow denotes the c.1525delT in exon 26 of the COL2A1 gene.
4. Discussion
We report a father with somatic mosaicism for a clinically significant COL2A1 mutation with two affected offspring. As far as we are aware, mosaicism for Stickler syndrome has not been previously documented. Although clinical variability has been reported [2,3], Stickler syndrome is generally thought to be fully penetrant. Faber et al. [2] reported on an affected individual with a novel COL2A1 gene mutation in which the father and paternal grandmother were considered to be unaffected but had the same mutation. Upon further collection of medical history of the father, Faber et al. [2] found subtle features consistent with Stickler syndrome; however, the paternal grandmother considered herself healthy and declined further evaluation. Importantly, mosaicism was not present in these mildly affected or seemingly unaffected individuals [2]. Therefore, it is possible that in some individuals the phenotype is so mild that affected status is difficult to determine. Although our proband’s father currently has no overt phenotypic manifestations of Stickler syndrome, it is possible that he could develop manifestations over time and hence routine follow up for the manifestations of Stickler syndrome is recommended.
Genotype–phenotype correlations for COL2A1 mutations have been reported [3,6]. As far as we are aware, the mutation in this family (c.1525delT) has not been previously reported or associated with any specific phenotypic expression or severity. We presume that the lack of any obvious phenotypic manifestations in the father is due to a low degree of mosaicism and theoretically higher amount of total functional protein product. It is unknown what degree of mosaicism in various tissues is needed to produce a particular manifestation. We assume that there is at least gonadal mosaicism given that he has two affected children with the same mutation.
When counseling a family with a child who has Stickler syndrome, one should consider both low level somatic and/or germline mosaicism in a parent when discussing recurrence risk. Potentially, the recurrence risk could be as high as 1-in-2 or 50% for each pregnancy. This family’s presentation supports the consideration of parental testing for the known mutation even if parents are phenotypically unaffected. Recurrence risk is still difficult to provide solely on analysis from parental peripheral blood; analysis of gonadal tissue could provide additional information although to our knowledge this is not clinically available. The recurrence risk for Stickler syndrome in phenotypically unaffected parents is likely higher than the background population risk due to the chance of gonadal mosaicism.
Supplementary Material
Acknowledgments
We thank the family for their participation and Austin Stevens for his help in sample collection, the National Center for Research Resources and the National Center for Advancing Translational Sciences National Institutes of Health Grant UL1RR025764, and the Children’s Health Research Center and Clinical Genetics Research Program at the University of Utah. Dr. Stevenson is supported by the Doris Duke Charitable Foundation Clinical Scientist Development Award, and Thrasher Research Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the above granting agencies.
Appendix A. Supplementary material
Supplementary material related to this article can be found online at doi:10.1016/j.ejmg.2012.03.006.
References
- 1.Donoso LA, Edwards AO, Frost AT, Ritter R, Ahmad N, Vrabec T, Rogers J, Meyer D, Parma S. Clinical variability of Stickler syndrome: role of exon 2 of the collagen COL2A1 gene. Surv Ophthalmol. 2003;48:191–203. doi: 10.1016/s0039-6257(02)00460-5. [DOI] [PubMed] [Google Scholar]
- 2.Faber J, Winterpacht A, Zabel B, Gnoinski W, Schinzel A, Steinmann B, Superti-Furga A. Clinical variability of Stickler syndrome with a COL2A1 haploinsufficiency mutation: implications for genetic counseling. J Med Genet. 2000;37:318–320. doi: 10.1136/jmg.37.4.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liberfarb RM, Levy HP, Rose PS, Wilkin DJ, Davis J, Balog JZ, Griffith AJ, Szymko-Bennett YM, Johnston JJ, Francomano CA, Tsilou E, Rubin BI. The Stickler syndrome: genotype/phenotype correlation in 10 families with Stickler syndrome resulting from seven mutations in the type II collagen gene locus COL2A1. Genet Med. 2003;5:21–27. doi: 10.1097/00125817-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Snead MP, Yates JR. Clinical and molecular genetics of Stickler syndrome. J Med Genet. 1999;36:353–359. [PMC free article] [PubMed] [Google Scholar]
- 5.Stickler GB, Belau PG, Farrell FJ, Jones JD, Pugh DG, Steinberg AG, Ward LE. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc. 1965;40:433–455. [PubMed] [Google Scholar]
- 6.Hoornaert K, Vereecke I, Dewinter C, Rosenberg T, Beemer FA, Leroy JG, Bendix L, Björck E, Bonduelle M, Boute O, Cormier-Daire V, et al. Stickler syndrome caused by COL2A1 mutations: genotype–phenotype correlation in a series of 100 patients. Eur J Hum Genet. 2010;18:872–880. doi: 10.1038/ejhg.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richards AJ, McNinch A, Martin H, Oakhill K, Rai H, Waller S, Treacy B, Whittaker J, Meredith S, Poulson A, Snead MP. Stickler syndrome and the vitreous phenotype: mutations in COL2A1 and COL11A1. Hum Mutat. 2010;31:E1461–E1471. doi: 10.1002/humu.21257. [DOI] [PubMed] [Google Scholar]
- 8.Lee KH, Hayward P. Retrospective review of Stickler syndrome patients with cleft palate 1997–2004. ANZ J Surg. 2008;78:764–766. doi: 10.1111/j.1445-2197.2008.04645.x. [DOI] [PubMed] [Google Scholar]
- 9.Baker S, Booth C, Fillman C, Shapiro M, Blair MP, Hyland JC, Ala-Kokko L. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am J Med Genet A. 2011;155A:1668–1672. doi: 10.1002/ajmg.a.34071. [DOI] [PubMed] [Google Scholar]
- 10.Ala-Kokko L, Baldwin CT, Moskowitz RW, Prockop DJ. Single base mutation in the type II procollagen gene (COL2A1) as a cause of primary osteoarthritis associated with a mild chondrodysplasia. Proc Natl Acad Sci U S A. 1990;7:6565–6568. doi: 10.1073/pnas.87.17.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Annunen S, Korkko J, Czarny M, Warman ML, Brunner HG, Kaariainen H, Mulliken JB, Tranebjaerg L, Brooks DG, Cox GF, Cruysberg JR, Curtis MA, Davenport SL, Friedrich CA, Kaitila I, Krawczynski MR, Latos-Bielenska A, Mukai S, Olsen BR, Shinno N, Somer M, Vikkula M, Zlotogora J, Prockop DJ, Ala-Kokko L. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999;65:974–983. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Camp G, Snoeckx RL, Hilgert N, van den Ende J, Fukuoka H, Wagatsuma M, Suzuki H, Smets RM, Vanhoenacker F, Declau F, Van de Heyning P, Usami S. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet. 2006;79:449–457. doi: 10.1086/506478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Der Hout AH, Verling E, Beemer FA, Buys CH, Hofstra RM, Scheffer H. Occurrence of deletion of a COL2A1 allele as the mutation in Stickler syndrome shows that a collagen type II dosage effect underlies this syndrome. Hum Mutat. 2000;20:236. doi: 10.1002/humu.9061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.