

Abstract
During physical exercise and stress, the sympathetic system stimulates cardiac contractility via β-adrenergic receptor activation, resulting in protein kinase A (PKA)-mediated phosphorylation of the cardiac ryanodine receptor, RyR2, at Ser2808. Hyperphosphorylation of RyR2-S2808 has been proposed as a mechanism contributing to arrhythmogenesis and heart failure. However, the role of RyR2 phosphorylation during β-adrenergic stimulation remains controversial. We examined the contribution of RyR2-S2808 phosphorylation to altered excitation–contraction coupling and Ca2+ signaling using an experimental approach at the interface of molecular and cellular levels and a transgenic mouse with ablation of the RyR2-S2808 phosphorylation site (RyR2-S2808A). Experimentally challenging the communication between L-type Ca2+ channels and RyR2 led to a spatiotemporal de-synchronization of RyR2 openings, as visualized using confocal Ca2+ imaging. β-Adrenergic stimulation re-synchronized RyR2s, but less efficiently in RyR2-S2808A than in control cardiomyocytes, as indicated by comprehensive analysis of RyR2 activation. In addition, spontaneous Ca2+ waves in RyR2-S2808A myocytes showed significantly slowed propagation and complete absence of acceleration during β-adrenergic stress, unlike wild type cells. Single channel recordings revealed an attenuation of luminal Ca2+ sensitivity in RyR2-S2808A channels upon addition of PKA. This suggests that phosphorylation of RyR2-S2808 may be involved in RyR2 modulation by luminal (intra-SR) Ca2+ ([Ca2+]SR). We show here by three independent experimental approaches that PKA-dependent RyR2-S2808 phosphorylation plays significant functional roles at the subcellular level, namely, Ca2+ release synchronization, Ca2+ wave propagation and functional adaptation of RyR2 to variable [Ca2+]SR. These results indicate a direct mechanistic link between RyR2 phosphorylation and SR luminal Ca2+ sensing.
Keywords: Excitation–contraction coupling, Ryanodine receptor phosphorylation, Sarcoplasmic reticulum, PKA, Ca2+ imaging
1. Introduction
During physical exercise or periods of emotional stress, the enhanced sympathetic tone leads to release of epinephrine, which activates cardiac β1-adrenergic receptors. This produces an increase in cytosolic cAMP levels and further activation of protein kinase A (PKA), which in turn is responsible for the phosphorylation of key proteins involved in cardiac excitation–contraction coupling (E–C coupling) to boost the contractile machinery for enhanced cardiac function. PKA-dependent phosphorylation of the L-type Ca2+ channel (CaV1.2) results in an increased Ca2+ influx for triggering Ca2+-induced Ca2+ release (CICR) and loading the sarcoplasmic reticulum (SR) with Ca2+. Phosphorylation of phospholamban (PLB), an inhibitory accessory protein of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), leads to its dissociation from SERCA, thereby relieving SERCA suppression. Together, these modifications lead to enhanced SR Ca2+ load and larger systolic Ca2+ release, underlying the positive inotropic effect during β-adrenergic stimulation. In vitro [1–4] and in vivo [5,6] data clearly indicate that the SR Ca2+ release channel/ryanodine receptor (RyR2) is also a target for PKA phosphorylation, but the functional output of this posttranslational modification has not been clearly defined and remains highly controversial [3,7–11]). Since both enhanced L-type Ca2+ current (ICaL) and PLB phosphorylation inherently cause substantial changes in E–C coupling, it has been difficult to dissect the functional consequences of RyR2 phosphorylation during β-adrenergic stimulation.
Biochemical data indicate that the RyR2 channel contains three major phosphorylation sites for PKA and/or Ca2+/calmodulin dependent kinase (CaMKII), namely Ser2808, Ser2814, and Ser2030 (mouse nomenclature). Although there are still some disagreements on the role of Ser2814 and Ser2030 as targets of CaMKII and PKA, respectively, most of the controversies are centered on the mode of regulation (activation or inhibition), molecular mechanism, kinase specificity, and role in cardiac disease of Ser2808 phosphorylation. If 1) β-adrenergic stimulation of PKA leads to enhanced Ca2+ release and coordinated E–C coupling [6,12], and 2) RyR2-S2808 is a major PKA site (for references see [9,10,13], but also [5,14]), then, one might surmise that PKA phosphorylation of RyR2 increases channel activity. Indeed, single channel data have shown that PKA-dependent phosphorylation of RyR2 enhances the channel sensitivity to activating (“triggering”) Ca2+, thereby increasing open probability (Po) [15]. Dissociation of the accessory regulatory protein FKBP12.6 (a.k.a. calstabin-2) by PKA phosphorylation, presumably leading to the appearance of subconductance states in RyR2 [7], has also been postulated as a mechanism for increasing Ca2+ release and for loss of coordinated channel openings. The latter mode of regulation, although controversial, is mechanistically appealing because it might result in diastolic SR Ca2+ leak and pro-arrhythmic tendencies during chronic stress, such as in heart failure and catecholaminergic polymorphic ventricular tachycardia (CPVT), an arrhythmogenic syndrome caused by genetic mutations in RyR2 [16–18]. However, while some studies have found hyperphosphorylation of RyR2-S2808 in animal and human heart failure with an increased stoichiometry of PKA-phosphorylation from 0.9 in normal to 3.3 out of 4 possible sites per tetramer in failing myocytes [7,13,19], others have not supported such findings [1,5,20], and the role of Ser2808 phosphorylation remains unresolved.
Due to experimental difficulties in dissecting and analyzing the functional contribution of RyR2 phosphorylation to the inotropic effect during stress (or to pathophysiological alterations in disease), mouse models have been developed in which RyR2-S2808 have been genetically replaced by alanine (RyR2-S2808A) [1,9,21]. In these animal models, RyR2 can no longer be phosphorylated at this site. Since this transgenic mouse was found to develop no adaptive modifications of the Ca2+ signaling, it should allow to specifically study the physiological role of RyR2-S2808 and its importance under pathophysiological conditions. However, initial characterizations of these mice by several groups have yielded different results. While some studies showed no functional consequence of deletion of this phosphorylation site for E–C coupling during stress and no significant benefit for animal survival after induced hypertrophy [1,3,5,11], other studies reported overall blunted cardiac function, protection from rolipram-induced exercise-triggered arrhythmia and even cardioprotection after myocardial infarction [9,13,21]. Taken together, these findings cannot clarify the central question on the physiological role of RyR2-S2808 phosphorylation during β-adrenergic stimulation. Recently, a related report showed changes in cardiac function in a mouse model that simulates constitutive phosphorylation of RyR2 at this particular site (RyR2-S2808D) [22]. As a consequence of this modification, enhanced RyR2 oxidation and nitrosylation, increased diastolic SR Ca2+ leak and reduced SR Ca2+ load were observed, findings which suggest a potential role of chronic RyR2 hyperphosphorylation in the development of cardiac dysfunction.
In the present study, we combined several methodological approaches that interface cellular and molecular mechanisms and identified modifications of E–C coupling resulting from the ablation of the RyR2-S2808 phosphosite: we examined E–C coupling in wild type (WT) and transgenic S2808A cardiomyocytes under conditions that challenge the functional coupling of L-type Ca2+ channels and RyR2 channel proteins [23]. Furthermore, we tested the hypothesis that RyR2-S2808 phosphorylation may modulate RyR2 function via its luminal Ca2+ dependence. Since Ca2+ wave propagation velocity was recently suggested to depend on RyR2 sensitization by luminal Ca2+ [24], we compared Ca2+ wave propagation in WT and transgenic myocytes. Our results indicate that, although removal of the RyR2-S2808 phosphoepitope does not critically derange E–C coupling, RyR2-S2808A myocytes lack Ca2+ release synchronization (probably via an intra-SR Ca2+ sensing mechanism). They also display slower Ca2+ wave speeds (presumably by the same mechanism) and completely lack Ca2+ wave acceleration during β-adrenergic stimulation [25]. Furthermore, direct recording of single RyR2 channels shows that ablation of the RyR2-S2808 phosphosite decreases the sensitivity of RyR2 to SR luminal Ca2+.
2. Material and methods
2.1. Experimental animals
Wild type (WT) and RyR2-S2808A+/+ mice were raised and studied according to the regulations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals (1996) and the Institutional Animal Care and Use Committees of the University of Wisconsin—Madison. All experiments were approved by the State Veterinary Office of Bern, Switzerland, and the Association for Assessment and Accreditation of Laboratory Care International.
2.2. Experimental procedures
All methods used in this study were described previously. Hearts were enzymatically dissociated to obtain single cardiomyocytes. SR-enriched microsomes were prepared from freshly homogenized ventricles. Electrophysiological measurements (patch clamp recordings and lipid bilayer experiments) were recorded using Axopatch 200 amplifiers (Axon Instruments). Line-scan images of cytosolic Ca2+ signals were obtained using a confocal microscope (MicroRadiance, BioRad). Changes in fluorescence are shown as ΔF/F0. All experiments were performed at room temperature. Data are presented as mean ± SEM. Student’s t-test was used to compare the means between groups of experiments. P<0.05 was considered as significantly different.
An expanded Material and Methods section describing the electrophysiological protocols, Ca2+ wave experiments, Ca2+ leak measurements, Western blots, single channel recordings and data analysis is available in the online data supplement.
3. Results
3.1. Comparison of E–C coupling in WT and S2808A myocytes during β-adrenergic stimulation
During cardiac E–C coupling, opening of L-type Ca2+ channels triggers Ca2+-induced Ca2+ release from the SR via activation of the RyR2. During membrane depolarizations from −40 mV to the test potentials (−25 and 0 mV), ICaL and Ca2+ transients were simultaneously recorded, both in WT and RyR2-S2808A cardiomyocytes (for the complete stimulation protocol see Supplementary Fig. 1). At the end of each experiment, SR Ca2+ load was estimated by application of short caffeine pulses (for comparison, see Supplementary Fig. 2). Under control conditions, genetic ablation of the PKA-dependent phosphorylation site in RyR2-S2808A cells did not result in any obvious differences in ICaL and Ca2+ transient properties when compared with WT myocytes (Fig. 1A and Supplementary Fig. 3). This is also reflected in the E–C coupling “gain”, an operational term that defines the efficiency by which ICaL evokes SR Ca2+ release. As an additional strategy to test the system closer to the level of molecular communication between the DHPR and the RyR2 and to reveal even small changes of RyR2 Ca2+ sensitivity, we reduced extracellular Ca2+ and applied small depolarizations to detect minimal changes in signal transduction occurring in the dyadic cleft [26,27]. Fig. 1C summarizes the E–C coupling gain at the test potential of −25 mV in control conditions and at reduced external Ca2+ (1.8 and 0.5 mmol/L Ca2+, respectively) showing no differences between both groups. Next, we studied E–C coupling during β-adrenergic stimulation, to determine whether RyR2-S2808A yielded differences in E–C coupling. ICaL and Ca2+ transients were recorded during stimulation with isoproterenol (Iso, 1 μmol/L) under the same experimental conditions for normal and “challenged” E–C coupling as above. Fig. 1B shows the results of β-adrenergic stimulation with Iso on ICaL and Ca2+ transients in WT and RyR2-S2808A myocytes. Even under these stringent conditions, there was no difference in the E–C coupling efficiency between WT and RyR2-S2808A cardiomyocytes (Fig. 1C). However, a more detailed investigation of the Ca2+ transients revealed compelling evidence for the functional importance of RyR2-S2808 phosphorylation in the context of β-adrenergic activation.
Fig. 1.

Excitation–contraction (E–C) coupling in wild type (WT) and RyR2-S2808A (S2808A) myocytes. A, B, Depolarization of voltage-clamped cells from −40 to 0 mV (left side) or to −25 mV (right side) at control conditions (1.8 mmol/L [Ca2+]o; A) and during β-adrenergic stimulation with 1 μmol/L isoproterenol (Iso; B). Shown are the stimulation protocol (upper traces), Ca2+ current traces (ICaL; bottom traces), confocal line-scan images and line profiles of the Ca2+ transients as relative changes in cytosolic Ca2+ concentrations (in ΔF/F0) of fluo-3-loaded WT (black) and S2808A (red) cells, respectively. C, Statistical summary of E–C coupling gain obtained from the ratio of the maximal Ca2+ transient amplitudes and peak ICaL at control and during β-adrenergic stimulation in the presence of normal (1.8 mmol/L) and reduced (0.5 mmol/L) [Ca2+]o, showing no significant differences.
3.2. Synchronization of Ca2+ release during β-adrenergic stimulation in WT and RyR2-S2808A cardiomyocytes
Comparison of the global rising phase of the Ca2+ transient during CICR at negative test potentials and low external Ca2+ indicated similar profiles for WT and RyR2-S2808A myocytes. Figs. 2A and B show examples of line-scan images and scaled line profiles of the rising Ca2+ transient in control and during β-adrenergic stimulation, respectively. Close examination of the initial phase of the Ca2+ transient in RyR2-S2808A during β-adrenergic stimulation reveals a delay in the Ca2+ release onset of several release units along the cell (corresponding to sarcomeres). The shortening of this delay in WT and genetically modified cells in Iso is in agreement with recent studies reporting that β-adrenergic stimulation improves synchronization of Ca2+ release units during E–C coupling [12,28–30].
Fig. 2.
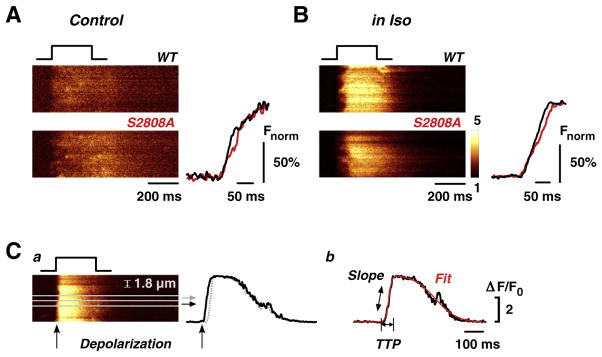
Comparison of global Ca2+ transients during small depolarizations at low external Ca2+ in control and during β-adrenergic stimulation. Global line-scan images and derived line profiles of Ca2+ transients at control (A) and during β-adrenergic stimulation (B) are shown during depolarization to −25 mV in low [Ca2+]o (0.5 mmol/L), where subtle changes in electromechanical coupling would be best seen, in WT (black) and S2808A (red) myocytes. For better comparison of the kinetics, line profiles have been normalized to their own peaks (Fnorm %). No differences in global Ca2+ signals were observed, although line-scan images of S2808A myocytes revealed slightly longer temporal delays in the onset of Ca2+ release during β-adrenergic stimulation. C, a) Variations in synchronization of Ca2+ release upon depolarization (see arrows in line-scan and profile) were analyzed by dividing the line-scan images into small bands of sarcomere width (1.8 μm); b) Ca2+ release events in each band profile were fitted and analyzed for time-to-peak (TTP, from start of depolarization to first peak Ca2+ release) and slope of rise (Slope).
We further investigated these differences in synchronization of Ca2+ release during β-adrenergic stimulation with a detailed analysis of the Ca2+ release kinetics at the subcellular level using the method described in Fig. 2C. Briefly, we subdivided each line-scan into small strips corresponding in width to subcellular sarcomeric compartments (1.8 μm, Fig. 2Ca). Each resulting band was fitted with two sigmoids to overcome the problems of high noise in the thin sections of the line scans. The first sigmoid describes the rising phase of the transient and the second corresponds to the decay phase. Further, the fitting function yields time-to-peak (TTP) and slope of activation (Fig. 2Cb). Importantly, for this analysis, TTP was defined as the time lapsed from the beginning of depolarization until the first peak in fluorescence in each sarcomere (see stimulation trace and two sample traces for rapid and delayed Ca2+ release in Fig. 2Ca) and not by the first increase in fluorescence as seen in the line-scan. The starting point to measure TTP is critical in determining Ca2+ release synchronization, since the first rise in fluorescence might be considerably delayed with regard to the start of depolarization.
Synchronization analysis was performed on all line-scan images from WT and RyR2-S2808A cells during depolarizations to −25 mV in low Ca2+ (0.5 mmol/L) under control conditions and during β-adrenergic stimulation. Figs. 3A and B summarize the consequences of eliminating RyR2-S2808 phosphorylation for sarcomeric synchronization of Ca2+ release. This analysis revealed two populations of release events in control conditions: an early (~80 ms) and a late (~280 ms) peak of release events (see Fig. 3A and Supplementary Fig. 4). In both WT and RyR2-S2808A, the mean peak time of a Gaussian fit to the distribution was comparable to each other. Upon β-adrenergic stimulation, Ca2+ release synchronized to a greater extent in WT cells, thereby essentially removing the population of delayed events. However, in RyR2-S2808A cells, the delayed events remained after Iso and, although the two populations were still present, a small shift in the mean peak time, from ~280 to 180 ms, occurred (Fig. 3B). An example of the acceleration of TTP in a WT cell is shown in Supplementary Fig. 5.
Fig. 3.

Ca2+ transient synchronization in WT and S2808A myocytes at control and during β-adrenergic stimulation. Detailed analysis of the rising phase of Ca2+ transients at depolarization to −25 mV in low [Ca2+]o. As shown in Fig. 2, line profiles of thin sections of the line-scans were analyzed for time-to-peak (TTP; A, B) and slope of rise (Slope, C, D) in control (A, C) and during β-adrenergic stimulation (B, D). The number of events per bin (Counts) is plotted as a function of TTP and Slope, respectively. Analysis of TTP revealed that under our experimental control conditions, there may be two temporally separated Ca2+ release populations (A), shown by double peak fits: one early release peaking near 80 ms, and a late release peaking near 290 ms. B, In WT, β-adrenergic stimulation synchronized these late release events to one peak near 100 ms. Synchronization was significantly less pronounced in S2808A cells, revealing still two temporally separated Ca2+ release populations. C, While there was no difference in Slope under control conditions, Iso treatment revealed a significantly slower rise in S2808A cells (D). Bar graphs of counts were fitted with multiple peak (A, B) and single (C, D) Gaussians. For statistical comparison, TTP and Slope values of S2808A cells were normalized to WT and displayed in histograms.
We also analyzed the slope of the Ca2+ transient on the level of sarcomeres. Shown is the number of events (counts) for each slope, and the total distribution of events has been fitted with a Gaussian function (Figs. 3C and D). In control conditions, we found no difference in the slope of WT and RyR2-S2808A Ca2+ transients. However, after β-adrenergic stimulation, WT slopes became steeper, resulting in faster rise of the Ca2+ transients when compared with basal conditions. Interestingly, the increase in slope of RyR2-S2808A transients was significantly smaller than that observed in WT cells (Fig. 3D). The peak of the Gaussian fit shifted from 0.038 to 0.27 (ΔF/F0)/s in WT, and from 0.038 to 0.12 (ΔF/F0)/s in RyR2-S2808A slopes. This shift was also noticed in the mean of all measured slopes (see histograms of normalized values). Overall, these differences suggested a substantial de-synchronization of SR Ca2+ release that was maintained during β-adrenergic stimulation in RyR2-S2808A myocytes.
3.3. Effect of RyR2-S2808A on diastolic SR Ca2+ leak
The lack of Ca2+ release synchronization in RyR2-S2808A cells during β-adrenergic stimulation may be linked to impaired Ca2+ sensitivity of RyR2 due to ablation of the Ser2808 phosphorylation site, which might also be manifest during diastole. Using a modification of the Shannon et al. protocol [31], diastolic SR Ca2+ leak was assessed in WT and RyR2-S2808A myocytes under basal and β-adrenergic stimulated conditions. This leak is thought to result mainly from spontaneous openings of RyR2s because the open probability (Po) of RyR2s is not zero even at resting [Ca2+]i, depends on SR Ca2+ load and RyR2 posttranslational modifications, and is detected as a small fluctuation in the fluorescence baseline or sometimes as Ca2+ sparks. An expanded explanation of the protocol is given in the Supplementary data file.
Diastolic SR Ca2+ leak was calculated from the difference in [Ca2+]i during 0 Na+/0 Ca2+ without and with tetracaine. Figs. 4A and B show the leak protocol and respective line-scans of WT myocytes and the corresponding line profiles of WT (black traces) and RyR2-S2808A myocytes (red traces) in control and with 1 μmol/L Iso. The time points used for measuring differences in diastolic Ca2+ leak are indicated with blue arrows. As expected, under basal conditions (“Control”), there was no difference in SR Ca2+ leak between WT and RyR2-S2808A myocytes (Fig. 4A). After β-adrenergic stimulation, PKA-mediated phosphorylation of the L-type Ca2+ channel enhances CICR (Fig. 4B), and activation of SERCA and RyR2 may lead to increased diastolic Ca2+ leak. Leak analysis did not reveal any difference in SR Ca2+ leak during β-adrenergic stimulation (Fig. 4B). However, a slightly elevated leak in WT cells may have lowered SR Ca2+ content until the leak matched again Ca2+ uptake via SERCA. In addition, we cannot exclude the possibility of small differences in diastolic Ca2+ concentrations between WT and RyR2-S2808A cells, which would influence both leak and SR Ca2+ content. The statistical summary of the measured Ca2+ leak is shown in Fig. 4C. Fig. 4D summarizes the total SR Ca2+ content in WT and RyR2-S2808A myocytes, as assessed by caffeine application. Under basal conditions, there is no difference in the SR Ca2+ content between the two groups. However, β-adrenergic stimulation significantly reduced SR Ca2+ content in WT cells compared to RyR2-S2808A cells, which could be the result of a slightly elevated leak. In comparison, SR Ca2+ load at the end of the E–C coupling experiments did not show significant differences between the two animal groups (please see Supplemental Fig. 2). This is most likely due to different experimental conditions. Most importantly, during E–C coupling experiments, cells were under whole-cell voltage clamp control with a defined SR Ca2+ pre-loading protocol, where the influence of a small diastolic leak would be minimal.
Fig. 4.

Comparison of sarcoplasmic reticulum (SR) Ca2+ leak at control and during β-adrenergic stimulation. A, B Fluo-3 AM-loaded cells were field-stimulated to steady-state Ca2+ transient amplitudes in control solution (label 1, 1.8 mmol/L Ca2+), then solution was rapidly switched to 0 Na+/0 Ca2+ containing solution (2) to minimize loss of cellular Ca2+ and to prevent external Ca2+ influx, thereby creating a closed system. Addition of tetracaine (1 mmol/L, 3) blocked RyR2-mediated SR Ca2+ leak, shown by a reduction in basal Ca2+ levels. Total SR Ca2+ content was assessed by caffeine application (10 mmol/L, 4) before return to control solution (5). Shown are representative line profiles of WT (black, with corresponding line-scan images) and S2808A (red) myocytes in control (A) and during β-adrenergic stimulation with Iso (1 μmol/L; B). Blue arrows indicate areas taken for measurement of SR Ca2+ leak. Leak is highlighted in blue. C, Statistical comparison of SR Ca2+ leak showed no difference. D, SR Ca2+ load was significantly higher in S2808A than in WT cells during β-adrenergic stimulation.
Thus, the observation that SR Ca2+ content is not reduced in RyR2-S2808A cells after β-adrenergic stimulation (compared to basal conditions) is consistent with the notion that Ca2+ sensing may be altered in RyR2-S2808A cells. The relatively long intervals involved in release synchronization shown in Fig. 3B point toward a slow rate-limiting step in the process of delayed SR Ca2+ release activation. This could possibly be mediated by SERCA-dependent SR Ca2+ uptake and subsequent RyR2 sensitization by luminal Ca2+, which is known to occur within this time window [32].
3.4. Influence of RyR2 phosphorylation on the speed of Ca2+ wave propagation
In order to further test our hypothesis that RyR2-S2808A cells may have Ca2+ sensitivity different from that of WT cells, we investigated the characteristics of “spontaneous” Ca2+ waves in WT and RyR2-S2808A myocytes. We elicited Ca2+ waves by elevating external [Ca2+] to 10 mmol/L. Fig. 5A (left panels) shows line-scan images of high [Ca2+]o-triggered Ca2+ waves in WT and RyR2-S2808A myocytes. The wave propagation under basal conditions was 98±5 μm/s (n=17) in WT cells and 73±6 μm/s (n=6) in RyR2-S2808A cells. Thus, wave velocity was 25% slower in RyR2-S2808A cells. This difference in Ca2+ signal propagation and the inability of PKA to phosphorylate this site in RyR2-S2808A cells suggest that this site may already be phosphorylated to a certain extent in WT cells under control conditions, as previously reported [7,10]. During β-adrenergic stimulation, wave speed increased by 14% in WT cells, to 113±5 μm/s, indicating enhanced sensitivity to activating Ca2+ (Fig. 5A, right upper panel). Strikingly, in RyR2-S2808A myocytes, wave velocity was unaffected by β-adrenergic stimulation (from 73±6 μm/s to 75±6 μm/s, n=7; Figs. 5A, right lower panel, B and C) despite the fact that global effects of β-adrenergic stimulation on the wave transients were clearly evident. The effect of β-adrenergic stimulation on the wave transients is best shown after digital deskewing of the Ca2+ wave front (see Supplementary Fig. 6). Fig. 5B shows representative traces of aligned Ca2+ wave transients under basal conditions and during β-adrenergic stimulation in WT and RyR2-S2808A cells. Significant acceleration of transient decay due to SERCA stimulation and enhancement of Ca2+ transient amplitude are evident in both WT and RyR2-S2808A cells (Fig. 5B), confirming a similar extent of PKA activation in both cell types. Taken together the specific lack of wave-speed modulation in RyR2-S2808A cells is consistent with an alteration of luminal Ca2+ sensitivity in these cells, but could also be caused by indirect changes of cytosolic Ca2+ sensitivity.
Fig. 5.

Ca2+ wave propagation analysis before and during β-adrenergic stimulation. A, Representative line-scan images of high (10 mmol/L) [Ca2+]o-induced Ca2+ waves in WT and S2808A cells before (left images) and during stimulation with Iso (right images). Please note the differences in the wavefront, representative for the propagation velocity of the Ca2+ wave, between WT and S2808A under both conditions. B, Line profiles of the Ca2+ waves shown in A after de-skewing of the wavefront. Faster transient kinetics by SERCA stimulation is obvious in both genotypes. C, Statistical comparison of wave velocity before and during β-adrenergic stimulation. Ca2+ wave propagation is significantly slowed in S2808A cells by 25% in high [Ca2+]o and does not increase upon Iso-stimulation.
3.5. Direct recording of WT and RyR2-S2808A channel activity in lipid bilayers
Voltage-dependent Ca2+ transients and spontaneous Ca2+ release events are meaningful assays of RyR2 activity, but their interpretation is complicated by incompletely controlled luminal and cytosolic factors that also influence channel activity. We reconstituted SR vesicles into planar lipid bilayers and recorded single-channel activity under controlled conditions to directly assess the effect of phosphorylation on isolated RyR2 channels. SR vesicles were first treated for 10 min with 10 U/mL protein phosphatase (PP) 1 to remove any residual phosphorylation present in RyR2 channels, as native RyR2 already shows basal phosphorylation at Ser2808 [7,10]. Completely dephosphorylated channels represent the starting point for these experiments. Using Western blots, we provide evidence that RyR2 channels were dephosphorylated by PP1 at Ser2808 under these conditions (Fig. 6A).
Fig. 6.

Differential response of WT and RyR2-S2808A channels to activation by luminal Ca2+. A, Western blots of dephosphorylated (PP1-treated) channels at the beginning of the bilayer experiment and after phosphorylation with PKA, as performed in the bilayer. B, Representative 2-second single-channel recordings of PP1-treated WT and RyR2-S2808A channels obtained in 5 μmol/L cytosolic Ca2+ (“Control, 5 μM Ca2+”) and after the indicated additions. Openings are represented as downward deflections in all traces. All recordings are from the same channel. C, Plot of Po after indicated additions to the cytosolic side of the channel. Sixty-second files from 6 WT channels (left) and 6 RyR2-S2808A channels (right) were used to obtain average Po under the conditions illustrated in B. Each average point corresponds to 5 s of activity. D, Summary of Po changes in 6 individual WT and 6 RyR2-S2808 channels after addition of 1 mmol/L of CaCl2 to the trans (SR luminal) side of the channel.
Dephosphorylated WT and RyR2-S2808A channels exhibited robust channel activity at cis (corresponding to cytosolic side) and trans (corresponding to luminal side) [Ca2+]=5 μmol/L (“Control, 5 μM Ca2+”), as shown in the representative traces of Fig. 6B. On average, the probability of the channel being open (Po) was similar for WT and RyR2-S2808A channels (0.384±0.08, n=6, and 0.393±0.091, n=6, respectively), as expected if the phosphorylation state were identical in both types of channels. Decreasing cytosolic [Ca2+] to 100 nmol/L (pCa 7) and adding 2 mmol/L MgATP (0.35 mmol/L free Mg2+) deactivated channels almost completely (Po ~0.001 in both groups), with only brief openings being detected sporadically. Addition of 1 μg/mL of the catalytic subunit of PKA failed to increase Po in WT and RyR2-S2808A channels (Po ~0.001); however, an increase of channel activity was seen upon further addition of 1 mmol/L CaCl2 to the SR luminal side of the channel. WT channels displayed consistently higher Po (0.062±0.031) than RyR2-S2808A channels (0.020±0.005; last traces of Fig. 6B). The latter was the only difference between WT and RyR2-S2808A channels during the course of the experiment, represented in Fig. 6C for a single channel and summarized in Fig. 6D for 6 channels. Again, using Western blots, we show that PKA effectively phosphorylates the Ser2808 phospho-epitope under the conditions used in the bilayer experiments (Fig. 6A, “pCa7, MgATP, PKA”). We therefore assign the difference in WT and RyR2-S2808A channel activity at 1 mmol/L luminal Ca2+ to the phosphorylation of Ser2808 in WT channels. Thus, PKA-dependent phosphorylation of Ser2808 increases the sensitivity of the channel to luminal Ca2+. In intact cells, this effect is reflected in improved Ca2+ release synchronization and Ca2+ wave acceleration, resulting from enhanced SR Ca2+ loading during β-adrenergic stimulation.
4. Discussion
Sympathetic increase in contractility is largely attributable to increased Ca2+ influx via the L-type Ca2+ channel, faster Ca2+ re-uptake into the SR and higher SR Ca2+ load by SERCA stimulation. Despite biochemical data confirming PKA-dependent phosphorylation of RyR2, its functional contribution to the inotropic effect of β-adrenergic stimulation remains to be elucidated. For the most part, in vitro assays have shown that phosphorylation of Ser2808, a major PKA-dependent RyR2 phosphorylation site, leads to increased channel activity. However, due to intrinsic difficulties associated with isolating the effect of RyR2 phosphorylation from the activating effect of triggering Ca2+ (ICaL) and the SR Ca2+ load (both of which increase during β-adrenergic stimulation), studies in intact cardiomyocytes have not yet convincingly demonstrated that Ser2808 phosphorylation contributes to enhance Ca2+ release during β-adrenergic stimulation.
The availability of a mouse model in which the Ser2808 phosphosite has been genetically ablated by substitution with alanine, represents an ideal experimental model to study the role of Ser2808 phosphorylation during catecholaminergic stimulation. However, intensive characterizations of this model by different groups have led to confiicting data claiming either no phenotype in RyR2-S2808A animals [1,3,5] or impaired exercise capacity, decreased responsiveness to β-adrenergic stimulation and cardioprotection [8,9,11,13].
4.1. E–C coupling and Ca2+ release synchronization during β-adrenergic stimulation
In the present study, we examined E–C coupling and Ca2+ signaling in RyR2-S2808A cardiomyocytes with a protocol that interfaces molecular and subcellular mechanisms, thereby covering several layers of complexity. Our initial results are consistent with findings by Benkusky et al. [1] that CICR in RyR2-S2808A myocytes is normal in control conditions and even during β-adrenergic stimulation. Thus, E–C coupling at the cellular level is not noticeably compromised by the lack of Ser2808 phosphorylation.
However, under conditions where we specifically challenged signal transduction between the L-type Ca2+ channels and the RyR2s, we discovered clear impairment of RyR2-S2808A cells to synchronize SR Ca2+ release by β-adrenergic stimulation. The recruitment of triggered Ca2+ release by RyR2-S2808A cells was significantly compromised, leading to prolonged local (i.e. sarcomeric) delays in the onset of Ca2+ release. These delays had little influence on the average Ca2+ release amplitude, since in these experimental conditions, Ca2+ release consisted largely of cumulated local Ca2+ release events with low overall release amplitude. For this reason, there was also no detectable difference in the E–C coupling gain (which only takes into account peak amplitudes, not the temporal variations in peak location). However, onset of Ca2+ release, which was best detected in the time delay from stimulation to peak Ca2+ amplitude and the slope of Ca2+ rise, was significantly slower in RyR2-S2808A cardiomyocytes compared with WT cells. In this context, we made another intriguing observation: when analyzing the temporal distribution of Ca2+ release events under control conditions, we identified two distinct release populations in both groups of mice. In β-adrenergic stimulated WT cells, the delayed release events merged with the early ones into a single Ca2+ release event population, whereas in RyR2-S2808A cells, the delayed release population did not fully blend with the early one, but still formed a distinct Ca2+ release population with significant delay relative to the first peak. Our data suggest that phosphorylation of RyR2 at Ser2808 is involved in the process of synchronization, presumably by enhancing Ca2+ sensitivity of phosphorylated RyR2s.
4.2. Luminal SR Ca2+ sensing mechanism of RyR2
Regulation of Ca2+ release and release synchronization is very complex and strongly depends on the interaction between RyR2 and SR Ca2+ content. Under normal conditions, SR Ca2+ load is determined by the rate of Ca2+ leak through RyR2s and Ca2+ re-uptake by the SERCA pump. The interaction between SR Ca2+ load and RyR2 activity is tightly regulated and controlled by luminal sensors that monitor Ca2+ levels and modulate RyR2 Ca2+ sensitivity. Recently, changes in SR Ca2+ content mediated by SERCA have been suggested to modulate cytosolic RyR2 Ca2+ sensitivity by an SR luminal mechanism, presumably involving binding of Ca2+ to calsequestrin and signaling to RyR2 mediated by junctin and triadin [32–37]. Unlike intra-sarcomeric Ca2+ diffusion over <1 μm, which is fast, luminal Ca2+ signaling mechanisms could readily account for delays of SR Ca2+ release in the range of approximately 100–200 ms, as observed here (see late peaks during depolarizations in Fig. 3A). These time delays of Ca2+ release suggest delayed openings of RyR2s, which could be activated by sensitization through slow filling of the SR by SERCA during the plateau of the depolarizing test pulse. According to our results, we propose that β-adrenergic stimulation re-synchronizes RyR2 recruitment through Ser2808 phosphorylation, leading to concerted Ca2+ release, initially via enhanced luminal Ca2+ sensing (Fig. 3B). Both processes (namely modulation of RyR2 Ca2+ sensitivity and synchronization of Ca2+ release) are intertwined since increased SR Ca2+ load leads to RyR2 activation, possibly indirectly by enhancing its cytosolic Ca2+ sensitivity [15].
The consequences of Ser2808 removal in RyR2-S2808A cells are therefore impaired RyR2 recruitment and diminished ability to respond to higher SR Ca2+ load. This interpretation is further supported by our SR Ca2+ leak experiments, where SR Ca2+ content was assessed during long periods of β-adrenergic stimulation. Enhanced Ca2+ sensitivity and receptor phosphorylation are thought to increase Po of RyR2s, promoting greater Ca2+ leak from the SR [31]. Thus, at the same SR Ca2+ load, a slightly larger leak would be expected in WT cells when compared with RyR-S2808A cells. Over time, this enhanced SR leak would lead to a lower SR Ca2+ content in WT cells, until a new leak-uptake balance is reached — and the leak is again almost the same, as observed in our experiments (see Figs. 4A and B). The observed reduction in Ca2+ content in WT compared to RyR2-S2808A cells during β-adrenergic stimulation is consistent with the notion that in WT cells the RyR2 phosphorylation promoted a small SR Ca2+ leak that slowly accumulated into a noticeable SR Ca2+ depletion [38].
Thus, this finding favors our proposal that phosphorylated RyR2s are more sensitized for release, leading to lower SR Ca2+ levels. This would explain why RyR2-S2808A cells retained significantly higher SR Ca2+ loads during β-adrenergic stimulation. A recent study by Kashimura et al. investigated another RyR2 mutation (RyR2-R4496C+/−) that promotes the generation of Ca2+ waves and delayed afterdepolarizations during catecholaminergic stimulation [39]. This model for CPVT shows the opposite phenotype from the RyR2-S2808A channel, leading to an abnormally sensitive “leaky” channel and reduced SR Ca2+ content. The authors conclude that in this model the critical parameter for the Ca2+ wave threshold during β-adrenergic stimulation is determined by SR Ca2+ content. Ca2+ wave initiation even required more Ca2+ in the SR than in control. However, it is unclear how this apparent “desensitization” of the RyR2s would affect Ca2+ wave speed and whether it was related to RyR2 phosphorylation. Furthermore, it remains to be determined whether the two putative sensitizing mechanisms (mutation and RyR2 phosphorylation) modify the Ca2+ wave threshold (i.e. the steady-state Ca2+ sensitivity of the RyR2) and the Ca2+ trigger sensitivity in the same direction and to the same extent. In RyR2-S2808A, the mutation is directly affecting the phosphorylation site of the channel, ablating any change in Ca2+ sensitivity mediated by this site.
4.3. Ca2+ wave propagation at changed SR Ca2+ load
Our conjecture that Ser2808 phosphorylation may be crucial for intra-SR Ca2+ sensing was further tested in a different set of experiments, where we studied Ca2+ wave propagation in intact myocytes. We have previously reported that Ca2+ wave propagation also depends on luminal Ca2+ loading [24], thus linking RyR2 activity to luminal Ca2+ levels. Interestingly, in the present study Ca2+ wave velocity was reduced in RyR2-S2808A cells even under basal conditions, which is consistent with previous reports showing basal PKA-dependent phosphorylation of RyR2 [7,10], and thus, with a general lack of phosphorylation at the mutated Ser2808 site. The significant reduction in wave propagation speed leads to the conclusion that RyR2-S2808A channels may be less sensitive to Ca2+. This notion bodes well with the fact that during β-adrenergic stimulation (which significantly increased wave propagation speed in WT myocytes), the wave front was not accelerated in RyR2-S2808A cells and remained at the same reduced speed as under basal conditions. The enhanced RyR2 sensitivity to luminal Ca2+ in normal compared to RyR2-S2808A channels after β-adrenergic stimulation, which we observed in the bilayer experiments on isolated RyR2 channels, could thus lead to activation of available RyR2s at lower SR Ca2+ levels, due to store overload-induced Ca2+ release, SOICR [35].
Thus, our Ca2+ wave experiments provide several lines of evidence suggesting that an SR luminal mechanism could be involved in synchronization of Ca2+ release: 1) SERCA stimulation enhanced Ca2+ wave amplitude and kinetics upon β-adrenergic stimulation similarly in WT and RyR2-S2808A cells, yet the latter did not change Ca2+ wave speed. Malfunction of the same mechanism could also underlie failure of synchronization during EC-coupling. It follows then that the larger SR Ca2+ release and the subsequent activation of RyR2-S2808A channels by cytosolic Ca2+ alone do not lead to faster Ca2+ waves in Iso. 2) Phosphorylation of Ser2808 seems to be required for faster waves. 3) Changes of the mechanism of RyR2 sensitization by luminal Ca2+ after Ser2808 phosphorylation are a likely mechanism and would be blunted in the transgenic mice.
4.4. Direct and indirect modulation of RyR2 by luminal Ca2+
Our data based on the recordings of cellular Ca2+ release signals support the notion of an SR luminal mechanism modulating RyR2 function and cytosolic Ca2+ sensitivity based on the state of S2808 phosphorylation. This conclusion is confirmed by direct recordings of WT and RyR2-S2808A channels, in which we tested the luminal Ca2+ sensitivity of WT and mutant RyR2. Previous studies with single RyR2-S2808 channels reconstituted into lipid bilayers exclusively addressed changes in channel activity at constant and/or low luminal Ca2+ levels [1,5], or used recombinant channels devoid of accessory proteins [40]. Such studies could not detect variations in RyR2 luminal Ca2+ sensitivity and therefore often did not reveal deficiencies in RyR2-S2808A channels during stimulation with PKA [1,40]. Based on the cellular results described above, we designed experiments not only to compare differences in channel activity brought about by Ser2808 phosphorylation, but also in response to variable luminal (trans) Ca2+ levels, mirroring the changes in SR Ca2+ load that occurred in our maneuvers with intact cells. Only under these settings a differential modulatory effect of luminal Ca2+ on RyR2 function could be unveiled between WT and RyR2-S2808A channels.
5. Conclusion
In summary, we present molecular and cellular evidence for a functional role of RyR2-S2808 phosphorylation in Ca2+ signaling and in the RyR2 ability to sense changes in luminal Ca2+. While this function does not seem to manifest itself noticeably during normal EC-coupling, it may become important under cardiac conditions where the EC-coupling mechanism is compromised or during SR Ca2+ overload. Our findings are consistent with a mechanism whereby RyR2 phosphorylation at Ser2808 leads to a change in its ability to be sensitized for cytosolic Ca2+ by elevations of luminal Ca2+, and imply that Ser2808 may be involved in the mechanisms that lead to functional modulation of RyR2 during stress. They may also explain some discrepancies in the literature, inasmuch as RyR2s behave differently under conditions where the cytosolic Ca2+ sensitivity is tested without control of luminal Ca2+. However, because of the tight functional interdependence of luminal and cytosolic Ca2+ sensitivity of the RyR2s, it is not possible to unequivocally distinguish between the two mechanisms. We propose that in situations of enhanced SR Ca2+ content, such as physiological stress or in pathological conditions, this phosphorylation-enhanced luminal Ca2+-sensing mechanism leads to sensitization of RyR2s and to synchronization of CICR. However, while increasing cardiac performance by revving up Ca2+ release, this sensitization of RyR2 may also have the potential to generate a significant pro-arrhythmic substrate. Our study also helps to resolve the importance of Ser2808 under pathological conditions: if the controversial hyperphosphorylation of Ser2808 indeed leads to heart failure [7,11], our results imply that the mechanism responsible could be phosphorylation-enhanced RyR2 sensitization.
Supplementary Material
Acknowledgments
We wish to thank Ghislaine Rigoli for excellent technical assistance and Fang Liu and Nancy Benkusky for help with the Western blots and lipid bilayer experiments.
Sources of funding
This work is supported by grants from the Swiss National Science Foundation (31-132689 and 31-109693 to E.N.) and the NIH (RO1-HL055438 and PO1-HL094291 to H.H.V.).
Abbreviation
- RyR2-S2808A
Transgenic mouse in which RyR2 cannot be phosphorylated on serine 2808
Appendix A. Supplementary material
Supplementary data to this article can be found online at doi:10.1016/j.yjmcc.2012.03.015.
Footnotes
Disclosure statement
None.
References
- 1.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, et al. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–29. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- 2.Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase a produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006;98:1506–13. doi: 10.1161/01.RES.0000227506.43292.df. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–16. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 4.Takasago T, Imagawa T, Furukawa K, Ogurusu T, Shigekawa M. Regulation of the cardiac ryanodine receptor by protein kinase-dependent phosphorylation. J Biochem. 1991;109:163–70. doi: 10.1093/oxfordjournals.jbchem.a123339. [DOI] [PubMed] [Google Scholar]
- 5.MacDonnell SM, Garcia-Rivas G, Scherman JA, Kubo H, Chen X, Valdivia H, et al. Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res. 2008;102:65–72. doi: 10.1161/CIRCRESAHA.108.174722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reiken S, Wehrens XH, Vest JA, Barbone A, Klotz S, Mancini D, et al. Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation. 2003;107:2459–66. doi: 10.1161/01.CIR.0000068316.53218.49. [DOI] [PubMed] [Google Scholar]
- 7.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12. 6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 8.Sarma S, Li N, van Oort RJ, Reynolds C, Skapura DG, Wehrens XH. Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:13165–70. doi: 10.1073/pnas.1004509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–8. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao B, Zhong G, Obayashi M, Yang D, Chen K, Walsh MP, et al. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Makarewich CA, Kubo H, Wang W, Duran JM, Li Y, et al. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res. 2012;110:831–40. doi: 10.1161/CIRCRESAHA.111.255158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogrodnik J, Niggli E. Increased Ca2+ leak and spatiotemporal coherence of Ca2+ release in cardiomyocytes during beta-adrenergic stimulation. J Physiol. 2010;588:225–42. doi: 10.1113/jphysiol.2009.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, et al. Phosphorylation of the ryanodine receptor mediates the cardiac fight or fiight response in mice. J Clin Invest. 2010;120:4388–98. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278:38593–600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 15.Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 17.Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol. 2002;190:1–6. doi: 10.1002/jcp.10031. [DOI] [PubMed] [Google Scholar]
- 18.Eisner DA, Kashimura T, O’Neill SC, Venetucci LA, Trafford AW. What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J Mol Cell Cardiol. 2009;46:474–81. doi: 10.1016/j.yjmcc.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Lehnart SE, Wehrens XH, Kushnir A, Marks AR. Cardiac ryanodine receptor function and regulation in heart disease. Ann N Y Acad Sci. 2004;1015:144–59. doi: 10.1196/annals.1302.012. [DOI] [PubMed] [Google Scholar]
- 20.Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–22. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- 21.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–87. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ullrich ND, Niggli E. Impaired Ca2+ release synchronization in RyR2-S2808A mouse cardiomyocytes during beta-adrenergic stimulation. Biophys J. 2010;98:550a. [Google Scholar]
- 24.Keller M, Kao JP, Egger M, Niggli E. Calcium waves driven by “sensitization” wave fronts. Cardiovasc Res. 2007;74:39–45. doi: 10.1016/j.cardiores.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Ullrich ND, Niggli E. Cardiac ryanodine receptor phosphorylation at Ser2808 is involved in intra-SR calcium sensing. Biophys J. 2011;100:353a. [Google Scholar]
- 26.McCall E, Ginsburg KS, Bassani RA, Shannon TR, Qi M, Samarel AM, et al. Ca2+ flux, contractility, and excitation–contraction coupling in hypertrophic rat ventricular myocytes. Am J Physiol. 1998;274:H1348–60. doi: 10.1152/ajpheart.1998.274.4.H1348. [DOI] [PubMed] [Google Scholar]
- 27.Ullrich ND, Fanchaouy M, Gusev K, Polakova E, Shirokova N, Niggli E. Changes of EC-coupling and RyR calcium sensitivity in dystrophic mdx mouse cardiomyocytes. Biophys J. 2009;96:10a–1a. [Google Scholar]
- 28.Ginsburg KS, Bers DM. Modulation of excitation–contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol. 2004;556:463–80. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng H. Beta-adrenergic stimulation synchronizes intracellular Ca2+ release during excitation–contraction coupling in cardiac myocytes. Circ Res. 2001;88:794–801. doi: 10.1161/hh0801.090461. [DOI] [PubMed] [Google Scholar]
- 30.Heinzel FR, Macquaide N, Biesmans L, Sipido K. Dyssynchrony of Ca2+ release from the sarcoplasmic reticulum as subcellular mechanism of cardiac contractile dysfunction. J Mol Cell Cardiol. 2011;50:390–400. doi: 10.1016/j.yjmcc.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak–load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 32.Szentesi P, Pignier C, Egger M, Kranias EG, Niggli E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ Res. 2004;95:807–13. doi: 10.1161/01.RES.0000146029.80463.7d. [DOI] [PubMed] [Google Scholar]
- 33.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–8. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008;77:245–55. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- 35.Xiao B, Tian X, Xie W, Jones PP, Cai S, Wang X, et al. Functional consequence of protein kinase A-dependent phosphorylation of the cardiac ryanodine receptor: sensitization of store overload-induced Ca2+ release. J Biol Chem. 2007;282:30256–64. doi: 10.1074/jbc.M703510200. [DOI] [PubMed] [Google Scholar]
- 36.Ramay HR, Liu OZ, Sobie EA. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res. 2011;91:598–605. doi: 10.1093/cvr/cvr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prosser BL, Ward CW, Lederer WJ. Subcellular Ca2+ signaling in the heart: the role of ryanodine receptor sensitivity. J Gen Physiol. 2010;136:135–42. doi: 10.1085/jgp.201010406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–8. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 39.Kashimura T, Briston SJ, Trafford AW, Napolitano C, Priori SG, Eisner DA, et al. In the RyR2(R4496C) mouse model of CPVT, beta-adrenergic stimulation induces Ca2+ waves by increasing SR Ca2+ content and not by decreasing the threshold for Ca2+ waves. Circ Res. 2010;107:1483–9. doi: 10.1161/CIRCRESAHA.110.227744. [DOI] [PubMed] [Google Scholar]
- 40.Stange M, Xu L, Balshaw D, Yamaguchi N, Meissner G. Characterization of recombinant skeletal muscle (Ser-2843) and cardiac muscle (Ser-2809) ryanodine receptor phosphorylation mutants. J Biol Chem. 2003;278:51693–702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.