

Abstract
X chromosome dosage compensation is required in male Drosophila to increase gene expression from the single X to equal that of both female X chromosomes. Although this is a sex-specific process, the MSL dosage compensation complex is thought to interface with non-sex-specific factors for both targeting and function. Therefore, a biochemical approach to MSL-associated factors is needed to complement genetic studies based on male-specific lethality. Here, we applied chromatin interacting protein-mass spectrometry (ChIP-MS) to identify MSL interactions on crosslinked chromatin, rather than focusing solely on complexes released from the DNA. Using this approach we identified MSL-enriched histone modifications, CG1832, a zinc finger protein implicated in initial MSL localization, and CG4747, a putative H3K36me3 binding protein. We found that CG4747 is associated with the bodies of active genes, coincident with H3K36me3, and is mis-localized in the set2 mutant lacking H3K36me3. CG4747 loss-of-function in vivo results in partial mis-localization of MSL complex to autosomes, and RNAi in cell culture confirms that CG4747 and SET2 function together to facilitate targeting of MSL complex to active genes. Our results demonstrate that the combination of crosslinking, affinity-purification, and mass spectrometry is a promising avenue for discovery of functional interactions on the chromatin template.
Keywords: Chromatin immunoprecipitation, mass spectrometry, histone modifications
INTRODUCTION
Eukaryotic genomes are highly annotated by specific chromosomal proteins and histone modifications along active genes, regulatory elements, or silent regions. An ongoing challenge is to decipher the rules that establish and maintain chromatin organization. Drosophila dosage compensation occurs via histone acetylation and transcriptional upregulation of the single male X chromosome to equal the output of both female X chromosomes1,2. Proteins that are specifically implicated in dosage compensation were discovered in genetic screens, as essential in males and dispensable in females3,4. The five proteins, MSL1, MSL2, MSL3, MOF, and MLE, are collectively called the MSL proteins based on their male-specific lethal mutant phenotype.
The MSL proteins associate specifically with active genes and acetylate H4K16ac on the male X chromosome5,6, and this targeting is proposed to occur in a multi-step process (reviewed in ref. 7). Initially, the MSL proteins are thought to recognize the X chromosome through co-transcriptional assembly at the roX1 and roX2 ncRNA genes, and by binding MSL recognition elements (MREs), which are sequences enriched at initial binding sites termed ‘chromatin entry sites’ (CES). The complex is then proposed to spread in cis to most active genes on the X to achieve its wild type binding pattern. This second step appears to be largely sequence-independent, as the complex can spread to active autosomal genes if attracted to the autosome by a roX RNA transgene8,9, or if autosomal genes are inserted on the X10. Therefore, general chromatin marks on active genes, such as histone H3K36me3, can facilitate MSL binding to X-linked genes, even though the modification itself is not X specific, but is found on all chromosomes9,11.
The five MSL proteins function together to achieve dosage compensation. MSL1 and MSL2 are essential for complex formation12,13. MSL3 is a chromodomain protein that binds chromatin and is implicated in recognition of methylated histones14–16. MOF is a MYST family histone acetyl-transferase that acetylates histone H4 lysine 16 (H4K16ac), resulting in the enrichment of this modification on active genes on the male X4,17–20. MLE is an RNA/DNA helicase21–23. All five MSL proteins are interdependent for their enriched X chromosomal localization, in support of the idea that they form a protein complex12,18,24. JIL-1, a histone H3 serine 10 kinase, is likewise implicated in dosage compensation based on its enrichment on the male X chromosome, which is genetically dependent on the MSL complex25,26.
The four proteins, MSL1, MSL2, MSL3 and MOF form a stable complex confirmed by biochemical purification27 and reconstitution with recombinant subunits14. However, in the absence of genetic analysis, the MLE helicase and JIL-1 kinase would not be linked to the MSL complex27. The interaction of MLE with the core MSL complex is highly sensitive to extraction conditions20,28. Therefore, we hypothesized that interactions of MLE, JIL-1, and other interesting factors with the core complex are not stably maintained under the conditions used to remove the complex from DNA. Therefore, we sought a method to identify such weak or transient yet functional interactions, including those that might only occur on chromatin. In addition, we sought to quantitate histone modifications associated with chromatin complexes in an unbiased rather than a candidate approach.
The trade-off between removing chromatin bound proteins from the DNA, to allow purification, and the resulting loss of weak or transient interactions with key partners has been addressed previously. One solution, developed in yeast, is to employ light sonication and wash solubilized chromatin under very mild conditions, to preserve protein interactions as much as possible29,30. Another approach in yeast is to use light crosslinking, again with sonication and washing under mild conditions31. We adopted a third solution, in which robust crosslinking is used to capture protein-protein interactions32–35. A key aspect of this approach, pioneered by the Huang and Kaiser groups, is the use of a 75-amino acid sequence from bacteria as an affinity tag that is recognized by endogenous biotinylation enzymes in both yeast and human32,33. The strong interaction of biotin with streptavidin allows stringent washing conditions to significantly decrease non-specific interactions.
By optimizing this approach for ChIP-MS of nuclear chromatin complexes in Drosophila S2 cells, we successfully identified MLE, JIL-1, and active histone marks (in particular H4K16ac), based on their enrichment in mass spectrometry of tagged MSL3 pulldowns. In addition, we identified novel candidates for MSL complex interaction on chromatin, including CG1832, a zinc finger protein recently implicated in initial MSL localization36, and CG4747, a putative H3K36me3 binding protein that we demonstrate facilitates MSL complex targeting to active genes. Therefore, ChIP-MS has significantly expanded our ability to capture key protein interactions involved in targeting MSL complex to the chromatin template.
RESULTS
Affinity purification of formaldehyde crosslinked MSL complex for protein analysis
In order to test the crosslinking - mass spectrometry approach in Drosophila, we inserted the HTB tag into genomic msl2 and msl3 transgenes, to generate transgenic flies expressing C-terminal MSL2-HTB or MSL3-HTB fusion proteins under their endogenous regulatory sequences. The HTB tag contains hexahistidine and biotinylation target sequences separated by a TEV protease cleavage site (Fig. 1a). The biotinylation target is a 75-amino acid sequence derived from a Propionibacterium shermanii transcarboxylase33,37. The MSL2-HTB and MSL3-HTB constructs rescued the respective msl2 or msl3 homozygous mutant male lethality, indicating that the fusion proteins are fully functional. We initially focused on MSL3-HTB by generating a stable Drosophila S2 cell line expressing the tagged protein. S2 cells were originally derived from embryos and are male in character demonstrating X chromosomal localization of the MSL complex5,6,12 and MSL-dependent transcriptional upregulation of active genes on the X chromosome38,39.
Figure 1. Use of ChIP-MS to identify proteins interacting with MSL3-HTB.

a. MSL2 (773 aa) and MSL3 (512 aa) proteins were C-terminally tagged with an HTB tag. His is a hexahistidine tag. Bio is a 75-amino acid (aa) sequence derived from a Propionibacterium shermanii transcarboxylase that is efficiently biotinylated in vivo in yeast and mammalian cells. TEV represents a cleavage site for the tobacco etch virus protease (9 aa) flanked by flexible linker regions (13 aa). RING: ring finger; CD: chromodomain.
b. Nuclei from S2 cells expressing HTB-tagged MSL3 were crosslinked, sonicated, and affinity-purified over streptavidin Dynabeads under denaturing conditions. Peptides were released by direct on-bead trypsin digestion and then identified by LC-MS/MS.
c. Streptavidin Dynabeads were boiled in reverse crosslinking buffer (Materials and Methods) and the eluate was loaded onto a 4–12% Tris-glycine gel followed by Western blot analysis. After MSL3-HTB pulldown, enrichment of MOF, MLE and JIL-1 was detected. MRG15 enrichment was not observed. IN, input; P, MSL3-HTB purification.
In adapting the crosslinking - mass spectrometry approach to chromatin proteins, we found that nuclear preparation prior to formaldehyde (FA) crosslinking significantly increased the subsequent chromatin concentration and thus protein yield after purification, and minimized the recovery of endogenous biotinylated proteins from the cytoplasm, allowing robust ChIP-MS in a single step. Crosslinked nuclei were sonicated to obtain soluble chromatin, and affinity purified using streptavidin coupled to magnetic beads in the presence of 6M urea and 0.2% SDS32 (Fig. 1b). To test for successful pulldown of MSL complex via MSL3-HTB, we performed Western blot analysis for MSL components (Fig. 1c). We observed strong enrichment of MOF in both the 1.7% and 3% FA crosslinked MSL3-HTB samples and no recovery from the non-HTB tagged sample, confirming efficient biotinylation of the tag in Drosophila cells.
The recovery of MOF was expected, as it is part of a soluble, core MSL complex previously seen to robustly co-IP and co-purify with MSL312,27. To determine whether FA crosslinking and streptavidin/biotin tagging also allowed efficient pulldown of MLE and JIL-1 with MSL3-HTB, we probed Western blots with their respective antibodies. Interestingly, we recovered MLE and JIL-1 at a level similar to that observed for MOF (Fig. 1c), validating the ChIP-MS approach. MRG15 is a chromodomain protein that localizes at transcription start sites in Drosophila40. No enrichment of MRG15 was observed, suggesting that our pulldowns were selective rather than non-specific. This selectivity was confirmed by mass spectrometry (see below).
Mass spectrometric analysis after single-step MSL3-HTB or MSL3-TAP affinity purification
Encouraged by the recovery of MOF, MLE, and JIL-1 with MSL3-HTB purifications, we proceeded to larger scale ChIP-MS. For each round of ChIP-MS, approximately 5X109 S2 cells were subjected to nuclear extraction, 3% FA crosslinking and subsequent purification with streptavidin coupled to magnetic beads. To determine our protein yield and purity, we boiled the beads in reverse crosslinking buffer, followed by SDS-PAGE. Colloidal blue staining demonstrated successful recovery of MSL3-HTB and a high level of complexity in the sample compared to purification from the negative control MSL3-TAP cells, lacking the biotinylated tag (Fig. 2a).
Figure 2. Characterization of MSL3 pulldowns to be analyzed by LC-MS/MS.

a. Colloidal blue staining shows efficient recovery of MSL3-HTB and a high level of protein complexity by single step streptavidin purification. IN, input; P, pulldown.
b. Colloidal blue-stained gel of MSL3-TAP purification with biotinylated IgG beads. MSL3-TAP and associated proteins were eluted with urea and SDS. Elution from IgG beads typically results in contamination of the eluate with free IgG (lane 2). We removed the biotinylated IgG using streptavidin magnetic beads. Both heavy and light chains were significantly depleted (lane 3).
The visualization of our results by colloidal blue staining suggested that the sample was of sufficient amount and purity for LC-MS/MS. Therefore, following streptavidin pulldown, we omitted the reverse cross-linking and gel electrophoresis steps and treated MSL3-HTB samples with direct on-bead trypsin digestion41,42 (Fig. 1b). All proteins identified in negative control cells lacking the HTB tag (Supplementary Table 1) were designated as false positives and removed from the MSL3-HTB pulldown list. Replicate ChIP-MS pulldowns with MSL3-HTB identified a number of proteins that consistently co-purified with MSL3-HTB (Table 1a). In addition to the core proteins MSL1, MSL2, MSL3 and MOF, we observed co-purification of MLE and JIL-1 with high total spectral counts, confirming the power of the crosslinking approach. Recovery of MSL2 was consistently lower than the other members of the MSL complex, but subsequent ChIP-MS of MSL2-HTB resulted in a highly overlapping list of proteins (Table 1a). Interestingly, even when MSL2-HTB was the bait protein, its recovery in terms of peptide count was lower than other members of the complex, suggesting a lower stoichiometry, or an intrinsic bias against recovery of MSL2 peptides in mass spectrometry.
Table 1. MSL3-associated proteins.
a. Proteins with the highest total peptide reads associated with MSL3-HTB after removal of proteins recovered in the negative control. Replicate experiments from single-step MSL3-HTB purifications with chromatin prepared at 3% formaldehyde crosslinking conditions showed similar results. ChIP-MS with MSL2-HTB and MSL3-TAP (using IgG purification) also recovered a highly overlapping list of proteins. MSL complex components and JIL-1 are highlighted in blue.
b. Proteins with the highest enrichment ratios after MSL3-HTB purification compared to chromatin input. Average peptide numbers from two MSL3-HTB purifications were used to calculate the relative ratio compared to input. A pseudocount of 0.5 was used to calculate the enrichment ratio.
| Table 1a | ||||||
|---|---|---|---|---|---|---|
| Gene | MSL3-HTB (#1) | MSL3-HTB (#2) | MSL3-HTB Avg | MSL2-HTB | MSL3-TAP | Molecular Feature |
| msl-3 | 122 | 136 | 129 | 60 | 62 | MSL complex |
| msl-1 | 82 | 123 | 103 | 93 | 55 | MSL complex |
| mle | 69 | 73 | 71 | 116 | 35 | MSL complex |
| mof | 72 | 66 | 69 | 59 | 29 | MSL complex |
| Top2 | 53 | 48 | 51 | 13 | 27 | Topoisomerase |
| CG7946 | 29 | 36 | 33 | 14 | 28 | PWWP domain |
| CG4747 | 22 | 32 | 27 | 16 | 8 | PWWP domain |
| CG9007 | 20 | 29 | 25 | 16 | 0 | SET domain |
| Hcf | 17 | 29 | 23 | 38 | 5 | Chromatin binding |
| JIL-1 | 13 | 27 | 20 | 20 | 18 | H3S10 kinase |
| NURF301 | 10 | 24 | 17 | 11 | 7 | Chromatin remodeling |
| CG12717 | 15 | 16 | 16 | 12 | 2 | Peptidase |
| SF2 | 12 | 19 | 16 | 2 | 3 | RNA splicing |
| msl-2 | 15 | 14 | 15 | 33 | 1 | MSL complex |
| FK506-bp1 | 13 | 16 | 15 | 4 | 4 | FK506 binding |
| Z4 | 14 | 14 | 14 | 3 | 4 | Zn finger |
| Ssrp | 10 | 17 | 14 | 0 | 2 | FACT complex |
| Ote | 8 | 19 | 14 | 0 | 1 | Transcription regulation |
| Spt16 | 8 | 17 | 13 | 0 | 5 | FACT complex |
| rump | 9 | 9 | 9 | 4 | 1 | RNA binding |
| pont | 11 | 5 | 8 | 2 | 3 | ATPase |
| glo | 9 | 7 | 8 | 0 | 2 | RNA localization |
| CG4038 | 10 | 4 | 7 | 1 | 0 | RNA binding |
| Wdr82 | 9 | 5 | 7 | 0 | 1 | COMPASS complex |
| CtBP | 8 | 5 | 7 | 2 | 2 | Transcription regulation |
| CG1832 | 8 | 2 | 5 | 6 | 3 | Zn finger |
| Table 1b | |||
|---|---|---|---|
| Gene | MSL3-HTB Avg | IN | Avg peptide/IN |
| msl-3 | 129 | 1 | 86 |
| msl-1 | 103 | 1 | 69 |
| CG12717 | 16 | 0 | 32 |
| mle | 71 | 8 | 8.4 |
| mof | 69 | 8 | 8.2 |
| msl-2 | 15 | 2 | 6.0 |
| Wdr82 | 7 | 1 | 5.0 |
| CG9007 | 25 | 5 | 4.5 |
| CG1832 | 5 | 1 | 3.7 |
| JIL-1 | 20 | 9 | 2.2 |
In addition to using the biotin tag, we also tested the applicability of crosslinking for the conventional TAP tag by performing affinity purification of MSL3-TAP using IgG linked to beads5. One major hurdle when using the protein A tag comes from excess IgG release during the elution step (Fig. 2b, lane 2), which interferes with mass spectrometry analysis. To eliminate excess IgG, we employed a depletion strategy. We coupled IgG to magnetic beads and then subjected them to in vitro biotinylation on free amines. After affinity purification and elution of MSL3-TAP and its interacting proteins under denaturing conditions, we could selectively deplete the biotinylated IgG with streptavidin beads (Fig. 2b, compare lanes 2 and 3). For mass spectrometry analysis, the IgG-depleted eluate was further subjected to desalting and detergent removal, followed by trypsin digestion and preparation for LC-MS/MS. The modified ChIP-MS of MSL3-TAP was less efficient in protein recovery but yielded similar protein candidates as that of ChIP-MS of MSL3-HTB (Table 1a).
Mass spectrometry identification of histone modifications associated with MSL complex
MSL complex is known to catalyze site-specific acetylation on histone H4 (H4K16ac), and proposed to utilize H3K36me3 to facilitate localization to active genes4,9,16,17. Both observations were initially based on the availability of antibodies that recognize these site-specific modifications in vivo. A particularly powerful use of ChIP-MS would be to identify the constellation of histone post-translational modifications (PTM) associated with specific protein complexes, without relying on antibodies and candidate approaches. However, analysis of modified histones cannot be achieved through simple trypsin digestion due to the high number of lysine residues in the N-terminal tails, as well as the difficulty of distinguishing partially digested peptides that are acetylated and methylated at multiple lysine residues43. Therefore, rather than using direct on-bead digestion, we sought to reduce the complexity of the samples by isolating intact histones through gel electrophoresis prior to LC-MS/MS. We found that direct loading of MSL3-HTB pulldowns on SDS-PAGE resulted in a large amount of the streptavidin monomer overlapping with histone H4 (data not shown). Therefore, we used S2 cells expressing MSL3-TAP rather than MSL3-HTB for ChIP-MS of modified histones.
To test whether histones treated with FA crosslinking are amenable to quantitative histone PTM mass spectrometry analysis, we quantitatively assessed uncrosslinked histones from S2 cells prepared by acid extraction and those prepared by gel electrophoresis of the crosslinked MSL3-TAP chromatin input. FA-crosslinked chromatin from S2 cells expressing MSL3-TAP was boiled in reverse crosslinking buffer, electrophoresed on a polyacrylamide gel and then stained with colloidal blue (Fig. 3a). The gel region containing histones was excised and histones were extracted. We found that the relative abundance of histone PTMs was comparable in acid-extracted histones and the FA crosslinked MSL3-TAP input (data not shown). This confirms that gel-extracted FA crosslinked histones can yield reliable and accurate quantitative data. We then performed MSL3-TAP pulldowns with IgG beads, excised the 4 histone bands from a colloidal blue-stained gel (Fig. 3a) and performed quantitative mass spectrometry for histone PTMs (Fig. 3b, Supplementary Table 2). When compared to input, H4K16ac is the most enriched PTM detected in our MSL3-TAP pulldowns. This is consistent with previous polytene and ChIP data showing its enrichment on the male X and role in dosage compensation4,9,17,19,44–46. H3K36me3 is also enriched, supporting previous studies demonstrating that this mark is involved in MSL complex targeting9,11. Other enriched PTMs include H3K79 methylation and H3K36me1 and me2, which are also marks reported to localize to the bodies of actively transcribed genes with a 3′ bias47,48. H3K79 methylation is proposed to play a positive role in transcriptional elongation (reviewed in ref. 49), and therefore could potentially play an additional role in MSL function.
Figure 3. Mass spectrometric identification of histones recovered from MSL3-TAP purification.
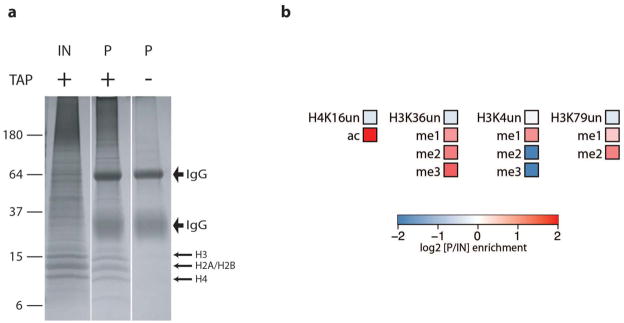
a. MSL3-TAP chromatin input and pulldown from single-step IgG purification were boiled in reverse crosslinking buffer, electrophoresed on an 18% Tris-glycine gel and stained with colloidal blue. The gel regions containing histones were excised and histones were extracted for mass spectrometry analysis of histone PTM. No histones were recovered after IgG purification of the no-tag control. IN, input; P, pulldown.
b. Heat maps showing the log2 of the enrichment ratio (P/IN) of histone PTMs associated with MSL3-TAP purification at positions: H3K4, H3K36, H3K79 and H4K16. H4K16ac, a modification deposited by MOF on the male X chromosome is highly enriched. There is also enrichment of histone PTMs known to localize at 3′ bodies of active genes such as H3K4me1, H3K36me2 and me2 and H3K79 me1 and 2. un: unmodified.
Reciprocal pulldowns confirm association of candidate MSL complex interactors
FA crosslinking stabilizes short-range protein-protein as well as protein-nucleic acid interactions. Therefore, we expected to identify key functional interactions, and also enrichment based on common localization to active chromatin. Many newly identified MSL-associated proteins in Table 1a are relatively abundant proteins that play a general role in active transcription (e.g. Top2, Hcf, and NURF30150–53), consistent with MSL localization to active gene bodies on the male X. Among the top abundant candidates, we selected CG4747, a PWWP domain-containing protein for further analysis. CG4747 is the Drosophila homolog of N-PAC/GLYR1, a human protein implicated in H3K36me3 binding. N-PAC/GLYR1 was identified in a mass spectrometry screen for proteins from HeLa cells that selectively bound H3K36me3 modified vs. unmodified histone H3 tail in vitro54. H3K36me3 is a histone modification thought to facilitate MSL targeting to active genes. It is laid down by the SET2 methyltransferase, which is recruited by elongating RNA polymerase II55–57. H3K36me3 is enriched in active gene bodies with a 3′ bias, and MSL binding to active genes on X is diminished in a set2 mutant, which lacks the H3K36me3 mark9,11. Co-purification of MSL complex with CG4747 might suggest a trivalent interaction58. Also co-purified was CG7946, a Drosophila homolog of mammalian Psip1/p52 (Table 1a). Psip1/p52 contains a closely related PWWP domain also demonstrated to bind methylated H3K3654,59–63. Psip1/p52 is thought to couple transcription to mRNA splicing through its binding to H3K36me3 and SF2, a splicing factor also identified in our ChIP-MS with MSL3 (Table 1a)59,60.
In considering additional candidates for further analysis, we employed an alternative way to examine our ChIP-MS list by calculating relative enrichment after affinity purification compared to the chromatin input (Table 1b). Using these criteria, core MSL subunits and MLE, which are highly enriched together on the X chromosome, gave a range of pulldown/input relative ratios of 6–86 fold after ChIP-MS. In contrast, JIL-1, which is a component of active chromatin on all chromosomes and is enriched approximately two-fold on the X, gave a more modest pulldown/input relative ratio of 2.2. By using the relative ratio over input as a parameter rather than total peptide recovery, we could identify CG1832/CLAMP, a low abundance zinc finger protein that was recently proposed to play a key role in initial recruitment of MSL complex to the MRE sequence36. CLAMP was not discovered in previous purifications, validating the utility of the ChIP-MS approach. The analysis of relative enrichment also called our attention to CG12717, a predicted sumo protease, CG9007, a SET domain protein related to MLL5 in humans64, and Wdr82, a known component of the Drosophila dSET1 COMPASS complex, that methylates histone H3 at lysine 4 and is associated with active transcription65–67. In addition to its involvement in COMPASS, the yeast and mammalian homologs of Wdr82 also play a role in transcription termination and mRNA processing, which are thought to account for its essential function in yeast68–70. Given MSL localization to active gene bodies with a 3′ bias, MSLs could interact with Wdr82 at these later steps in the transcription process. Consistent with this hypothesis, dSET1, the H3K4 methyltransferase of COMPASS thought to function mainly near 5′ ends of genes, was not enriched in our pulldowns.
To validate the specificity of the MSL3-HTB ChIP-MS, we tagged Wdr82 and CG4747 with C-terminal tags containing the biotinylation target sequence to examine reciprocal pulldowns. The tags were incorporated into genomic transgenes to preserve endogenous regulatory sequences for each gene. We created stable S2 cell lines expressing the tagged proteins and performed ChIP-MS via the biotin-streptavidin interaction. The reciprocal pulldowns confirmed the specificity of our MSL3 ChIP-MS, as we observed recovery of all MSL components and JIL-1 with both Wdr82 and CG4747 (Table 2). ChIP-MS of CG4747 suggests many associations in the nucleus, consistent with its relative abundance. Interestingly, of the MSL related components, JIL1 is most enriched, consistent with the close correlation of JIL-1 ChIP with H3K36me3 genome-wide71. ChIP-MS of Wdr82 is consistent with its known roles in 5′ COMPASS complex and 3′ processing and termination. In addition, MSL3, Wdr82, and CG4747 all showed a strong interaction with putative Drosophila homologues of the human MLL5 complex (CG9007, Smr, and ebi). Our results suggest that follow-up of reciprocal candidates could lead to a rich composite picture of active chromatin, that could be further integrated with localization maps derived from classical ChIP.
Table 2.
ChIP-MS of biotin tagged- CG4747 and Wdr82.All components of MSL complex and JIL-1 were reciprocally recovered with CG4747 and Wdr82 ChIP-MS. Selected additional proteins are categorized by complex or localization.
| Wdr82 (Total peptides) | CG4747 (Total peptides) | |
|---|---|---|
| Wdr82 | 27 | 0 |
| CG4747 | 9 | 156 |
| MSL complex | - | |
| msl-3 | 8 | 1 |
| msl-1 | 5 | 1 |
| mle | 5 | 2 |
| mof | 3 | 5 |
| JIL-1 | 3 | 19 |
| msl-2 | 2 | 1 |
| Set1/COMPASS | - | |
| dSet1 | 28 | 1 |
| dCXXC1 | 7 | 1 |
| Ash2 | 8 | 2 |
| Wds | 5 | 4 |
| Hcf | 47 | 52 |
| 3′ processing | - | |
| pAbp | 41 | 19 |
| CstF64 | 31 | 17 |
| Pcf11 | 11 | 2 |
| Cpsf73 | 8 | 0 |
| CstF50 | 5 | 0 |
| Bodies of active genes | ||
| CG7946 | 7 | 31 |
| SF2 | 15 | 2 |
| Rpd3 | 7 | 7 |
| MLL5 complex homology | ||
| CG9007 | 29 | 35 |
| Smr | 19 | 10 |
| ebi | 3 | 4 |
Colocalization of CG4747 with H3K36me3 on active genes
To investigate whether CG4747 colocalizes with MSL complex and H3K36me3 in vivo, we generated transgenic flies containing CG4747 tagged at the C-terminus with biotin and protein A tags (CG4747-TAP). We immunostained larval salivary gland polytene chromosomes with PAP antibody to detect the protein A moiety and found that tagged CG4747 displayed strong localization to interbands, indicating a potential role in active transcription (Fig. 4 a, b). Given the robust signal on polytene chromosomes, we performed chromatin immunoprecipitation coupled with next generation sequencing (ChIP-seq) from male and female larvae expressing CG4747-TAP. We found that the CG4747 binding pattern correlates with that of H3K36me3 genome-wide, and localizes to active gene bodies with a 3′-bias (Pearson linear correlation coefficient = 0.62, p<1e-10) (Fig. 5). Similar to what was observed for H3K36me3, X chromosome and autosomes were equally enriched for CG4747 (Fig. 5c). On the X chromosome, MSL3-TAP co-localized with CG4747 (Fig. 5a), consistent with MSL spreading to active genes and their chromatin marks in cis.
Figure 4. CG4747 colocalization with H3K36me3 is dependent on Set2.

a. Immunostaining for CG4747-TAP detected with PAP antibody (red) and H3K36me3 detected with a mouse monoclonal antibody (green). The mouse monoclonal antibody does not cross-react with the protein A tag. Hoechst staining of DNA is shown in blue. On wild-type larval salivary gland polytene chromosomes, CG4747 displays an interband binding pattern and colocalizes with H3K36me3. This pattern is disrupted in Set21 mutants, which lack H3K36me3.
b. Enlargement of CG4747-TAP and H3K36me3 binding patterns on boxed regions of polytene chromosomes shown in A.
Figure 5. CG4747 colocalizes with H3K36me3 and MSL3 on the X chromosome.

a. Representative ChIP-seq profiles of CG4747-TAP, H3K36me3 and MSL3-TAP on the X chromosome. H3K36me3 ChIP-seq data were obtained from the modENCODE consortium40 and MSL-TAP ChIP-seq results were described previously80. CG4747-TAP colocalization with H3K36me3 is observed on X chromosome and autosomes, whereas CG4747-TAP and MSL3-TAP colocalize only on the X chromosome. Top row genes are transcribed from left to right, and bottom row genes are transcribed from right to left. Numbers along the x-axis refer to chromosomal position, and the units are base pairs along the chromosomes. The y-axis shows the log2 ratio of IP/input ChIP-seq signal.
b. A high level of correlation was observed between ChIP-seq data from CG4747-TAP and H3K36me3. Red dots represent loci on the X chromosome (sampled from a 500 bp grid); blue represent autosomes, and bold green represent previously identified chromatin entry sites (CES)80. The correlation coefficients (Pearson linear correlation), together with the corresponding P-values are shown for all positions, X chromosome, and CES separately.
c. The metagene profiles show that CG4747-TAP (blue) and H3K36me3 (red) bind to bodies of genes with a 3′ bias. X chromosome (solid) and autosomes (dotted) are equally enriched.
The colocalization of CG4747 with H3K36me3 supports the hypothesis that CG4747 is an H3K36me3 binding protein. To test this possibility genetically, we crossed the CG4747-TAP transgene into the Set21 mutant background. Set2 is an essential gene in Drosophila, but mutant larvae survive to the third instar stage, allowing examination of their polytene chromosomes9. We found that CG4747-TAP displayed aberrant localization in the Set21 mutant background (Fig. 4 a, b). Instead of displaying a clearly defined interband immunostaining pattern, CG4747-TAP coated the polytene chromosomes, but without a discernable banding pattern. This diffuse staining may be attributable to the maternal contribution of Set2, or the relative abundance of CG4747, whose PWWP domain may have DNA-binding activity72,73. Its correct targeting to interbands, however, appears dependent on Set2, and thus strongly implicates H3K36me3. As a control for the specificity of this defect, we immunostained polytene chromosomes for the Z4 chromatin protein, which showed a robust interband staining pattern in both wild-type and the Set21 mutant (Supplementary Fig. 1).
Robust MSL targeting in S2 cells is dependent on Set2 and CG4747
To test the hypothesis that CG4747 acts synergistically with Set2+ to facilitate MSL targeting, we performed RNAi against these two genes in S2 cells, followed by MSL ChIP for selected target genes and chromatin entry sites (Fig. 6). For ChIP assays we use anti-MSL2 to detect the MSL complex because it gives the most robust enrichment. Although MSL2 localization is clearly diminished in Set21 mutant larvae9, RNAi for Set2 had little effect in S2 cells, perhaps due to incomplete knockdown of Set2 by RNAi or the difficulty in removing MSL complex once it is established. CG4747 RNAi alone decreased MSL2 localization (p= 4.8e-7, paired Wilcoxon test), and when both genes were knocked down, there was a stronger decrease in MSL targeting (p= 4.8e-7, paired Wilcoxon test). Notably, MSL2 binding at both target genes and previously identified chromatin entry sites were significantly depleted with Set2 and CG4747 double RNAi. Taken together, these results indicate that CG4747 participates with H3K36me3 in attracting MSL complex to active genes.
Figure 6. MSL2 localization is facilitated by CG4747.
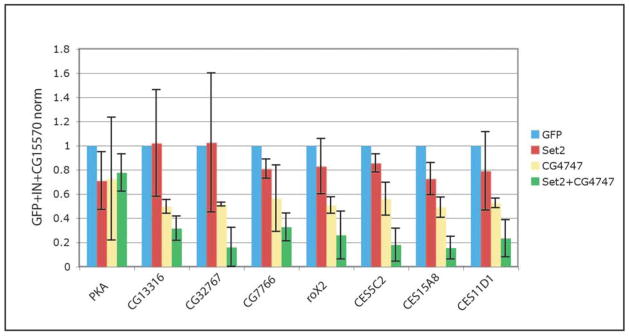
ChIP-qPCR shows that MSL2 localization is significantly decreased after double RNAi for Set2 and CG4747 (p= 4.8e-7, paired Wilcoxon test). Set2 or CG4747 RNAi alone caused a less severe decrease in MSL2 targeting. qPCR signal is normalized to input, CG15570 (an X-linked unbound gene) and the GFP RNAi control. PKA: autosomal unbound gene. CG13316, CG32767, CG7766: known MSL target genes. roX2, CES5C2, CES15A8, and CES11D1: chromatin entry sites.
Loss of CG4747 function causes aberrant MSL targeting on polytene chromosomes
In parallel, to investigate whether MSL targeting is dependent on CG4747 in the developing organism, we isolated a mutant allele of CG4747 by imprecise P-element excision of a neighboring P[EP] transposon (see Methods). We obtained deletion CG47473, which eliminates the putative promoter regions and 5′ UTRs of both CG4747 and the neighboring divergently transcribed gene Sister of Yb (SoYb). SoYb is a TUDOR domain-containing protein implicated in the piRNA pathway in the female germline74. We found that CG47473 causes late larval lethality in both males and females with few adult escapers (1–5%), and that female adult escapers are sterile. Analysis of RNA by quantitative RT-PCR of homozygous CG47473 male larvae showed lack of expression for both SoYb (CG31755) and CG4747 (data not shown).
To investigate whether the CG47473 deletion affects MSL targeting we immunostained homozygous mutant male polytene chromosomes with anti-MSL2 antibodies. We found that X chromosome localization was still evident, but in conjunction with unusual, mis-localization of MSL complex to ectopic sites on autosomes (Fig. 7a). Since we did not isolate a mutant that deleted solely the CG4747 gene, we tested whether either CG4747 RNAi or SoYb RNAi could cause the same phenotype. We immunostained polytene chromosomes from male larvae expressing either a CG4747 or SoYb RNAi hairpin driven by actin-5C-GAL4 (A5C-GAL4)75, and found that A5C-GAL4>4747i males showed ectopic MSL binding sites on autosomes while A5C-GAL4>SoYbi did not (Fig. 7 c, d). These results confirm that the abnormal MSL targeting observed in CG47473 male polytene chromosomes is due to loss of CG4747, not SoYb. Taken together, our results support a model in which CG4747 facilitates MSL recognition of active genes on the X chromosome. In the absence of CG4747, retention of MSL complex on the X chromosome in cis may be less efficient, resulting in release and ectopic binding on autosomes.
Figure 7. MSL complex localization is disrupted in CG47473 mutant males.
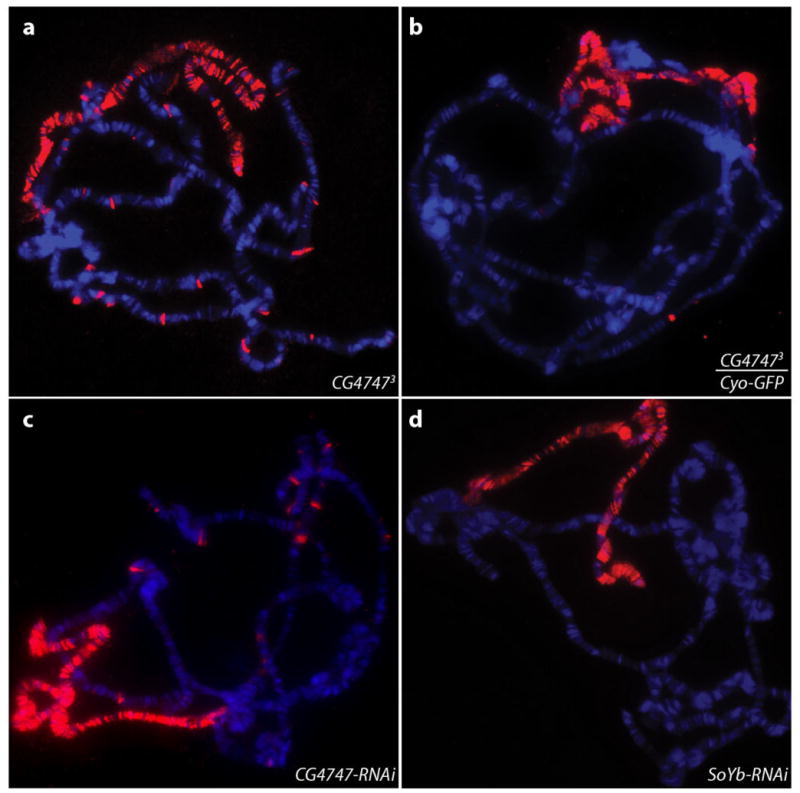
a. MSL2 immunostaining (red) of salivary gland polytene chromosomes from CG47473 mutant male larvae shows ectopic autosomal binding sites. DNA stained by Hoechst is shown in blue. For each genotype, more than 20 nuclei from multiple salivary glands were examined.
b. MSL2 staining of CG47473/Cyo-GFP males shows a wild-type pattern MSL complex localization pattern. Few autosomal binding sites are seen.
c. MSL2 staining of male larvae expressing a CG4747 RNAi hairpin driven by A5C-GAL4 shows ectopic autosomal binding sites similar to those observed in CG47473 mutant male larvae.
d. Wild-type MSL pattern is observed with MSL2 staining of male larvae expressing a SoYb RNAi hairpin driven by A5C-GAL4.
DISCUSSION
In this study, we successfully optimized an affinity-purification method to identify interacting proteins and histone modifications associated with the Drosophila MSL complex. By combining crosslinking and a biotinylation targeting sequence, we were able to preserve transient or low affinity interactions that may only be captured on the intact chromatin template. The biotinylation tag allows stringent washing conditions to significantly decrease non-specific interactions. Additionally, the high affinity of biotin-streptavidin and the compact structure of streptavidin allows direct on-bead trypsin digestion, thus comprehensive analysis by LC-MS/MS32,33. Since the biotinylation of the target sequence is catalyzed by the cells’ endogenous biotin ligase, there is no need to introduce an exogenous biotin ligase gene, such as in the BirA/biotinylation tag system76,77. This provides the advantage of convenient adaptation in both cells and potentially whole organisms. Improvements such as development of a dual tag that will function well on crosslinked material should enable ChIP-MS to be adapted to crude tissues and developmental stages in the future.
Our studies of CG4747 revealed that it colocalizes with H3K36me3 on chromatin and that its concentration in interbands of polytene chromosomes is dependent on the function of Set2. This strongly suggests that CG4747 is indeed an H3K36me3 binder, as is the homologous human protein, N-PAC/GLYR154, and an additional human protein with a closely related PWWP domain, Psip/p5260. We demonstrated that CG4747 is crucial for robust targeting of MSL complex in both Drosophila S2 cells and male salivary gland polytene chromosomes, providing another functional link between H3K36me3 and MSL targeting of active genes. We hypothesize that the ectopic autosomal sites observed in CG4747 knockdown in vivo are a consequence of diffusion of MSL complex if it fails to be efficiently captured on the X chromosome.
Previous studies suggested that MSL3, through its chromodomain, is responsible for the recognition of H3K36me39,16. Our analysis indicates that CG4747 also binds to H3K36me3 and recruits MSL complex. These hypotheses are not mutually exclusive as there could be a trivalent interaction between MSL3, CG4747 and the multiple histone H3 tails on one or neighboring nucleosomes58. Furthermore, in vitro the isolated MSL3 chromodomain interacts best with H4K20me1 peptides78,79; the culmination of such interactions and others may function together to stabilize MSL complex on its functional substrates, active X-linked genes.
Our success using ChIP-MS on the MSL complex supports the general applicability of this method for unbiased analysis of chromatin-associated proteins. In addition to identifying interacting partners of a chromatin complex, we also successfully performed quantitative histone post-translational modification mass spectrometry with the crosslinking method, using a protein A rather than HTB tag. Histone H4K16ac was the most enriched PTM with MSL3 pulldown, along with other histone modifications with known localization to the bodies of actively transcribed genes. As the ability to analyze post-translational modifications by mass spectrometry continues to improve, this approach has the exciting possibility of allowing a comprehensive and unbiased survey of the combinatorial histone modifications associated with any chromatin complex.
METHODS
Plasmids and cloning
The pGS-mw[+] vector was kindly provided by Dr. G. Schotta81 and used as a backbone to make a new vector, pFly, which contains P-element ends, a single attB site and is marked with mini-white+. To introduce genomic constructs of the proteins of interest and tag sequences into pFly, we employed a similar cloning strategy as described previously5. C-terminal tags were introduced between the coding region and 3′UTR while disrupting the native stop codon, achieving in-frame expression of the proteins of interest and tags. The HTB tag and Bio sequences were PCR-amplified from the pFA6a-HTB-Kmc plasmid from Dr. P. Kaiser33. The sequences of the Protein A and TEV cleavage site were obtained from the pBS1479 vector82. Complete sequences of plasmids are available upon request.
S2 cell transfection
Stable transformants were generated as previously described5.
ChIP-MS chromatin preparation
Chromatin preparation was prepared as previously described35 with modifications. S2 cells were grown in CCM3 media (ThermoFisher) to reach a density of 106 to 107 cells/ml. 1L of cells was washed in PBS once and resuspended in 200 ml nuclear extraction buffer (10% sucrose; 10 mM NaCl; 3 mM MgCl2; 20 mM HEPES, pH=7.9; 1 mM PMSF) and dounced 5 times with a loose pestle and 5 times with a tight pestle. 200 ml of nuclei were added to 800 ml of PBS containing formaldehyde, reaching a final concentration of 3%, and crosslinked for 30 min at room temperature (RT) (or 1.7% formaldehyde for 10 min at RT). Nuclei were centrifuged at 3,000xg for 10 min at 4°C, washed in PBS four times, equilibrated in sucrose buffer (0.3 M sucrose; 1% Triton X-100; 3 mM CaCl2; 2 mM MgOAc; 10 mM HEPES, pH=7.9; 1 mM PMSF), and dounced with a tight pestle. Nuclei were equilibrated in glycerol buffer (25% glycerol; 0.1 mM EDTA; 0.1 mM EGTA; 5 mM MgOAc; 10 mM HEPES, pH=7.9; 1 mM PMSF) and resuspended in the same volume of glycerol buffer. The pellet was frozen in liquid nitrogen and stored at −80°C or used immediately for sonication. Approximately 3×109 nuclei were centrifuged at 2,500xg for 10 min at 4°C, resuspended in PBS-Triton and dounced lightly to remove clumps. Two PBS washes were performed followed by lysis in lysis buffer (0.2% SDS; 0.1% sarkosyl; 2 mM EDTA; 1 mM EGTA; 50 mM NaCl; 10 mM HEPES, pH=7.9; 1 mM PMSF). Samples were sonicated (Micro-tip, Misonix-3000) at Power setting 7 (36–45 Watts), 15 s constant pulse, and 45 s pause for a 7 min total processing time. Chromatin was collected by centrifugation at 16,000xg for 15 min at RT and preserving the supernatant while discarding the pellet.
For mass spectrometry analysis of input, Pierce detergent removal spin columns (Thermo) and Amicon Ultra-0.5 ml centrifugal filters-10kD (Millipore) were used to remove detergent and salt. Recovered proteins were resuspended in 100 nM ammonium bicarbonate (pH=8.0) and incubated with 10 ng/μl trypsin (Promega) at 37°C overnight before further processing.
Streptavidin pulldown
The chromatin solution was adjusted to contain final concentrations of 6M urea, 0.2% SDS, 0.2 M NaCl and 100 mM Tris, pH=7.4. For 10 ml of adjusted chromatin, 400 μl of MyOne Streptavidin C1 Dynabeads (Invitrogen) pre-equilibrated in urea pulldown buffer (6 M urea; 0.2% SDS; 0.2 M NaCl; 100 mM Tris, pH=7.4) were added for overnight incubation at RT. Beads were washed with urea wash buffer (6 M urea; 2% SDS; 0.2 M NaCl; 100 mM Tris, pH=7.4) two times at RT, low-Tween TEN buffer (0.005% Tween-20; 0.5 mM EDTA; 150 mM NaCl; 10 mM Tris, pH=7.4) three times at RT, lysis buffer (0.2%SDS; 2 mM EDTA; 50 mM NaCl; 10 mM HEPES, pH=7.9) three times at 42°C, and low-Tween TEN buffer for two times at RT. Beads were then moved to 1.5 mL tubes and washed six times with detergent-free buffer (0.2 M NaCl; 100mM Tris, pH=7.4) at RT. For SDS-PAGE, beads were boiled in 200 μl of reverse crosslinking buffer (2% SDS; 0.5 M 2-mercaptoethanol; 250 mM Tris, pH=8.8) and incubated at 100°C for 25 min. The proteins were separated using a 4–12% Tris-glycine acrylamide gel (Invitrogen) and analyzed by colloidal blue staining (Invitrogen) or Western blotting.
For mass spectrometry analysis, beads were incubated with 10 ng/μl trypsin (Promega) in 100 nM ammonium bicarbonate, pH=8.0 and 10% acetonitrile at 37°C overnight before further processing.
IgG pulldown of MSL3-TAP
The chromatin solution was adjusted to contain final concentrations of 0.1% SDS; 0.5% TritonX-100; 2 mM EDTA; 250 mM NaCl, 20 mM Tris, pH=8. For 10 ml of adjusted chromatin, 400 μl of IgG-conjugated Dynabeads M-270 (Invitrogen, protocol: Moazed lab) pre-equilibrated in 250 mM NaCl Buffer (0.1% SDS; 1% TritonX-100; 2 mM EDTA; 250 mM NaCl; 50 mM Tris, pH=8) were added for overnight incubation at 4°C. Beads were washed with 250 mM NaCl Buffer two times, 500 mM NaCl buffer (0.1% SDS; 1% TritonX-100; 2 mM EDTA; 500 mM NaCl; 50 mM Tris, pH=8) 6 times and PBST (0.1% TritonX-100) two times.
For histone PTM analysis, beads were boiled in 80 μl reverse crosslinking buffer and incubated at 100°C for 25 min. The samples were separated using an 18% Tris-glycine acrylamide gel (Invitrogen) and analyzed by colloidal blue staining (Invitrogen) to visualize histones. Gel slices containing histones were excised and subjected to histone PTM mass spectrometry.
For analysis of interacting proteins, IgG-coupled magnetic beads were biotinylated using the EZ-Link Sulfo-NHS-Biotin and Biotinylation Kit (Thermo). MSL3-TAP purification was performed followed by elution with 600 μl of urea pulldown buffer at RT for 30 minutes. For depletion of IgG, the above eluate was incubated with 200 μl of MyOne Streptavidin C1 Dynabeads (Invitrogen) for 3 hours at RT. Pierce detergent removal spin columns (Thermo) and Amicon Ultra-0.5 ml centrifugal filters-10kD (Millipore) were used to remove detergent and salt. Recovered affinity purified proteins were resuspended 100 nM ammonium bicarbonate, pH=8.0 and incubated with 10 ng/μl trypsin (Promega) at 37°C overnight before further processing.
Western blotting
Western blotting was conducted using the WesternBreeze immunodetection kit (Invitrogen). Primary antibodies were used at the following dilutions: 1) anti-MLE (1:2,000) (SDI); 2) anti-MOF (1:2,000) (SDI); 3) anti-JIL-1 (1:,2,000)(SDI); 4) anti-MRG15 (1:2,000)(SDI).
Mass spectrometry
Digested peptides were extracted and desalted using Vivapure C18 micro spin columns (Sartorius Stedim). Subsequently, peptides were lyophilized, dissolved in 5% acetonitrile/5% formic acid, and loaded onto a reversed phase microcapillary column (100 mm I.D.) packed with 18 cm of Maccel C18AQ resin (3 mm, 200 A, The Nest Group, Inc). Peptides were eluted using a gradient of 4%–26% acetonitrile in 0.125% formic acid over 120 min and detected in a hybrid linear ion trap-orbitrap mass spectrometer (LTQ-Orbitrap Discovery, ThermoFisher). Precursors selected for MS/MS fragmentation were corrected for errors in monoisotopic peak assignment, and tandem MS spectra were searched using version 28 of the Sequest algorithm. Searches occurred against the FlyBase database (Dmel Release 5.9) containing all Drosophila melanogaster proteins in both forward and reversed directions. Variable modifications included oxidation of methionine (15.994946). The precursor mass tolerance was set to 25 ppm, and two missed cleavages were allowed. The target-decoy method was used to estimate false discovery rates (FDR)83, and linear discriminant analysis (LDA) was employed to filter peptides to an initial 1% peptide-level FDR, indicated by the number of decoy sequences in the filtered data set. Subsequently, peptides were assembled into proteins and further filtered to a protein-level FDR of 1%, as described in Huttlin et al.84. After applying these filters, final peptide-level FDRs were below 0.30% for all runs.
Quantitative mass spectrometry of histones
After colloidal blue staining (Invitrogen), the histone bands were excised and destained overnight in 40% EtOH, 10% AcOH solution. The gel pieces were rinsed in NH4HCO3 for 5 min and then covered in 100% MeCN on a shaker for 10 mins. The samples were dried in a vacuum centrifuge, covered in a 1:1 solution of 100 mM NH4HCO3 and propionic anhydride, and vortexed for 15 mins. After two rounds of propionylation, the dried gel pieces were rehydrated with 12.5 ng/μl of trypsin diluted in 100 mM NH4HCO3 and left overnight at RT. The resulting tryptic peptides were extracted from gel slices as previously described85 and further subjected to two rounds of propionylation in-solution as described86. Histone peptides were desalted and sequenced by nano-HPLC/MS-MS as previously described87. Briefly, samples were loaded via an Eksigent autosampler (Eksigent Technologies) onto a 75 μm C18 reverse phase capillary column and resolved on a 110-minute 1–100% buffer B gradient (buffer A = 0.1 mol/L AcOH, buffer B = 70% MeCN in 0.1 mol/L AcOH at 0.070 ml/min on an Agilent HPLC system). The peptides were directly electrosprayed into and analyzed by an LTQ-Orbitrap (ThermoFisher). Peak abundance for each modified peptide was calculated by manual chromatographic peak integration of full MS scans using Qual Browser software (ThermoFisher). Precursor ions of low-level modifications were selected for targeted analysis to increase identification of these PTMs. The relative abundance of each modified state was quantified by expressing the area under the curve for each peak as a percentage of the total histone peptide.
S2 cell RNAi
Methods for dsRNA synthesis and RNAi treatment are described on www.flyrnai.org.
Chromatin immunoprecipitation and analysis by qPCR
Chromatin preparation from S2 cells and larvae and ChIP assays were performed as described5,46. Primer sequences for MSL target genes were also described previously9,46,80. 0.4 μl of anti-MSL2 serum was used for each IP containing 40–100 μg of chromatin. We calculated the ΔCt for each IP relative to input and the X-linked CG15570 gene, which is not bound by MSL complex. The relative MSL2 enrichment was calculated by normalizing to the GFP RNAi sample, which acts as a negative control. We performed RNAi experiments in S2 cells in triplicate and calculated the standard deviations between replicates. MSL2 ΔCt relative to input and CG15570 was used to perform paired Wilcoxon test with the assumption that each locus tested is independent.
ChIP-seq library preparation, sequencing, and analysis
Immunoprecipitated DNA was isolated as described in Alekseyenko et al. (2008)80. 300 ng of DNA was used for ChIP-seq library preparation. DNA amplification was performed using the NEBNext ChIP-seq library prep master mix set for Illumina (NEB) and Illumina Truseq adapter indexes (Illumina) with minor modifications. One μl of 1:25 dilution of each Illumina Truseq adapter index oligo was used per 300 ng ChIP DNA. Adapter ligated products were PCR amplified for 12 cycles. Purified DNA was sequenced on the Illumina Hiseq platform.
The sequenced reads were aligned to the dm3 genome assembly using bowtie, recording only reads that can be aligned uniquely. The enrichment profiles along the genome (smoothed maximum likelihood estimates) were estimated using SPP package88. For illustrating metagene enrichment profiles (Fig. 5c), only genes longer than 1 Kbp were taken into account.
Immunostaining of polytene chromosomes
Polytene chromosomes were isolated and processed for immunostaining as described8. Primary antibodies were used at the following dilutions: 1) PAP antibody (1:200) (Sigma); 2) mouse anti-H3K36me3 (1:1,000) (Active Motif); 3) mouse anti-Z4 (1:1,000)89; 4) rabbit anti-MSL2 serum (1:500). Secondary antibodies used were 1) anti-rabbit AlexaFluor555 (1:500) (Invitrogen); 2) anti-mouse AlexaFluor488 (1:500) (Invitrogen). Hoechst dye (Invitrogen) was used to stain DNA.
Generation of CG4747 deletion alleles
A series of CG4747 mutant alleles was generated by imprecise P-element excision of the P[EP] transposon from the y1 w*; P{w[+mC]=EP}G2585 stock, where this element was integrated at 2L:10,002 (Dmel release=r5.44). Briefly, jump-start males were obtained from a cross of y1 w*; P{w[+mC]=EP}G2585 females and wg[Sp-1]/CyO; ry[506] Sb[1] P{ry[+t7.2]=Delta2-3}99B/TM6 males. These were mated to If/Cyo females. P-element excision events were screened among the male progeny that had lost expression of the white+ marker gene from P[EP]. Such flies were crossed to If/Cyo females and balanced stocks were established. To identify stocks with excisions, we isolated DNA using the DNeasy tissue DNA extraction kit (Qiagen), and performed PCR with primers flanking the P[EP] insertion. The CG47473 allele is a deletion of 2L: 10001515-10004009 (Dmel release=r5.47).
Drosophila stocks and transgenesis
The following were obtained from the Bloomington Stock Center: y1 w*; P{Act5C-GAL4}17bFO1/TM6B, Tb1 (Bloomington stock 3954); y1 w*; P{w[+mC]=EP}G2585 (Bloomington stock 28446); wg[Sp-1]/CyO; ry[506] Sb[1] P{ry[+t7.2]=Delta2-3}99B/TM6 (Bloomington stock 2535).
CG4747 RNAi line (y1 sc* v1; P{TRiP.HMS00568}attP2; Bloomington stock 33696) and SoYb RNAi line (TRiP stock GL01045) were obtained from the TRiP/Drosophila RNAi Screening Center at Harvard Medical School.
CG4747-TAP transgenic flies were generated using φC31 integrase-mediated attB/attP recombination at cytological location 76A2 (PBac{yellow[+]-attP-9A}VK00013; Bloomington stock 9732) (BestGene Inc.). Transgenics were identified by mini-white expression and balanced to obtain homozygous lines.
Supplementary Material
Acknowledgments
We thank Dr. G. Schotta (Ludwig-Maximilians Universität) for the pGS-mw[+] vector. The anti-Z4 antibody was a generous gift from Dr. H. Saumweber (Humboldt University). We thank E. Gerace from the Moazed lab (Harvard Medical School) for providing the protocol for coupling IgG to magnetic beads. We thank the Bloomington Stock Center and TRiP at Harvard Medical School (NIH/NIGMS R01-GM084947) for providing fly stocks used in this study. We are grateful to M. Gelbart for support and expertise, E. Smith for technical assistance, and A. Ciccia, B. Adamson and A. Plachetka for critical reading of the manuscript. This work was supported by grants from the NIH to M.I.K. (GM45744), P.V.K. (K25AG037596), and S.J.E. (GM44664). A.E.H.E. is supported by fellowships from The Jane Coffin Childs Foundation and The American Society for Radiation Oncology. B.A.G. is supported by a National Science Foundation Early Faculty CAREER award, and NIH award number DP2OD007447 from the Office of the Director.
Footnotes
AUTHOR CONTRIBUTIONS
C.I.W., A.A.A and A.A.G. performed ChIP-MS experiments; A.E.H.E. performed LC-MS/MS to identify MSL-interacting proteins; G.L. and L-M.P.B. performed quantitative mass spectrometry of histone PTM; C.I.W. performed all other experiments; P.V.K. performed all bioinformatics analyses; S.J.E., B.A.G. and M.I.K. supervised analyses; C.I.W. and M.I.K. prepared the manuscript in consultation with all co-authors.
References
- 1.Lucchesi JC, Kelly WG, Panning B. Chromatin remodeling in dosage compensation. Annu Rev Genet. 2005;39:615–651. doi: 10.1146/annurev.genet.39.073003.094210. [DOI] [PubMed] [Google Scholar]
- 2.Conrad T, Akhtar A. Dosage compensation in Drosophila melanogaster: epigenetic fine-tuning of chromosome-wide transcription. Nat Rev Genet. 2011;13:123–134. doi: 10.1038/nrg3124. [DOI] [PubMed] [Google Scholar]
- 3.Belote JM, Lucchesi JC. Control of X chromosome transcription by the maleless gene in Drosophila. Nature. 1980;285:573–575. doi: 10.1038/285573a0. [DOI] [PubMed] [Google Scholar]
- 4.Hilfiker A, Hilfiker-Kleiner D, Pannuti A, Lucchesi JC. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J. 1997;16:2054–2060. doi: 10.1093/emboj/16.8.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev. 2006;20:848–857. doi: 10.1101/gad.1400206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilfillan GD, et al. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev. 2006;20:858–870. doi: 10.1101/gad.1399406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gelbart ME, Kuroda MI. Drosophila dosage compensation: a complex voyage to the X chromosome. Development. 2009;136:1399–1410. doi: 10.1242/dev.029645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelley RL, et al. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell. 1999;98:513–522. doi: 10.1016/s0092-8674(00)81979-0. [DOI] [PubMed] [Google Scholar]
- 9.Larschan E, et al. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell. 2007;28:121–133. doi: 10.1016/j.molcel.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Gorchakov AA, Alekseyenko AA, Kharchenko P, Park PJ, Kuroda MI. Long-range spreading of dosage compensation in Drosophila captures transcribed autosomal genes inserted on X. Genes Dev. 2009;23:2266–2271. doi: 10.1101/gad.1840409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell O, et al. Transcription-coupled methylation of histone H3 at lysine 36 regulates dosage compensation by enhancing recruitment of the MSL complex in Drosophila melanogaster. Mol Cell Biol. 2008;28:3401–3409. doi: 10.1128/MCB.00006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Copps K, et al. Complex formation by the Drosophila MSL proteins: role of the MSL2 RING finger in protein complex assembly. EMBO J. 1998;17:5409–5417. doi: 10.1093/emboj/17.18.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott MJ, Pan LL, Cleland SB, Knox AL, Heinrich J. MSL1 plays a central role in assembly of the MSL complex, essential for dosage compensation in Drosophila. EMBO J. 2000;19:144–155. doi: 10.1093/emboj/19.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morales V, et al. Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J. 2004;23:2258–2268. doi: 10.1038/sj.emboj.7600235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buscaino A, Legube G, Akhtar A. X-chromosome targeting and dosage compensation are mediated by distinct domains in MSL-3. EMBO Rep. 2006;7:531–538. doi: 10.1038/sj.embor.7400658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sural TH, et al. The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nat Struct Mol Biol. 2008;15:1318–1325. doi: 10.1038/nsmb.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turner BM, Birley AJ, Lavender J. Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei. Cell. 1992;69:375–384. doi: 10.1016/0092-8674(92)90417-b. [DOI] [PubMed] [Google Scholar]
- 18.Gu W, Szauter P, Lucchesi JC. Targeting of MOF, a putative histone acetyl transferase, to the X chromosome of Drosophila melanogaster. Dev Genet. 1998;22:56–64. doi: 10.1002/(SICI)1520-6408(1998)22:1<56::AID-DVG6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 19.Akhtar A, Becker PB. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol Cell. 2000;5:367–375. doi: 10.1016/s1097-2765(00)80431-1. [DOI] [PubMed] [Google Scholar]
- 20.Smith ER, et al. The drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol. 2000;20:312–318. doi: 10.1128/mcb.20.1.312-318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee CG, Chang KA, Kuroda MI, Hurwitz J. The NTPase/helicase activities of Drosophila maleless, an essential factor in dosage compensation. EMBO J. 1997;16:2671–2681. doi: 10.1093/emboj/16.10.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morra, Smith, Yokoyama, Lucchesi The MLE subunit of the Drosophila MSL complex uses its ATPase activity for dosage compensation and its helicase activity for targeting. Mol Cell Biol. 2007 doi: 10.1128/MCB.00995-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izzo A, Regnard C, Morales V, Kremmer E, Becker PB. Structure-function analysis of the RNA helicase maleless. Nucleic Acids Res. 2008;36:950–962. doi: 10.1093/nar/gkm1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyman LM, Copps K, Rastelli L, Kelley RL, Kuroda MI. Drosophila male-specific lethal-2 protein: structure/function analysis and dependence on MSL-1 for chromosome association. Genetics. 1997;147:1743–1753. doi: 10.1093/genetics/147.4.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin Y, et al. JIL-1: a novel chromosomal tandem kinase implicated in transcriptional regulation in Drosophila. Mol Cell. 1999;4:129–135. doi: 10.1016/s1097-2765(00)80195-1. [DOI] [PubMed] [Google Scholar]
- 26.Jin Y, Wang Y, Johansen J, Johansen KM. JIL-1, a chromosomal kinase implicated in regulation of chromatin structure, associates with the male specific lethal (MSL) dosage compensation complex. J Cell Biol. 2000;149:1005–1010. doi: 10.1083/jcb.149.5.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendjan S, et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell. 2006;21:811–823. doi: 10.1016/j.molcel.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Smith ER, et al. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol. 2005;25:9175–9188. doi: 10.1128/MCB.25.21.9175-9188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert JP, Mitchell L, Rudner A, Baetz K, Figeys D. A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol Cell Proteomics. 2009;8:870–882. doi: 10.1074/mcp.M800447-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert JP, et al. Defining the budding yeast chromatin-associated interactome. Mol Syst Biol. 2010;6:448. doi: 10.1038/msb.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smart SK, Mackintosh SG, Edmondson RD, Taverna SD, Tackett AJ. Mapping the local protein interactome of the NuA3 histone acetyltransferase. Protein Sci. 2009;18:1987–1997. doi: 10.1002/pro.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guerrero C, Tagwerker C, Kaiser P, Huang L. An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol Cell Proteomics. 2006;5:366–378. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Tagwerker C, et al. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivocross-linking. Mol Cell Proteomics. 2006;5:737–748. doi: 10.1074/mcp.M500368-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Tardiff DF, Abruzzi KC, Rosbash M. Protein characterization of Saccharomyces cerevisiae RNA polymerase II after in vivo cross-linking. Proceedings of the National Academy of Sciences. 2007;104:19948–19953. doi: 10.1073/pnas.0710179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Déjardin J, Kingston RE. Purification of proteins associated with specific genomic Loci. Cell. 2009;136:175–186. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larschan E, et al. Identification of Chromatin-Associated Regulators of MSL Complex Targeting in Drosophila Dosage Compensation. PLoS Genet. 2012;8:e1002830. doi: 10.1371/journal.pgen.1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cronan JE. Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J Biol Chem. 1990;265:10327–10333. [PubMed] [Google Scholar]
- 38.Hamada FN, Park PJ, Gordadze PR, Kuroda MI. Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev. 2005;19:2289–2294. doi: 10.1101/gad.1343705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Straub T, Gilfillan GD, Maier VK, Becker PB. The Drosophila MSL complex activates the transcription of target genes. Genes Dev. 2005;19:2284–2288. doi: 10.1101/gad.1343105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kharchenko PV, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kho Y, et al. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc Natl Acad Sci USA. 2004;101:12479–12484. doi: 10.1073/pnas.0403413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rybak JN, et al. In vivo protein biotinylation for identification of organ-specific antigens accessible from the vasculature. Nat Methods. 2005;2:291–298. doi: 10.1038/nmeth745. [DOI] [PubMed] [Google Scholar]
- 43.Plazas-Mayorca MD, et al. One-pot shotgun quantitative mass spectrometry characterization of histones. J Proteome Res. 2009;8:5367–5374. doi: 10.1021/pr900777e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith ER, Allis CD, Lucchesi JC. Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J Biol Chem. 2001;276:31483–31486. doi: 10.1074/jbc.C100351200. [DOI] [PubMed] [Google Scholar]
- 45.Kind J, et al. Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell. 2008;133:813–828. doi: 10.1016/j.cell.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 46.Gelbart ME, Larschan E, Peng S, Park PJ, Kuroda MI. Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat Struct Mol Biol. 2009;16:825–832. doi: 10.1038/nsmb.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pokholok DK, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 48.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mondal N, Parvin JD. DNA topoisomerase IIalpha is required for RNA polymerase II transcription on chromatin templates. Nature. 2001;413:435–438. doi: 10.1038/35096590. [DOI] [PubMed] [Google Scholar]
- 51.Xiao H, et al. Dual functions of largest NURF subunit NURF301 in nucleosome sliding and transcription factor interactions. Mol Cell. 2001;8:531–543. doi: 10.1016/s1097-2765(01)00345-8. [DOI] [PubMed] [Google Scholar]
- 52.Badenhorst P, Voas M, Rebay I, Wu C. Biological functions of the ISWI chromatin remodeling complex NURF. Genes Dev. 2002;16:3186–3198. doi: 10.1101/gad.1032202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guelman S, et al. Host cell factor and an uncharacterized SANT domain protein are stable components of ATAC, a novel dAda2A/dGcn5-containing histone acetyltransferase complex in Drosophila. Mol Cell Biol. 2006;26:871–882. doi: 10.1128/MCB.26.3.871-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vermeulen M, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 55.Strahl BD, et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002;22:1298–1306. doi: 10.1128/mcb.22.5.1298-1306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carrozza MJ, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 57.Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 58.Huh JW, et al. Multivalent di-nucleosome recognition enables the Rpd3S histone deacetylase complex to tolerate decreased H3K36 methylation levels. EMBO J. 2012;31:3564–3574. doi: 10.1038/emboj.2012.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ge H, Si Y, Wolffe AP. A novel transcriptional coactivator, p52, functionally interacts with the essential splicing factor ASF/SF2. Mol Cell. 1998;2:751–759. doi: 10.1016/s1097-2765(00)80290-7. [DOI] [PubMed] [Google Scholar]
- 60.Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8:e1002717. doi: 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maurer-Stroh S, et al. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci. 2003;28:69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- 62.Vezzoli A, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010;17:617–619. doi: 10.1038/nsmb.1797. [DOI] [PubMed] [Google Scholar]
- 63.Dhayalan A, et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339:240–249. doi: 10.1016/j.ydbio.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee JH, Skalnik DG. Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A Histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol Cell Biol. 2008;28:609–618. doi: 10.1128/MCB.01356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu M, et al. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol. 2008;28:7337–7344. doi: 10.1128/MCB.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mohan M, et al. The COMPASS family of H3K4 methylases in Drosophila. Mol Cell Biol. 2011;31:4310–4318. doi: 10.1128/MCB.06092-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng H, He X, Moore C. The essential WD repeat protein Swd2 has dual functions in RNA polymerase II transcription termination and lysine 4 methylation of histone H3. Mol Cell Biol. 2004;24:2932–2943. doi: 10.1128/MCB.24.7.2932-2943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dichtl B, Aasland R, Keller W. Functions for S. cerevisiae Swd2p in 3′ end formation of specific mRNAs and snoRNAs and global histone 3 lysine 4 methylation. RNA. 2004;10:965–977. doi: 10.1261/rna.7090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nedea E, et al. The Glc7 phosphatase subunit of the cleavage and polyadenylation factor is essential for transcription termination on snoRNA genes. Mol Cell. 2008;29:577–587. doi: 10.1016/j.molcel.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 71.Regnard C, et al. Global analysis of the relationship between JIL-1 kinase and transcription. PLoS Genet. 2011;7:e1001327. doi: 10.1371/journal.pgen.1001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qiu C, Sawada K, Zhang X, Cheng X. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat Struct Biol. 2002;9:217–224. doi: 10.1038/nsb759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen T, Tsujimoto N, Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol Cell Biol. 2004;24:9048–9058. doi: 10.1128/MCB.24.20.9048-9058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Handler D, et al. A systematic analysis of Drosophila TUDOR domain-containing proteins identifies Vreteno and the Tdrd12 family as essential primary piRNA pathway factors. EMBO J. 2011;30:3977–3993. doi: 10.1038/emboj.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ni JQ, et al. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods. 2011;8:405–407. doi: 10.1038/nmeth.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet. 2005;37:1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- 77.Strübbe G, et al. Polycomb purification by in vivo biotinylation tagging reveals cohesin and Trithorax group proteins as interaction partners. Proc Natl Acad Sci USA. 2011;108:5572–5577. doi: 10.1073/pnas.1007916108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moore SA, Ferhatoglu Y, Jia Y, Al-Jiab RA, Scott MJ. Structural and biochemical studies on the chromo-barrel domain of male specific lethal 3 (MSL3) reveal a binding preference for mono or dimethyl lysine 20 on histone H4. J Biol Chem. 2010 doi: 10.1074/jbc.M110.134312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim D, et al. Corecognition of DNA and a methylated histone tail by the MSL3 chromodomain. Nat Struct Mol Biol. 2010 doi: 10.1038/nsmb.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alekseyenko AA, et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134:599–609. doi: 10.1016/j.cell.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schotta G, Reuter G. Controlled expression of tagged proteins in Drosophila using a new modular P-element vector system. Mol Gen Genet. 2000;262:916–920. doi: 10.1007/pl00008659. [DOI] [PubMed] [Google Scholar]
- 82.Puig O, et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 83.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 84.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Analytical chemistry. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 86.Garcia BA, et al. Organismal differences in post-translational modifications in histones H3 and H4. J Biol Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 87.Zee BM, Levin RS, DiMaggio PA, Garcia BA. Global turnover of histone post-translational modifications and variants in human cells. Epigenetics & Chromatin. 2010;3:22. doi: 10.1186/1756-8935-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saumweber H, Symmons P, Kabisch R, Will H, Bonhoeffer F. Monoclonal antibodies against chromosomal proteins of Drosophila melanogaster: establishment of antibody producing cell lines and partial characterization of corresponding antigens. Chromosoma. 1980;80:253–275. doi: 10.1007/BF00292684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.