

Abstract
Current strategies for the treatment of Alzheimer's disease (AD) involve tackling the formation or clearance of the amyloid-beta peptide (Aβ) and/or hyper-phosphorylated tau, or the support and stabilization of the remaining neuronal networks. However, as we gain a clearer idea of the large number of molecular mechanisms at work in this disease, it is becoming clearer that the treatment of AD should take a combined approach of dealing with several aspects of the pathology. The concept that we also need to protect specific sensitive targets within the cell should also be considered. In particular the role of protecting the function of a specific mitochondrial protein, amyloid binding alcohol dehydrogenase (ABAD), will be the focus of this review. Mitochondrial dysfunction is a well-recognized fact in the progression of AD, though until recently the mechanisms involved could only be loosely labeled as changes in `metabolism'. The discovery that Aβ can be present within the mitochondria and specifically bind to ABAD, has opened up a new area of AD research. Here we review the evidence that the prevention of Aβ binding to ABAD is a drug target for the treatment of AD.
Keywords: ABAD, mitochondria, Alzheimer's disease, drug discovery
INTRODUCTION
The past decade has seen many advances in dementia research towards the development of new innovative therapies for Alzheimer's disease (AD). However, these efforts have yet to lead to a major breakthrough in AD treatment. The AD research community still debates the relative significance of tau and amyloid-beta (Aβ) pathologies in the progression of this disease and with regard to the latter whether this is intracellular or extracellular [1]. There is also debate concerning the relative importance of “nature and nurture” with regard to active research programs using genome wide analysis to identify genetic risk factors versus changes in lifestyle [2]. The truth is likely to be a combination of all of these events. What is therefore clear is that a future treatment for AD will not consist of a one-drug therapy, but will include a combination of different approaches.
Despite the best efforts of the pharmaceutical industry, some in the science community are now seeking a radical rethink in how drugs to treat this disease will be developed. In particular, many new and recent attempts can become thwarted in the early stages of development when trying to solve all the technical issues of target-based drug design in one go [3]. Instead, methods such as lead-oriented synthesis of new compounds have recently been suggested as more promising alternative routes [4]. This is accompanied by an increasing number of authors stressing the importance of understanding the detailed molecular mechanism of action of a potential new drug and of using appropriate phenotypic assays in its validation and design [5]. It could be argued that we are only starting to understand the molecular changes that are occurring within neurons in AD. However, we would predict that future treatments will consist of targeting three aspects:
-
(i)
The formation or clearance of Aβ and/or hyper-phosphorylated tau; though debate rages on whether dismantling a protein aggregate would be a good thing, and as yet targeting the production or synthesis of these toxic proteins has proven to be difficult [6–8].
-
ii)
The support and stabilization of the remaining neuronal networks. This includes the current drug therapies such as cholinesterase inhibitors and memantine [9].
-
iii)
The protection of specific sensitive targets. By identifying the particularly important sensitive targets in AD, it may be possible to protect them against the toxicities of Aβ and/or tau.
It is in regard to this last point which provides the setting of this review and in particular leads to the role of protecting a specific mitochondrial protein (amyloid-binding alcohol dehydrogenase (ABAD)) as an example of how the new criteria of drug-design can be applied.
Mitochondrial dysfunction is a well-recognized fact in the progression of AD, though until recently the mechanisms involved could be loosely labeled as changes in `metabolism' [10]. It is now apparent that there are subtle and complex molecular factors at work in mitochondria and of particular importance is the fact that the observed mitochondrial damage is potentially reversible, as has been shown in various animal models [11–13]. It is also now known that not all mitochondria are created equal and as such synaptic mitochondria have been found to be more vulnerable to the stresses associated with AD pathology [14–18].
The function of mitochondria is to create energy and provide homeostasis and so energy production and metabolism have been known for many years to be defective in AD pathology [10, 19]. Changes in mitochondrial enzymes involved in the electron transport chain and citric acid cycle have been previously noted by many groups [9, 20–22] and as such these observations fit with the idea of mitochondrial dysfunction and oxidative stress as central aspects of AD pathogenesis [23]. Accordingly, clinicians have used the metabolism of glucose as a particular useful method of detecting specific metabolic changes in the brains of AD patients [24, 25]. In addition, although administration of simple antioxidants, such as vitamin E, in the management of AD has so far been unsuccessful [26], the development and application of mitochondria-targeted antioxidant compounds has emerged as a promising approach for the treatment of neurodegenerative diseases [13, 27]. However, until recently what has not been so clear are the molecular mechanisms causing mitochondrial damage.
Recent research efforts have introduced the concept that the accumulation of Aβ inside the cells and specifically in mitochondria might be a likely explanation for the observed mitochondrial damage in AD (reviewed in [28]). The accumulation of intracellular Aβ (of both Aβ(1–40) and Aβ(1–42)) has been shown several times [29–37] and indeed, unlike extracellular plaques, has been shown to correlate with the disease progression [38, 39]. In 2004, it was published that Aβ could be found inside mitochondria in both animal models and significantly also in human AD sufferers [34]. This therefore begs the question what is the intra-mitochondrial Aβ doing? At present two molecular binding targets for Aβ have been identified within mitochondria, namely ABAD and cyclophilin D [15, 40]. What is particularly significant about these two proteins, is that both show an increase in expression levels in AD and at the same time have been shown to bind Aβ with nanomolar affinity [34, 37]. This is in contrast to the changes observed in other mitochondrial proteins involved in metabolism which have been found to have reduced expression levels and activity in AD and for which a specific interaction with Aβ has not been described.
AMYLOID BINDING ALCOHOL DEHYDROGENASE
It was the identification of an intracellular protein via originally a yeast two hybrid screen for Aβ binding molecules, that opened up a new area of mitochondria focused AD research [41]. From this initial screen the mitochondrial protein ABAD has subsequently been shown to bind Aβ via a plethora of different techniques ranging from biophysical approaches (NMR, SPR) through to immunoprecipitation experiments from both AD transgenic mice and human AD brains [34, 42, 43], where ABAD expression levels have also been found to be up-regulated [41]. Initial studies indicated that the binding of Aβ could occur at nanomolar affinity; however, it was not until micromolar levels of Aβ were reached, that changes in ABAD activity were also observed [44, 45]. ABAD contains a Rossman fold [46] for the binding of nucleotides and it has been shown to be able to catalyze, with the help of NAD+/NADH, the reduction of aldehydes and ketones and oxidation of alcohols for energy production utilizing different substrates. As reviewed by Muirhead et al. [47], these substrates range from simple alcohols and amino acid metabolites [48] through to fatty acid metabolites and steroids [28]. Of interest, deficient turnover of one of these substrates, isoleucine, due to inherited point mutations in ABAD, is associated with neurological abnormalities [48].
ABAD activity can be manipulated in two ways, under conditions of stress (it increases) [49] or in the presence of Aβ (it decreases) [34, 50]. With regard to the latter, the change in activity due to the binding of Aβ to ABAD has been previously presented as a rather digital response, that is, as Aβ binds to ABAD it switches its activity off with respect to a number of simple substrates [44, 45]. However, recent data from our laboratories suggests that this rather digital view may be an over simplification (FGM, unpublished data). An up-regulation of ABAD has been suggested to have protective effects in models of Parkinsonism [51] and metabolic stress [49]. However, in AD models, it has been shown that the presence of active ABAD [34, 52], but not an inactive mutant of the enzyme [45], can in fact enhance mitochondrial dysfunction and oxidative stress in vitro [34, 41] and in vivo [34, 52] (Fig. 1). Moreover, studies have now identified additional consequences of Aβ binding to ABAD in living organisms. Specifically, there are a number of genes that are switched on and appear to be controlled by the binding of Aβ to ABAD. Examples include the antioxidant protein peroxiredoxin-2 [53] and the presynaptic endocytic protein endophilin-1 (also referred to as endophilin A1) [54], both of which have been found to be up-regulated in the human AD brain as well [53, 54]. Other proteins have also been shown to be increased such as creatine kinase B, and heat shock protein 70 (FGM and SDY unpublished data) both of which have now been linked by other groups to AD progression [55–57]. The ability to control the expression of an array of other proteins could suggest that it is not a simple question of switching off activity, but potentially that the binding of Aβ to ABAD causes the enzyme to change its ability to utilize certain substrates, which in turn would affect other proteins directly or indirectly. It has been challenging to pin point the exact in vivo substrates of ABAD in the brain, though our recent work suggests that there are indeed subtle changes in lipid metabolism in response to altered ABAD activity in cells (FGM and SDY unpublished data).
Fig. (1).
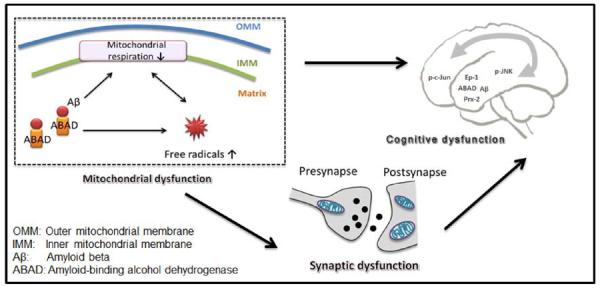
Working Hypothesis. Interaction of ABAD with Aβ enhances generation/accumulation of free radicals and impairs mitochondrial respiratory function. This leads to synaptic dysfunction and aggravates cognitive dysfunction.
CONSEQUENCES OF ABAD AND Aβ BINDING
A number of molecular and cellular changes occur after ABAD and Aβ interact within the mitochondria [28], but as mentioned above, in living organisms it can result in protein expression changes in the brain. The two best described proteins, peroxiredoxin-2 and endophilin-1, are examples of the complexity of the biochemical pathways that become activated in AD and both can be linked to synaptic activity (Fig. 1).
Peroxiredoxin-2 is known to be an antioxidant protein, which has the ability to prevent Aβ induced toxicity [53]. Its expression has also been shown to be elevated in both transgenic AD animals and the post-mortem human brain [53]. Peroxiredoxin-2 has further been linked to the mechanisms at work in the parkinsonian brain [58–60]. However, it would appear that it is not a simple question of just elevated levels of this protein, as it has been shown that there is a critical residue that can be phosphorylated (Thr89) which would lead to the inactivation of the enzyme's activity [59]. This is thought to be the case in the parkinsonian brain [59], but it remains to be seen whether this is the case in the AD brain, too. Indeed, one of the kinases known to phosphorylate this residue in peroxiredoxin-2 is CDK5 [59], which in turn has been shown to have elevated levels of activity in the AD brain [61, 62], thus suggesting peroxiredoxin-2 may indeed be phosphorylated and therefore inactivated in AD. In addition, peroxiredoxin-2 activity can also be controlled directly by oxidation [63], for example induced by the administration of the dopaminergic toxin 6-hydroxydopamine [58] or S-nitrosylation [60]. Therefore it appears that the actual state of post-translational modification of this protein in the brain affected by Parkinson's disease or AD, may be an important aspect.
The second protein that has been shown to be elevated in the AD brain in response to ABAD binding to Aβ is endophilin-1. This is a member of a family of proteins which together have an expanding number of functions, ranging from synaptic vesicle endocytosis, mitochondrial function and receptor trafficking [64]. Specifically, this family of proteins was predicted to be involved in neurodegenerative diseases [65] prior to them now being shown to be involved in AD [54], Parkinson's disease [66], spinocerebellar ataxia 2 [67], and Huntington's disease [64]. In the case of AD, it was the family member endophilin-1 that has been directly shown to be involved. Initially, it was shown that increased levels of endophilin-1 can be linked to increased activation levels of the stress kinase c-Jun N-terminal kinase (JNK) in both HEK293 cells [68] and primary cortical neurons [54] and increased JNK-activity has been known to be a feature of AD pathology [69]. The activation by endophilin-1 occurs potentially through a germinal center like kinase (GLK)-mediated pathway [68]. GLK is part of the germinal center kinases family and their activation is thought to be via the binding of SH2/SH3 adapter binding proteins which are associated with membranes [70]. Endophilin-1 fulfills both of these criteria by affecting lipid membrane curvature for synaptic vesicle formation and by containing a C-terminal SH3 domain [65]. With this in mind, it is interesting to note that endophilin-1 as well as peroxiredoxin-2 have been identified in the same vesicle complexes as the scaffold protein Sunday driver/JNK-interacting protein 3 (JIP3) by immunoprecipitation studies on synaptosomes from the mouse cortex [71]. The recruitment and activation of GLK can lead to the activation of the MAP3K, MEKK1 [72]. Previous findings indicated that MEKK1 interacts with inactive MKK4 to form a MEKK1-MKK4 complex. Active MEKK1 can phosphorylate and activate MKK4 (and/or MKK7), resulting in its dissociation from the complex. The free and active MKK4 then specifically interacts with JNK. Once activated, JNK can translocate to the nucleus or other target sites to phosphorylate downstream effectors, thereby affecting important aspects of neuronal function such as neurite outgrowth, mitochondrial function, synaptic plasticity and apoptosis (all reviewed in [69]). This suggests that through recruiting GLK via its SH3-domain, endophilin-1 might be able to co-localize important components of the JNK-signaling cascade and facilitate its activation. This might further be influenced by the presence of peroxiredoxin-2 [71], which has also been implicated in the activation of JNK-signaling in a cancer cell model [73].
However, it is possible that endophilin-1 may have another direct effect on synaptic signaling as it can lead to the increased probability of glutamate release [74]. This implies that an elevated level of endophilin-1 at the synapse can interfere with normal neurotransmitter signaling. Indeed, it is possible to speculate that an increase in endophilin-1 may have different effects in different locations, as endophilin-1 has been found both in the pre-synaptic neuron [75–77] but also the post-synaptic density [78], where it has recently been given a role in dendritic development [79]. In either case, the relative presence or absence of other signaling molecules will determine which pathways endophilin-1 can influence inside the cell. In addition, the function of endophilin-1 has also been found to be affected by local levels of calcium [80], which are known to be disturbed in AD [81]. This coupled with the recent discovery that pre-synaptic mitochondria expressing ABAD and Aβ are more sensitive than their soma compatriots [14] implies, that spatial localization of these events could be of paramount importance as they may also influence endophilin-1 function through altered ABAD activity affecting lipid metabolism or mitochondrial dysfunction and calcium homeostasis. Therefore, the interaction of ABAD and Aβ within mitochondria is able to elevate endophilin-1 protein [54] and mRNA expression levels (Yan, unpublished results). How this occurs is unknown, but it is likely to have far-reaching consequences for synaptic function (Fig. 2).
Fig. (2).
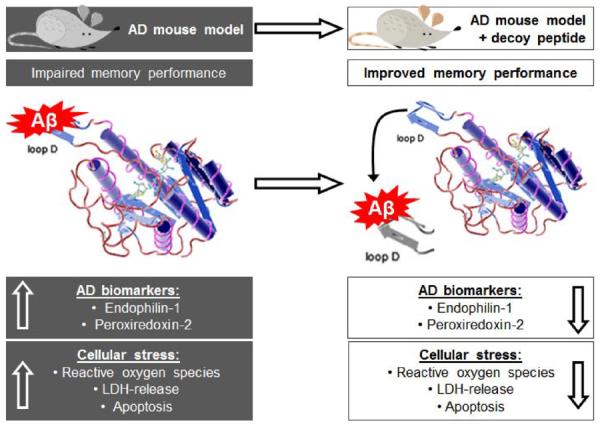
Synaptic dysfunction caused by the ABAD-Aβ interaction. Binding of Aβ affects the enzymatic function of ABAD, thereby causing unfavorable changes in lipid metabolism and mitochondrial respiration. This ultimately leads to increased reactive oxygen species (ROS) production and impaired calcium (Ca2+) retention and provokes the up-regulation of peroxiredoxin-2 (Prx-2) and endophilin-1 (Ep-1). 1) Prx-2 is able to degrade ROS but its function can be affected by elevated Ca2+ leading to its phosphorylation and inactivation and the accumulation of ROS. 2) Ep-1 plays a role in glutamatergic (glut) synaptic transmission by functioning in synaptic vesicle endocytosis, which is directly regulated by Ca2+ binding to Ep-1 [80]. Ep-1 can also be involved in 3) JNK-activation as well as 4) signaling events in the post-synapse leading to dendritogenesis. The correct balance of all of these events in the synapse, which is disturbed by the ABAD-Aβ interaction, is of crucial importance for synaptic plasticity and memory formation.
EVIDENCE THAT ABAD IS A DRUG TARGET
The specific up-regulation of ABAD in human AD and interaction with Aβ [34, 41] which caused mitochondrial dysfunction [52] and other cellular effects also present in the human AD brain [53, 54], suggested that the ABAD-Aβ interaction might be a potential drug target. Using the knowledge gained from understanding how ABAD and Aβ bind to each other, a series of publications have now further validated this view. The crystal structure of ABAD with Aβ bound produced an insight into their interaction. Unfortunately, the exact contact sites could not be established due to distortion of the crystal structure in this area [34], but it was found that a region called loop D encompassing residues 92 to 120 was the region of ABAD that could bind Aβ and this could be confirmed by various biophysical experiments [34, 82]. However, the most convincing data that this interaction was significant, came from the ability of a peptide based on the sequence of this region to work as a decoy in vivo (Fig. 2). Initial experiments showed that modifying the decoy peptide by addition of peptide sequences from the cell-membrane transduction domain of the human immunodeficiency virus-1 (HIV-1) tat protein [83, 84], enabled the ABAD decoy peptides (ABAD-DP) to cross cell membranes and prevent Aβ toxicity in neuronal cultures [34, 85]. Others showed that linking this peptide region with thioredoxin 1 and then expressing the chimeric protein in cell cultures, also protected against Aβ toxicity [85]. It was then the ability of using modified decoy peptides in living organisms that introduced the fascinating possibility that the ABAD and Aβ interaction could be a drug target. Adding a mitochondrial targeting sequence to the decoy peptide facilitated its localization in mitochondria after intraperitoneal injection and transport through the blood brain barrier. It was then possible to show in 6 month old transgenic animals expressing elevated levels of ABAD and Aβ, that the observed increases in the expression levels of peroxiredoxin-2 and endophilin-1, could be reversed [53, 54]. Even more significant was the finding that using the same approach, it was possible to reverse the behavioral changes in these animals as well [11]. Another important aspect of these studies was, that the reversal of behavioral changes could be achieved by both purified peptide injected intraperitoneally and also in a transgenic animal model expressing the ABAD region 92–120 [11]. This approach also showed an additional potentially protective error; positive effect as the level of mitochondrial Aβ was reduced in the treated animals which correlated with an increase in the Aβ-degrading enzyme PreP (presequence peptidase) [11].
Therefore, in keeping with the requirements for a suitable drug target (“ligandability” and “druggability”) recently reviewed by Hann et al [86], it has been shown by both biochemical and physiological determinates that preventing Aβ binding to ABAD can be achieved (ligandability) and has a significant positive effect (druggability). However the question remains whether it is possible to design drugs around such a complex target.
APPROACHES: BLOOD BRAIN BARRIERS, CELLULAR ASSAYS AND PEPTIDES
As indicated above, ABAD is a sensitive site in AD that can be protected from its interaction with Aβ, which could be beneficial in AD treatment. Classically, protein-protein interactions have been avoided as therapeutic targets because of perceived difficulties in developing compounds [87]. To overcome the perceived problems, then it is necessary to be imaginative.
For example, crossing the blood brain barrier is a perennial problem when dealing with small molecules; however, the studies above have shown that intraperitoneally injected peptides are capable of crossing the blood brain barrier and targeting the correct site in animal models [11, 53, 54]. Other recent imaginative work has shown that other approaches can have the same capability. A study in mice revealed, that modified retroviruses showed an increased efficiency of gaining access to the central nervous system when mannitol was peritoneally injected before intravenous administration of the viruses [88]. Another recent breakthrough has been the use of targeted exosomes, again injected intravenously, which allowed biological relevant compounds (in this case siRNA) encapsulated in these membranes, to be successfully delivered into the brain [89]. Therefore there may be new horizons for getting compounds across the blood brain barrier.
For good reasons, the development of small molecules as drugs is the standard method in academia [3]. However, coupled to this approach has been the classical method of devising a high throughput in vitro enzyme assay, but this approach has recently been one of the pitfalls for the pharmaceutical industry, and so there has been a push to develop more cell based phenotypic assays [5]. For ABAD and Aβ the ability of the tested compounds to prevent this interaction has been monitored at the cellular level by measuring the protection provided by them in terms of for example preserving mitochondrial integrity, cytochrome C release and respiration [12, 34, 85]. More direct methods may also be possible with the advent of the first generation of ABAD targeted substrate mimics such as the fluorogenic cyclohexenylamine naphthalene alcohol (CHANA)-(−) [47, 90], which, under specific conditions, could be used to measure the prevention of Aβ induced inhibition of ABAD activity in living cells as a read out for compound testing.
Use of the loop D of ABAD has proven to be successful as described above, but in drug terms a 40 amino acid peptide (20 amino acids from ABAD, approximately 20 amino acids from targeting sequences) would be thought of as too large a molecule to become a drug. However, it could be thought of as a template. At present the minimal size of the loop D ABAD and its constituent parts that provide protection is not known. In addition, it might be possible to design smaller mitochondrial targeting peptides [91], and also modify the amino acids as simple peptide isosteres and heterocyclic peptide isosteres to increase rigidity of the peptidomimetic, or the use of cyclic peptides which increase cellular stability. Indeed, there are examples of modified peptides that have been produced to act on mitochondrial proteins such as antamanide, a 10 amino acid cyclic peptide that targets cyclophilin D [92].
However, small molecule compounds that prevent Aβ binding to ABAD can also be considered as possible. For example, frentizole was identified by an ELISA-based screening assay, as a novel inhibitor of the ABAD-Aβ interaction, and was subsequently modified with the production of a novel benzothiazole urea, resulting in a 30-fold improvement in potency [42]. This compound was found to have potential undesirable immunosuppressive qualities [42]; however, this approach indicated that it might be possible to develop pharmacologically active and medically useful compounds targeting ABAD. Fragment-based drug discovery has been a popular strategy for this used in academia as well as pharmaceutical industry [3]. Two commonly employed screening technologies in this field are thermal shift analysis and saturated transfer difference NMR spectroscopy [93–95], which can identify compounds with millimolar affinity for the target. By grouping compounds, for example using structure activity relation (SAR) analysis, improved affinities for the target can then be achieved [93, 96]. Alternatively, fragments can also be combined, in order to effectively sample the available chemical space as proposed by Hann et al. [86].
Thus, though the ABAD-Aβ interaction is not a classical site for drug therapy, the biology dictates that this interaction may be a future target (Fig. 3). At the very least, the work that was inspired from the original yeast two hybrid screen has shown that it is possible to identify not just new drug targets but also novel biological pathways and events that occur in neurodegenerative diseases such as AD.
Fig. (3).
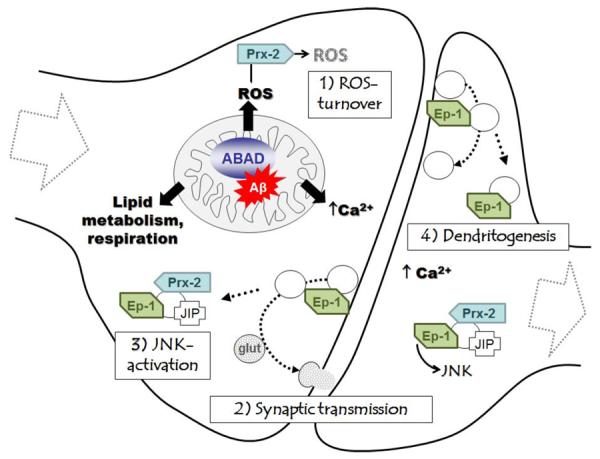
The ABAD decoy peptide approach. Left: Levels of Aβ are elevated in the AD mouse model (and the human AD brain), increasing its binding to loop D in ABAD. Endophilin-1 and peroxiredoxin-2 expression levels are increased in the AD mouse model as a result of the ABAD-Aβ interaction, which also causes neuronal cellular stress (increased ROS production, impaired metabolism, resulting in reduced plasma membrane integrity and LDH-release and apoptosis). Right: Administration of modified peptides based to the loop D amino acid sequence of ABAD are able to disrupt the ABAD-Aβ interaction in the AD mouse model in vivo, decrease the expression of endophilin-1 and peroxiredoxin-2 and restore cellular function and memory performance.
CONCLUSION
Multiple lines of evidence indicate that mitochondrial dysfunction is an early pathological feature of AD. Aβ can directly and indirectly interfere with mitochondria by affecting mitochondrial energy metabolism, oxidative stress, calcium homeostasis and mitochondrial dynamics in axons and the synapse, eventually leading to neuronal injury and cognitive impairment. Therefore, the mitochondria are a very important target in the pathogenesis of AD. Given the role of ABAD in Aβ-induced synaptic pathology, the development of small molecules that inhibit the interaction of ABAD with Aβ, as outlined in this review, is a novel innovative therapeutic strategy for the management of AD.
ACKNOWLEDGEMENTS
This work was supported by grants from National Institute on Aging (NIA), the Alzheimer Association, and the Alzheimer's Research UK and the Biotechnology and Biological Sciences Research Council.
Footnotes
CONFLICT OF INTEREST The author(s) confirm that this article content has no conflicts of interest.
REFERENCES
- [1].Gravitz L. Drugs: a tangled web of targets. Nature. 2011;475:S9–11. doi: 10.1038/475S9a. [DOI] [PubMed] [Google Scholar]
- [2].Deweerdt S. Prevention: activity is the best medicine. Nature. 2011;475:S16–17. doi: 10.1038/475S16a. [DOI] [PubMed] [Google Scholar]
- [3].Frearson JA, Collie IT. HTS and hit finding in academia – from chemical genomics to drug discovery. Drug Discovery Today. 2009;14:1150–1158. doi: 10.1016/j.drudis.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nadin A, Hattotuwagama C, Churcher I. Lead-Oriented Synthesis: A New Opportunity for Synthetic Chemistry. Angewandte Chemie International Edition. 2012;51:1114–1122. doi: 10.1002/anie.201105840. [DOI] [PubMed] [Google Scholar]
- [5].Swinney DC, Anthony J. How were new medicines discovered? doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- [6].Brody DL, Holtzman DM. Active and Passive Immunotherapy for Neurodegenerative Disorders. Annu Rev Neurosci 2011. 2008;31:175–193. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Citron M. Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci. 2004;5:677–685. doi: 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- [8].Citron M. Beta-secretase inhibition for the treatment of Alzheimer's disease--promise and challenge. Trends Pharmacol Sci. 2004;25:92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- [9].Palmer AM. Neuroprotective therapeutics for Alzheimer's disease: progress and prospects. Trends Pharmacol Sci. 2011;32:141–147. doi: 10.1016/j.tips.2010.12.007. [DOI] [PubMed] [Google Scholar]
- [10].Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- [11].Yao J, Du H, Yan S, Fang F, Wang C, Lue LF, et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer's disease. J Neurosci. 2011;31:2313–2320. doi: 10.1523/JNEUROSCI.4717-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yao J, Mei L, Luddy J, Chen X, Stern D, Arancio O, et al. Antagonizing ABAD–Aβ interaction maintains mitochondrial and neuronal function in an Aβ rich environment: Studies in neuronal cultures and transgenic mice. In: Neuroscience S.f., editor. Neuroscience 2004 Meeting. San Diego, California, USA: 2004. Program No 716.711. [Google Scholar]
- [13].Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, et al. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J Alzheimers Dis. 2010;20:S609–631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Du H, Yan SS. Mitochondrial permeability transition pore in Alzheimer's disease: cyclophilin D and amyloid beta. Biochim Biophys Acta. 2010;1802:198–204. doi: 10.1016/j.bbadis.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Du H, Guo L, Yan SS. Synaptic mitochondrial pathology in Alzheimer's disease. Antioxid Redox Signal. 2012;16:1467–1475. doi: 10.1089/ars.2011.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brown MR, Sullivan PG, Geddes JW. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- [19].Bowling AC, Beal MF. Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci. 1995;56:1151–1171. doi: 10.1016/0024-3205(95)00055-b. [DOI] [PubMed] [Google Scholar]
- [20].Gibson GE, Park LC, Zhang H, Sorbi S, Calingasan NY. Oxidative stress and a key metabolic enzyme in Alzheimer brains, cultured cells, and an animal model of chronic oxidative deficits. Ann NY Acad Sci. 1999;893:79–94. doi: 10.1111/j.1749-6632.1999.tb07819.x. [DOI] [PubMed] [Google Scholar]
- [21].Cottrell DA, Blakely EL, Johnson MA, Ince PG, Turnbull DM. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology. 2001;57:260–264. doi: 10.1212/wnl.57.2.260. [DOI] [PubMed] [Google Scholar]
- [22].Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- [23].Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- [24].Duara R, Grady C, Haxb J, Sundaram M, Cutler NR, Heston L, et al. Positron emission tomography in Alzheimer's disease. Neurology. 1986;36:879–887. doi: 10.1212/wnl.36.7.879. [DOI] [PubMed] [Google Scholar]
- [25].McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vina J, Lloret A, Giraldo E, Badia MC, Alonso MD. Antioxidant pathways in Alzheimer's disease: possibilities of intervention. Curr Pharm Des. 2011;17:3861–3864. doi: 10.2174/138161211798357755. [DOI] [PubMed] [Google Scholar]
- [27].Szeto HH. Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann N Y Acad Sci. 2008;1147:112–121. doi: 10.1196/annals.1427.013. [DOI] [PubMed] [Google Scholar]
- [28].Muirhead KEA, Borger E, Aitken L, Conway SJ, Gunn-Moore FJ. The consequences of mitochondrial amyloid β-peptide in Alzheimer's disease. Biochemical J. 2010;426:255–270. doi: 10.1042/BJ20091941. [DOI] [PubMed] [Google Scholar]
- [29].Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, et al. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- [30].Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res. 1998;52:691–698. doi: 10.1002/(SICI)1097-4547(19980615)52:6<691::AID-JNR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- [31].Buckig A, Tikkanen R, Herzog V, Schmitz A. Cytosolic and nuclear aggregation of the amyloid beta-peptide following its expression in the endoplasmic reticulum. Histochem Cell Biol. 2002;118:353–360. doi: 10.1007/s00418-002-0459-2. [DOI] [PubMed] [Google Scholar]
- [32].Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, et al. Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004;279:50310–50320. doi: 10.1074/jbc.M405600200. [DOI] [PubMed] [Google Scholar]
- [34].Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- [35].Martin BL, Schrader-Fischer G, Busciglio J, Duke M, Paganetti P, Yankner BA. Intracellular accumulation of beta-amyloid in cells expressing the Swedish mutant amyloid precursor protein. J Biol Chem. 1995;270:26727–26730. doi: 10.1074/jbc.270.45.26727. [DOI] [PubMed] [Google Scholar]
- [36].Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. Faseb J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- [37].Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001;125:489–492. doi: 10.5858/2001-125-0489-IAAPDO. [DOI] [PubMed] [Google Scholar]
- [40].Giorgio V, Soriano ME, Basso E, Bisetto E, Lippe G, Forte MA, et al. Cyclophilin D in mitochondrial pathophysiology. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2010;1797:1113–1118. doi: 10.1016/j.bbabio.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, et al. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 1997;389:689–695. doi: 10.1038/39522. [DOI] [PubMed] [Google Scholar]
- [42].Xie Y, Deng S, Chen Z, Yan S, Landry DW. Identification of small-molecule inhibitors of the A-ß-ABAD interaction. Bioorg Med Chem Lett. 2006;16:4657–4660. doi: 10.1016/j.bmcl.2006.05.099. [DOI] [PubMed] [Google Scholar]
- [43].Yan Y, Liu Y, Sorci M, Belfort G, Lustbader JW, Yan SS, et al. Surface plasmon resonance and nuclear magnetic resonance studies of ABAD-Abeta interaction. Biochemistry. 2007;46:1724–1731. doi: 10.1021/bi061314n. [DOI] [PubMed] [Google Scholar]
- [44].Oppermann UC, Salim S, Tjernberg LO, Terenius L, Jornvall H. Binding of amyloid beta-peptide to mitochondrial hydroxyacyl-CoA dehydrogenase (ERAB): regulation of an SDR enzyme activity with implications for apoptosis in Alzheimer's disease. FEBS Lett. 1999;451:238–242. doi: 10.1016/s0014-5793(99)00586-4. [DOI] [PubMed] [Google Scholar]
- [45].Yan SD, Shi Y, Zhu A, Fu J, Zhu H, Zhu Y, et al. Role of ERAB/L-3-Hydroxyacyl-coenzyme A Dehydrogenase Type II Activity in Aß-induced Cytotoxicity. J Biol Chem. 1999;274:2145–2156. doi: 10.1074/jbc.274.4.2145. [DOI] [PubMed] [Google Scholar]
- [46].Powell AJ, Read JA, Banfield MJ, Gunn-Moore F, Ya SD, Lustbader J, et al. Recognition of structurally diverse substrates by type II 3-hydroxyacyl-CoA dehydrogenase (HADH II)/amyloid-beta binding alcohol dehydrogenase (ABAD) J Mol Biol. 2000;303:311–327. doi: 10.1006/jmbi.2000.4139. [DOI] [PubMed] [Google Scholar]
- [47].Muirhead KEA, Froemming M, Li X, Musilek K, Conway SJ, Sames D, et al. (−)-CHANA, a Fluorogenic Probe for Detecting Amyloid Binding Alcohol Dehydrogenase HSD10 Activity in Living Cells. ACS Chemical Biology. 2010;5:1105–1114. doi: 10.1021/cb100199m. [DOI] [PubMed] [Google Scholar]
- [48].Ofman R, Jos Ruiter PN, Feenstra M, Duran M, Bwee PollThe T, Zschocke J, et al. 2-Methyl-3-Hydroxybutyryl-CoA Dehydrogenase Deficiency Is Caused by Mutations in the HADH2 Gene. Am J Hum Genet. 2003;72:1300–1307. doi: 10.1086/375116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yan SD, Zhu Y, Stern ED, Hwang YC, Hori O, Ogawa S, et al. Amyloid beta -Peptide-binding Alcohol Dehydrogenase Is a Component of the Cellular Response to Nutritional Stress. J Biol Chem. 2000;275:27100–27109. doi: 10.1074/jbc.M000055200. [DOI] [PubMed] [Google Scholar]
- [50].He X-Y, Wen G-Y, Merz G, Lin D, Yang Y-Z, Mehta P, et al. Abundant type 10 17[beta]-hydroxysteroid dehydrogenase in the hippocampus of mouse Alzheimer's disease model. Brain Res Mol Brain Res. 2002;99:46–53. doi: 10.1016/s0169-328x(02)00102-x. [DOI] [PubMed] [Google Scholar]
- [51].Tieu K, Perier C, Vila M, Caspersen C, Zhang H-P, Teismann P, et al. L3-hydroxyacyl-CoA dehydrogenase II protects in a model of Parkinson's disease. Anna Neurol. 2004;56:51–60. doi: 10.1002/ana.20133. [DOI] [PubMed] [Google Scholar]
- [52].Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, et al. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. Faseb J. 2005;19:597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- [53].Yao J, Taylor M, Davey F, Ren Y, Aiton J, Coote P, et al. Interaction of amyloid binding alcohol dehydrogenase/Abeta mediates up-regulation of peroxiredoxin II in the brains of Alzheimer's disease patients and a transgenic Alzheimer's disease mouse model. Mol Cell Neurosci. 2007;35:377–382. doi: 10.1016/j.mcn.2007.03.013. [DOI] [PubMed] [Google Scholar]
- [54].Ren Y, Xu HW, Davey F, Taylor M, Aiton J, Coote P, et al. Endophilin I expression is increased in the brains of Alzheimer disease patients. J Biol Chem. 2008;283:5685–5691. doi: 10.1074/jbc.M707932200. [DOI] [PubMed] [Google Scholar]
- [55].Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesber WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- [56].Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, et al. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- [57].Di Domenico F, Sultana R, Tiu GF, Scheff NN, Perluigi M, Cini C, et al. Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: An investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Res. 2010;1333:72–81. doi: 10.1016/j.brainres.2010.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hu X, Weng Z, Chu CT, Zhang L, Cao G, Gao Y, et al. Peroxiredoxin-2 Protects against 6-Hydroxydopamine-Induced Dopaminergic Neurodegeneration via Attenuation of the Apoptosis Signal-Regulating Kinase (ASK1) Signaling Cascade. J Neurosci. 2011;31:247–261. doi: 10.1523/JNEUROSCI.4589-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Qu D, Rashidian J, Mount MP, Aleyasin H, Parsanejad M, Lira A, et al. Role of Cdk5-Mediated Phosphorylation of Prx2 in MPTP Toxicity and Parkinson's Disease. Neuron. 2007;55:37–52. doi: 10.1016/j.neuron.2007.05.033. [DOI] [PubMed] [Google Scholar]
- [60].Fang J, Nakamura T, Cho DH, Gu Z, Lipton SA. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson's disease. Proc Natl Acad Sci USA. 2007;104:18742–18747. doi: 10.1073/pnas.0705904104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pei J-J, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer's disease neurofibrillary degeneration. Brain Res. 1998;797:267–277. doi: 10.1016/s0006-8993(98)00296-0. [DOI] [PubMed] [Google Scholar]
- [62].Swatton JE, Sellers LA, Faull RLM, Holland A, Iritani S, Bahn S. Increased MAP kinase activity in Alzheimer's and Down syndrome but not in schizophrenia human brain. Eur J Neurosci. 2004;19:2711–2719. doi: 10.1111/j.0953-816X.2004.03365.x. [DOI] [PubMed] [Google Scholar]
- [63].Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends in Biochemical Sciences. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- [64].Kjaerulff O, Brodin L, Jung A. The Structure and Function of Endophilin Proteins. Cell Biochem Biophys. 2010 doi: 10.1007/s12013-010-9137-5. [DOI] [PubMed] [Google Scholar]
- [65].Reutens AT, Glenn Begley C. Endophilin-1: a multifunctional protein. Int J Biochem Cell Biol. 2002;34:1173–1177. doi: 10.1016/s1357-2725(02)00063-8. [DOI] [PubMed] [Google Scholar]
- [66].Trempe JF, Chen CX, Grenier K, Camacho EM, Kozlov G, McPherson PS, et al. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol Cell. 2009;36:1034–1047. doi: 10.1016/j.molcel.2009.11.021. [DOI] [PubMed] [Google Scholar]
- [67].Nonis D, Schmidt MH, van de Loo S, Eich F, Dikic I, Nowock J, et al. Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal. 2008;20:1725–1739. doi: 10.1016/j.cellsig.2008.05.018. [DOI] [PubMed] [Google Scholar]
- [68].Ramjaun AR, Angers A, Legendre-Guillemin V, Tong X-K, McPherson PS. Endophilin Regulates JNK Activation through Its Interaction with the Germinal Center Kinase-like Kinase. J Biol Chem. 2001;276:28913–28919. doi: 10.1074/jbc.M103198200. [DOI] [PubMed] [Google Scholar]
- [69].Mehan S, Meena H, Sharma D, Sankhla R. JNK: a stress-activated protein kinase therapeutic strategies and involvement in Alzheimer's and various neurodegenerative abnormalities. J Mol Neurosci. 2011;43:376–390. doi: 10.1007/s12031-010-9454-6. [DOI] [PubMed] [Google Scholar]
- [70].Kyriakis JM. Signaling by the germinal center kinase family of protein kinases. J Biol Chem. 1999;274:5259–5262. doi: 10.1074/jbc.274.9.5259. [DOI] [PubMed] [Google Scholar]
- [71].Abe N, Almenar-Queralt A, Lillo C, Shen Z, Lozach J, Briggs SP, et al. Sunday Driver Interacts with Two Distinct Classes of Axonal Organelles. J Biol Chem. 284:34628–34639. doi: 10.1074/jbc.M109.035022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Diener K, Wang XS, Chen C, Meyer CF, Keesler G, Zukowski M, et al. Activation of the c-Jun N-terminal kinase pathway by a novel protein kinase related to human germinal center kinase. Proc Natl Acad Sci USA. 1997;94:9687–9692. doi: 10.1073/pnas.94.18.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lee KW, Lee DJ, Lee JY, Kang DH, Kwon J, Kang SW. Peroxiredoxin II restrains DNA damage-induced death in cancer cells by positively regulating JNK-dependent DNA repair. J Biol Chem. 2011;286:8394–8404. doi: 10.1074/jbc.M110.179416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Weston Matthew C, Nehring Ralf B, Wojcik, Sonja M, Rosenmund C. Interplay between VGLUT Isoforms and Endophilin A1 Regulates Neurotransmitter Release and Short-Term Plasticity. Neuron. 2011;69:1147–1159. doi: 10.1016/j.neuron.2011.02.002. [DOI] [PubMed] [Google Scholar]
- [75].Micheva KD, Kay BK, McPherson PS. Synaptojanin Forms Two Separate Complexes in the Nerve Terminal. Journal of Biological Chem. 1997;272:27239–27245. doi: 10.1074/jbc.272.43.27239. [DOI] [PubMed] [Google Scholar]
- [76].Ringstad N, Nemoto Y, De Camilli P. Differential Expression of Endophilin 1 and 2 Dimers at Central Nervous System Synapses. J Biol Chem. 2001;276:40424–40430. doi: 10.1074/jbc.M106338200. [DOI] [PubMed] [Google Scholar]
- [77].Schuske KR, Richmond JE, Matthies DS, Davis WS, Runz S, Rube DA, et al. Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron. 2003;40:749–762. doi: 10.1016/s0896-6273(03)00667-6. [DOI] [PubMed] [Google Scholar]
- [78].Bayes A, van de Lagemaat LN, Collins MO, Croning MDR, Whittle IR, Choudhary JS, et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Fu X, Yang Y, Xu C, Niu Y, Chen T, Zhou Q, et al. Retrolinkin cooperates with endophilin A1 to mediate BDNF-TrkB early endocytic trafficking and signaling from early endosomes. Molecular Biology of the Cell. 2011 doi: 10.1091/mbc.E11-04-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chen Y, Deng L, Maeno-Hikichi Y, Lai M, Chang S, et al. Formation of an endophilin-Ca2+ channel complex is critical for clathrin-mediated synaptic vesicle endocytosis. Cell. 2003;115:37–48. doi: 10.1016/s0092-8674(03)00726-8. [DOI] [PubMed] [Google Scholar]
- [81].Berridge MJ. Calcium hypothesis of Alzheimer's disease. Pflugers Arch. 2010;459:441–449. doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- [82].Milton NG, Mayor NP, Rawlinson J. Identification of amyloid-beta binding sites using an antisense peptide approach. Neuroreport. 2001;12:2561–2566. doi: 10.1097/00001756-200108080-00054. [DOI] [PubMed] [Google Scholar]
- [83].Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- [84].Cao G, Pei W, Ge H, Liang Q, Luo Y, Sharp FR, et al. In Vivo Delivery of a Bcl-xL Fusion Protein Containing the TAT Protein Transduction Domain Protects against Ischemic Brain Injury and Neuronal Apoptosis. J Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Yang X, Yang Y, Wu J, Zhu J. Stable expression of a novel fusion peptide of thioredoxin-1 and ABAD-inhibiting peptide protects PC12 cells from intracellular amyloid-ß. J Mol Neurosci. 2007;33:180–188. doi: 10.1007/s12031-007-0063-y. [DOI] [PubMed] [Google Scholar]
- [86].Hann MM, Keserü GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. 2012 doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- [87].Y., J.B.G.G.G.N.G.M.T.S.P.P.J.A.V.K.K.K. Protein Based Drug Discovery. International Journal of Drug Discovery. 2009;1:40–51. [Google Scholar]
- [88].Louboutin JP, Chekmasova AA, Marusich E, Chowdhury JR, Strayer DS. Efficient CNS gene delivery by intravenous injection. Nat Methods. 2010;7:905–907. doi: 10.1038/nmeth.1518. [DOI] [PubMed] [Google Scholar]
- [89].Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- [90].Froemming MK, Sames D. Harnessing Functional Plasticity of Enzymes: A Fluorogenic Probe for Imaging 17β-HSD10 Dehydrogenase, an Enzyme Involved in Alzheimer's and Parkinson's Diseases. Journal of the American Chemical Society. 2007;129:14518–14522. doi: 10.1021/ja072601x. [DOI] [PubMed] [Google Scholar]
- [91].Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Mitochondria-penetrating peptides. Chem Biol. 2008;15:375–382. doi: 10.1016/j.chembiol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- [92].Azzolin L, Antolini N, Calderan A, Ruzza P, Sciacovelli M, Marin O, et al. Antamanide, a derivative of Amanita phalloides, is a novel inhibitor of the mitochondrial permeability transition pore. PLoS One. 2011;6:e16280. doi: 10.1371/journal.pone.0016280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Schulz MN, Hubbard RE. Recent progress in fragment-based lead discovery. Curr Opin Pharmacol. 2009;9:615–621. doi: 10.1016/j.coph.2009.04.009. [DOI] [PubMed] [Google Scholar]
- [94].Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- [95].Carr RA, Congreve M, Murray CW, Rees DC. Fragment-based lead discovery: leads by design. Drug Discov Today. 2005;10:987–992. doi: 10.1016/S1359-6446(05)03511-7. [DOI] [PubMed] [Google Scholar]
- [96].Ciulli A, Abell C. Fragment-based approaches to enzyme inhibition. Curr Opin Biotechnol. 2007;18:489–496. doi: 10.1016/j.copbio.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]