

Abstract
Nucleosomes were reconstituted from recombinant histones and a 147-mer DNA sequence containing the damage reporter sequence 5′-…d([2AP]T[GGG]1TT[GGG]2TTT[GGG]3TAT)… with 2-aminopurine (2AP) at position 27 from the dyad axis. Footprinting studies with •OH radicals reflect the usual effects of “in” and “out” rotational settings, while, interestingly, the guanine oxidizing one-electron oxidant CO3•− radical does not. Site-specific hole injection was achieved by 308 nm excimer laser pulses to produce 2AP•+ cations, and superoxide via the trapping of hydrated electrons. Rapid deprotonation (~ 100 ns) and proton coupled electron transfer generates neutral guanine radicals, G(−H)• and hole hopping between the three groups of [GGG] on micro- to millisecond time scales. Hole transfer competes with hole trapping that involves the combination of O2•− with G(−H)• radicals to yield predominantly 2,5-diamino-4H-imidazolone (Iz) and minor 8-oxo-7,8-dihydroguanine (8-oxoG) end-products in free DNA (Misiaszek et al. J. Biol. Chem. 2004, 279, 32106). Hole migration is less efficient in nucleosomal than in the identical protein-free DNA by a factor of 1.2 – 1.5. The Fpg/piperidine strand cleavage ratio is ~1.0 in free DNA at all three GGG sequences and at the “in” rotational settings [GGG]1,3 facing the histone core, and ~2.3 at the “out” setting at [GGG]2 facing away from the histone core. These results are interpreted in terms of competitive reaction pathways of O2•− with G(−H)• radicals at the C5 (yielding Iz) and C8 (yielding 8-oxoG) positions. These differences in product distributions are attributed to variations in the local nucleosomal B-DNA base pair structural parameters that are a function of surrounding sequence context and rotational setting.
Introduction
Chronic inflammation induced by diverse infectious and environmental factors increases the risk of malignant cell transformations and the development of many human cancers.1, 2 The enhanced production of reactive oxygen and nitrogen species in inflammatory tissues gives rise to the formation of genotoxic DNA lesions. Guanine is the most easily oxidizable nucleic acid base3 and is thus the primary target of oxidative modifications under inflammatory conditions.4 The formation of oxidatively generated guanine lesions is a base-sequence dependent process initiated by the one-electron oxidation of nucleobases (“hole injection”). Localization of “holes” (guanine radical cations) is more efficient in runs of guanines according to 5′-G < 5′-GG < 5′-GGG5–8 and this phenomenon is correlated with the calculated guanine ionization potentials (IP) in the gas phase,5, 9 and also in the hydrated state in duplex DNA.10, 11 However, almost all experimental5–7, 12–14 and theoretical15–17 studies have explicitly focused on the base-sequence effects in protein-free B-form DNA duplexes.
In mammalian cells, DNA molecules are wrapped around octamer histone proteins thus forming nucleosome core particles (NCP) that are further assembled into chromatin. This basic repeating unit of the in vivo chromatin structure is assembled from a 147 base pair DNA molecule and an octamer of two copies of four histone proteins H2A, H2B, H3 and H4.18, 19 In the nucleosome, the DNA molecule is wrapped around the octamer core particle as a 1.65 turn superhelix and the strain required for wrapping the nucleosomal DNA is relieved by overwinding (10.0 bp/turn) in the extended DNA regions on each side of the axis of dyad symmetry and underwinding (10.7 bp/turn) in the central region.20 Thus, the structural parameters of the nucleosomal DNA are considerably varied from those of the free B-form DNA in solution (10.5 bp/turn) and can potentially affect the formation of guanine lesions in nucleosomal DNA. However, very little is known about the effects of the architecture of these self-assembled nucleosome structures on the formations and distributions of guanine lesions induced by hole injection into nucleosomal DNA.
In this work we studied the distributions of guanine lesions initiated by the site-selective one-electron oxidation of guanine in nucleosomes assembled from recombinant histones and the 147 base pair strong positioning 601 DNA sequence first described by Lowary and Widom.21 This sequence was modified by the insertion of the DNA damage reporter sequence, 5′-d([2AP]T[GGG]1TT[GGG]2TTT[GGG]3TAT) containing three GGG-sites and a single 2-aminopurine (2AP) residue. The 2AP nucleic base analog paired with a T base has an absorption band near 305 nm and was used for the site-selective photoionization of 2AP by a tandem two-photon absorption and ionization mechanism22 resulting in the formation of a 2AP radical (“hole”) and a hydrated electron. The latter is trapped by dissolved oxygen, thus generating superoxide (O2•−). The holes migrate to adjacent guanines and are trapped by reactions with O2•−, ultimately leading to 2,5-diamino-4H-imidazolone (Iz) and 8-oxo-7,8-dihydroguanine (8-oxoG)23 that are sensitive to cleavage by treatment with piperidine (only Iz) or with formamidopyrimidine DNA N-glycosylase, Fpg (both Iz and 8-oxoG).24 Analysis of the distributions of the guanine lesions quantified by high resolution polyacrylamide gel electrophoresis (PAGE) showed that the hole migration efficiency is somewhat smaller in nucleosomal than in free DNA, and indicates that the relative proportions of 8-oxoG and Iz lesions depend on the rotational settings in nucleosomal DNA.
Experimental
Materials
Analytical grade chemicals, HPLC grade organic solvents, and Milli-Q purified (ASTM type I) water were used throughout. The recombinant histones, H2A, H2B, H3.1 and H4, as well as Fpg protein were purchased from New England Biolabs (Ipswich, MA), while Proteinase K was obtained from 5 Prime Inc. (Gaithersburg, MD). The enzymes OptiKinase and T4 DNA ligase were obtained from USB Molecular Biology Reagents and Biochemicals (Cleveland, OH). The [γ-32P]ATP with activity of 6000 Ci/mmol was purchased from PerkinElmer Life Sciences (Boston, MA). The oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and purified by denaturing 12% PAGE (acrylamide/bisacrylamide = 19 : 1) in 7.5 M urea and 1×TBE buffer (8.9 mM Tris-Borate, 0.2 mM EDTA, pH 8.0).
Preparation of 2AP-containing DNA duplexes
A 65-nt-long oligonucleotide (a) with the reporter sequence, 5′-d([2AP]T[GGG]1TT[GGG]2TTT[GGG]3TAT) at the 3′-end of a and five fragments based on the strong 601 DNA nucleosome positioning sequence (b, c, d, e and f) defined in Figure 1 were 5′-phosphorylated with OptiKinase and ATP (100 mM, pH 7.5) at 37 °C for 45 min. The resulting six 5′-phosphorylated oligonucleotides were purified by denaturing 12% PAGE and mixed in a molar ratio of a : b : c : d : e : f = 1 : 1.5 : 1.5 : 2 : 2 : 2 and annealed in buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 5 mM DTT) by first rapid heating to 95 °C in ~ 2 min, followed by slow cooling to room temperature overnight, and then ligated in situ by T4 DNA ligase in buffer (66 mM Tris-HCl, pH 7.6, 6.6 mM MgCl2, 10 mM DTT, 66 μM ATP) at 16 °C for 16 h. The 147-nt-long strands thus obtained were purified by denaturing 12% PAGE using a 38×50 cm BioRad Sequencing cell (Melville, NY). The 147-mer bands were cut out, soaked overnight in an elution buffer (500 mM ammonium acetate, 10 mM magnesium acetate, 2 mM EDTA, pH 6.5), isolated by standard ethanol precipitation, and annealed in buffer (25 mM NaCl, 10 mM Tris-HCl, 2.5 mM EDTA, pH 8.0). The 5′-32P-end-labeled 2AP-modified DNA duplexes were prepared using the 65-nt-long sequence phosphorylated with OptiKinase and [γ-32P]ATP at the 5′-end. The integrity of DNA duplexes was confirmed by native 6% PAGE (acrylamide/bisacrylamide = 37.5 : 1) in 0.3×TBE buffer.
Figure 1.

Design of the 147 base pair-long 601 DNA containing the reporter sequence (marked in red) by in situ ligation of six oligonucleotides. The sequences d, e, and f are the natural complementary strands for the oligonucleotides a, b, and c. The base coinciding with the Dyad axis is marked in blue.
Refolding of histone octamers
The histone octamers were assembled from the recombinant histones as described by Luger et al.25, 26 Briefly, the H2A, H2B, H3.1 and H4 proteins were unfolded by replacing the histone storage buffer for the unfolding buffer (7 M Guanidine HCl, 20 mM Tris-HCl, pH7.5, 10 mM DTT) using Amicon Ultra Centrifugal Filter Devices (3 kDa MWCO). After incubation at room temperature for 30 min the histone samples were centrifugated at 10000 g (10 min, 4 °C) and the concentrations of the unfolded histone proteins were determined from their absorbances at 276 nm and the known extinction coefficients of each histone.25 The four histone proteins were mixed in a molar ratio of H2A : H2B : H3.1 : H4 = 1.2 : 1.2 : 1 : 1 and diluted with the unfolding buffer to obtain a total final protein concentration of ~1 mg/mL. The 2 mg histone sample was placed in a Pierce Slide-A-Lyzer Dialysis Cassette (3.5 kDa MWCO) and dialyzed against three changes of the refolding buffer (2 M NaCl, 20 mM Tris/HCl, pH 7.5, 1 mM EDTA, 1 mM DTT) at 4 °C. After centrifugation, the refolded octamer samples were concentrated to 200 – 250 μL using an Amicon Ultra Centrifugal Filter Device (10 kDa MWCO) and purified by FPLC (separation conditions: GE Healthcare Superdex 200 10/300 GL column, mobile phase - refolding buffer, flow rate of 0.2 mL/min, 4 °C). The purified octamer sample was concentrated to ~ 5 mg/mL protein, mixed with glycerol (1 : 1 v/v) and stored at −20 °C. The integrity of histone octamers was confirmed by SDS-PAGE 27 and mass spectrometry using a Bruker Daltonics ultrafleXtreme MALDI-TOF/TOF mass spectrometer. The 0.5 μL sample aliquots of the octamers (1 mg/mL) were mixed with the 1 μL aliquots of the matrix solution (α-cyano-4-hydroxycinnamic acid in 50% acetonitrile/0.1% TFA), spotted onto the MALDI target and air-dried before analysis. The mass spectra of the histone octamers were recorded in the positive linear mode; the m/z values were obtained using an external calibration with the Bruker Daltonics Protein Calibration Standard I.
Reconstitution of nucleosomes
The nucleosome core particles were assembled from the histone octamers and 147-bp DNA duplexes using the protocol developed by Luger et al.25, 26 Briefly, the 5 μg histone sample was dialyzed in a Pierce Slide-A-Lyzer MINI Dialysis Unit (10 kDa MWCO) against the refolding buffer (overnight, 4 °C) to remove glycerol. The histone sample was centrifuged at 10000 g (10 min, 4 °C) and its concentration was determined from the absorbance at 276 nm.25, 26 A 10 pmol 147-bp DNA duplex spiked with 5′-32P-end-labeled 2AP-modified 147-bp DNA was incubated with a stoichiometric quantity of the histone octamer in 2 M NaCl solution in a final volume of ~25 μL for 30 min at room temperature. The sample was dialyzed against the buffer solutions (10 mM Tris-HCl, pH 8.0, 2.5 mM EDTA) containing 1 M, 0.6 M, 0.1 M and 0.025 M NaCl for 2 – 3 h and finally overnight at 4 °C. To obtain uniformly positioned DNA, the nucleosomes were incubated for 30 min at 37 °C.26 After centrifugation, the nucleosome samples were analyzed by native 5% PAGE in 0.3×TBE buffer. In all further experiments described, the nucleosome solutions contained less than 5% unbound DNA.
Hydroxyl radical footprinting of nucleosomes
Hydroxyl radical footprinting was performed as described previously.28 Briefly, 2 μL aliquots of 20 mM sodium ascorbate, 1 mM FeSO4•7H2O and 2 mM EDTA, and 0.6% H2O2 were premixed and added within 5 s to 20 μL of the nucleosome sample. The reaction mixture was incubated for 3 min at room temperature and stopped by the addition of 3 μL aliquots of 50% (v/v) glycerol and 2 μL aliquot of 400 mM EDTA solution. The samples were extracted with 20 μL phenol/chloroform/3-methyl-1-butanol (25 : 24 : 1) mixture to remove the histone proteins. The 20 μL of free DNA sample was premixed with 1 μL aliquots of calf thymus DNA (0.25 mg/mL) and incubated with the footprinting reagents for 30 s. The oxidatively modified DNA was isolated by standard ethanol precipitation repeated twice and analyzed by denaturing 8% PAGE in 7.5 M urea in 1×TBA buffer.
Damage to nucleosomal DNA induced by photoionization of 2AP bases
The 15 μL samples of nucleosome samples (~ 5 pmol) containing 5′-32P-end-labeled DNA in 2×2 mm square Pyrex capillary tubes (Vitrocom, Inc., Mountain Lakes, NJ) were excited by a train of 1 to 50 nanosecond 308 nm XeCl excimer laser pulses (~12 ns width at half-height, ~80 mJ/pulse, 1 Hz). The number of pulses was adjusted in order to maintain the fractions of cleaved oligonucleotide strands below 20%.29 Under these conditions, each DNA molecule contains no more than one cleavable site (defined here as a single-hit condition).30
Analysis of DNA damage
The DNA strands in the irradiated samples were cleaved at sites of nucleobase modifications by either of two methods. (1) Standard hot piperidine treatment. The histone proteins in the irradiated nucleosome samples were first digested by treatment with 1 μL aliquots of Proteinase K (0.2 mg/mL) and 0.25% (w/v) SDS for 1.5 h at 37 °C. The irradiated, oxidatively damaged DNA was isolated by standard ethanol precipitation and was mixed with 100 μL of 1 M piperidine, heated at 90 °C for 30 min, vacuum dried, and the traces of piperidine were removed by lyophilization repeated twice. (2) Treatment with Fpg protein. The irradiated nucleosome samples were incubated with 5 units of Fpg protein in buffer (0.1 mg/mL BSA, 10 mM Bis-Tris Propane-HCl, 10 mM MgCl2, 1 mM DTT, pH 7.0) for 1.5 h at 37 °C. Then the histone proteins were digested by Proteinase K and the damaged DNA was isolated by standard ethanol precipitation. Alternatively, the histone proteins in the irradiated nucleosome samples were first digested by Proteinase K. The samples were then treated with 4 μL of Agilent StrataClean Resin to remove Proteinase K and the damaged DNA strands were cleaved by Fpg protein as described above.
The cleaved DNA fragments were analyzed by denaturing 8% PAGE in 7.5 M urea in 1×TBE buffer at 65 W for 2–3 h. The vacuum-dried gels were quantitatively assayed using a Storm 840 Phosphorimage System (GE Healthcare). The extent of cleavage was estimated from densitometric traces of the autoradiograms utilizing the Storm 840 software package.
Results and discussion
Design of nucleosomes containing site-specifically positioned 2AP bases
The nucleosome core particles were assembled from the recombinant histone octamers and the 147 base pair-long 601 DNA sequence first described by Lowary and Widom.21 This strong positioning sequence was modified by the replacement of the fragment with the 5′-(C27 - G9) base positions from the dyad axis for the oligonucleotide sequence 5′-d([2AP]T[GGG]1TT[GGG]2TTT[GGG]3TAT). The latter contains three GGG-sites and a single 2-aminopurine (2AP) nucleic base analog, that is designated here as the DNA damage reporter sequence (Figure 1).
The histone octamers were refolded from the recombinant histones25, 26 and their integrity was confirmed by SDS-PAGE (data not shown) and by MALDI-TOF mass spectrometry (Figure 2A). The purity and integrity of the nucleosomes were confirmed by native 5% PAGE in 0.3×TBE buffer (Figure 2B) and hydroxyl radical footprinting methods using the [FeII(EDTA)]23/H2O2/ascorbate system28 expected for good-quality nucleosome particles.20 The observed variations in hydroxyl radical-generated DNA strand cleavage reflect the “in” and “out” rotational settings of the nucleosomal DNA that correspond to the DNA sequences facing towards the histone core or away from it, respectively (Figure 2C). This effect is not observed in free DNA since the •OH radical-generated strand cleavage (after treatment with hot piperidine) is close to random.
Figure 2.

Reconstitution of nucleosomes. (A) Positive MALDI-TOF mass spectrum of histone octamers purified by FPLC. (B) Native 5% PAGE of nucleosome core particles (lane 1) and free 147 base pair-long 601 DNA containing the reporter sequence (lane 2) in 0.3×TBE buffer. (C) Histograms of hydroxyl radical footprints of nucleosomes and of free DNA.
Distributions of alkali-labile lesions generated by carbonate radicals
The modulation of the direct strand cleavage patterns in nucleosomal DNA is a clear indication that the histone proteins shield 2-deoxyribose residues from attack by •OH radicals.31 Here, we explore the effects of nucleotide rotational positions (relative to the histone core surface, Figure 1C) on the distributions of oxidatively generated nucleobase modifications induced by one-electron oxidation. In these experiments we used the biologically relevant carbonate radical anion, a one-electron oxidant.2, 4 Our previous experiments have shown that CO3•− radicals selectively oxidize guanine bases in DNA to yield a spectrum of stable base modifications; these DNA lesions can be detected as strand breaks induced by the standard hot piperidine treatment or by incubation with the Fpg protein.32–34
The CO3•− radicals were generated by continuous UV irradiation of air-equilibrated buffer solutions (pH 7.5) containing nucleosomes with the 2AP-containing 147-bp DNA sequences, Na2S2O8, and NaHCO3. In the presence of an excess of HCO3− anions, SO4•− radicals derived from the photo-induced dissociation of S2O82− anions oxidize bicarbonate, thus generating CO3•− radicals.32–34 The photochemically generated CO3•− radicals induce damage predominantly at the guanine sites in both nucleosomal and in free DNA (Figure 3).
Figure 3.

Representative histograms of autoradiographs of denaturing 8% gels showing the cleavage patterns generated by the one-electron oxidation of 5′-32P-end-labeled nucleosomal and free 2AP-containing modified DNA with photochemically generated CO3•− radicals. Following irradiation, the oxidatively modified DNA bases were converted to strand breaks by hot piperidine treatment.
In nucleosomal DNA, the rotational position of the GGG sequence does not affect the extent of damage initiated by solution-borne CO3•− radicals; the yield of the damage at the [GGG]2-site in the “out” rotational setting is close to the yields at the [GGG]1,3-sites oriented within the “in” rotational setting orientations. Thus, in contrast to the experiments with •OH radicals (Figure 2C), there is no observable modulation of alkali-labile strand cleavage patterns in nucleosomal DNA. Thus, the shielding of guanines in the “in” rotational setting observed with hydroxyl radicals is not evident when the same bases are positioned within the “in” rotational setting where the guanine bases are in closer contact with histone proteins than in the “out” settings.
Distributions of guanine lesions initiated by site-selective hole injection
The utilization of 2AP as a photosensitizer for the site-selective hole injection into DNA has been previously described by us.35, 36 The UV absorbance of 2AP at 308 nm is situated beyond the absorption threshold of DNA and thus photoexcitation by intense nanosecond 308 nm excimer laser pulses induces efficient ionization of 2AP bases by a tandem two-photon absorption mechanism.22 The 2AP radicals thus formed decay by electron transfer reactions from nearby guanine residues37 thus creating holes at [GGG]1, as well as at [GGG]2 and [GGG]3 by further electron transfer or ‘hole hopping’ events. Further transformations of these guanine radicals lead to stable oxidation products. After irradiation with the 308 nm XeCl laser pulses, the 2AP-containing 147-bp 601 DNA sequences in free or nucleosomal DNA were treated with hot piperidine to reveal strand breaks in each of the [GGG]1,2,3 sequences.24 In contrast, using the same irradiation conditions, the extent of cleavage of the 601 nucleosomal DNA without 2AP (the latter was substituted with A) was negligible (data not shown). This is clear evidence that the oxidation of guanines with the formation of alkali-labile lesions is triggered by the photoionization of 2AP in agreement with our previous experiments.22
Typical kinetic profiles calculated from gel autoradiographs show that the extent of oxidatively generated damage at all three GGG-sites increases as a function of energy dosage as shown in Figure 4. In experiments with 2AP-containing nucleosomal DNA, the overall cleavage levels were kept below 20% to emphasize single hit conditions.
Figure 4.

Kinetics of oxidatively generated damage at GGG-sites induced by hole injection in the nucleosomal 5′-32P-end-labeled 2AP-modified DNA strands. Oxidatively modified bases were transformed to DNA strand breaks by standard hot piperidine treatment. The cleavage percentages were calculated from the histograms of the autoradiographs of denaturating gels and normalized relative to the total DNA in the same lane.
Typical histograms observed with the 5′-32P-end-labeled, 2AP-modified DNA strands in nucleosomes and in free DNA duplexes are compared in Figure 5. The most efficient cleavage is observed at [GGG]1-sites separated by one T base from the 2AP residue. In both nucleosomal and free DNA, the distributions of alkali-labile lesions in the 5′-[G1G2G3]i -sites indicate that G2 is more reactive than G1 and that G3 is less reactive than G2. This relative selectivity of G-oxidation (G2 ≥ G1 > G3) has been demonstrated in free double-stranded DNA in 5′-..TGGGT.. sequence contexts exposed to one-electron oxidants38–40 and has been the subject of extensive theoretical analysis.15, 16 The extent of cleavage is the highest in the first GGG-triplet and then gradually decreases with increasing distance from the 2AP residue [GGG]1 > [GGG]2 > [GGG]3 (Figure 5). This is a clear indication that the holes injected into nucleosomal DNA at 2AP-sites migrate towards the 5′-end of the 2AP-containing DNA strand and that the oxidatively generated guanine damage decreases with increasing distance from the 2AP site.
Figure 5.

Representative histograms of autoradiographs of denaturing 8% gels showing the cleavage patterns generated by irradiation of the 2AP residue and hole injection into the nucleosomal and free 5′-32P-end-labeled DNA strands. The oxidatively modified bases were transformed to DNA strand breaks by standard hot piperidine treatment or by incubation with the Fpg protein.
We next investigated the cleavage patterns produced by Fpg-treatment of the irradiated nuclesomal 2AP-containing DNA. The Fpg protein can excise 8-oxoG lesions which are not sensitive to hot piperidine treatment.41–43 In order to assess whether 8-oxoG or other hot piperidine-resistant lesions that are substrates of Fpg are formed as a result of irradiation of free or nucleosomal 2AP-containing DNA with 308 nm laser pulses, the irradiated samples were treated with Fpg. Two approaches were used: the irradiated nucleosome samples were first treated with Fpg and then with Proteinase K (the latter was employed to digest histone proteins), or with Proteinase K followed by Fpg. The extent of cleavage of nucleosomal and free DNA at the three different GGG-sites induced by hot piperidine and by Fpg are compared in Figure 5. The extents of cleavage at the [GGG]2.3-sites relative to the [GGG]1-site (assigned a value of 100) is shown in Figure 6. The patterns of cleavage at the individual guanines G1, G2, and G3 are similar in both cases; the lowest extent of cleavage is observed at G3, and the damage is somewhat smaller at G3 than at G2. As in the case of the hot piperidine treatment, the Fpg treatment reveals the greatest amount of damage at [GGG]1 closest to the 2AP residue, and the lowest at [GGG]3, the most distant one (Figure 5). The extent of overall cleavage observed at [GGG]3 relative to [GGG]1 “in” sites, evaluated by the hot piperidine or Fpg approach, is in the range of (30–33%) in the case of free DNA, and 21–27% in the case of nucleosomal DNA (Figure 6). These ratios indicate that the hole migration distance over the 13 base pairs from the 2AP residue to [GGG]3 is ~ 1.2–1.5 times smaller in nucleosomal than in free DNA. However, the overall extent of hot piperidine-sensitive damage is about two times smaller at the [GGG]2 “out” group than at the “in” [GGG]1 and [GGG]3 groups. This effect is not observed at the analogous [GGG]2 group in free DNA (Figure 6). In nucleosomal DNA this effect does not depend on the order of treatment of the irradiated samples with Proteinase K and with Fpg.
Figure 6.

Relative yields of oxidatively generated damage at the three different GGG-sites initiated by hole injection in nucleosomal and in free 5′-32P-end-labeled 2AP-modified DNA strands. The oxidatively modified guanine bases were transformed to DNA strand breaks by either (i) standard hot piperidine treatment, or (ii) by incubation with Fpg-protein followed by digestion of histone proteins with Proteinase K, or (iii) by digestion of histone proteins with Proteinase K before incubation with Fpg-protein. The yields calculated from the integrated peak areas in the histograms represent the mean of four independent experiments.
One-electron oxidation of DNA by solvent-borne carbonate radical anions: Comparison with hydroxyl radicals
The CO3•− and •OH radicals are small biologically important oxyl radicals. The major sources of •OH radicals in cells are the one-electron reduction of hydrogen peroxide mediated by metal ions such as Fe2+ and Cu+ (Fenton chemistry), and ionizing radiation.44 The CO3•− radicals are known to be overproduced at sites of inflammation as a result of the decomposition of peroxynitrosocarbonate that is formed by the bimolecular combination of peroxynitrite and carbon dioxide.2, 4, 45
Reactions of hydroxyl radicals with DNA occur by two major mechanisms: (i) •OH radical addition to nucleobases, which leads to base modifications, and (ii) H-atom abstraction from 2-deoxyribose residues followed by formation of DNA strand breaks.24, 31, 46 The H-atom abstraction reactions generate C-centered radicals of 2-deoxyribose. Further fragmentation of these intermediates results in the cleavage of 2-deoxyribose and the formation of “direct” DNA strand breaks. The efficiencies of direct strand cleavage induced by •OH radicals are correlated with the solvent-accessibilities of the different 2-deoxyribose H-atoms and can provide unique information on the conformational properties of DNA and DNA-protein complexes.28, 47 In this method (footprinting), the binding sites of proteins are revealed by the absence of strand cleavage at nucleotide sites that are not accessible to hydroxyl radicals because the corresponding DNA sites are less accessible to solvent-borne oxidants. Typically, •OH radicals are generated by the [FeII(EDTA)]2−/H2O2/ascorbate system that efficiently produces direct strand breaks by the base sequence-independent cleavage of the 2-deoxyribose moieties.24, 28, 31
The nucleosomal DNA has a total of 14 contact points with the histone core that occur with a periodicity of 10 bp, equivalent to about one turn of the double helix (10.0–10.7 bp per turn in nucleosomes, vs 10.5 bp in free B-form DNA).20, 48, 49 The hydroxyl radical footprinting method for characterizing the structural integrity of nucleosomes is based on the principle that nucleotides facing towards the histone core (the “in” sites, Fig. 2C) are less accessible to hydroxyl radicals than the more solvent accessible “out” sites. Therefore, the strand cleavage patterns reflect the differences in the reactivities of the individual 2-deoxyribose residues to •OH radicals and the more reactive “out” and the less reactive “in” sites occur with a periodicity of ~10 nucleotides;20, 50 our results shown in Figure 2C are a reflection of this periodicity.
In this work we explored the question whether the same shielding effect is observed when a one-electron oxidant such as the CO3•− radical is employed that is capable of oxidizing guanine bases, but not 2-deoxyribose residues. The carbonate radical is a relatively mild one-electron oxidant with a reduction potential Eo(CO3•−/CO32−) = 1.59 V,51 and we have demonstrated that it can selectively oxidize guanine bases in DNA to form guanine neutral radicals; its midpoint reduction potential at pH 7 is E7[dG(−H)•, H+/dG] = 1.29 V vs. NHE.3 Further chemical transformations of radical intermediates yield a diverse spectrum of stable base modifications including alkali- and Fpg-labile lesions.32–34 We find that the selective one-electron oxidation of guanine bases in nucleosomal DNA with CO3•− radicals does not, after treatment with hot piperidine, result in the appearance of the same kind of strong periodic cleavage patterns (Figure 3) that are observed in the case of direct strand breaks produced by •OH radicals (Figure 2C). One contributing factor could be that a hole generated in an “out” GGG position can be transferred to an adjacent “in” position. However, judging from the differences in the levels of hot piperidine-labile lesions in [GGG]2 and [GGG]3, the efficiency of hole transfer from an “out” to a 3′-neighboring “in” GGG-site is of the order of ~ 50% (Figure 5). Therefore, hole transfer between ‘in” and ‘out” GGG sequences cannot account for the apparent complete lack of periodicity observed in nucleosomal DNA exposed to carbonate radicals (Figure 3).
DNA damage initiated by site-selective hole injection into DNA
The effects of hole redistribution after hole injection into DNA are related to the so-called DNA chemistry at a distance (reviewed in 12–14). These phenomena are interpreted in terms of a primary injection of holes into the DNA (the one-electron oxidation transfer step with guanine acting as an electron donor), migration of mobile intermediates along the DNA helix, the trapping of these intermediates at particular DNA bases, followed by their chemical transformation to stable end-products. Barton and co-workers have shown52 that photoexcitation of a rhodium complex linked to the 5′-terminus of nucleosomal DNA initiates the selective oxidation of guanine bases within a distance of up to 24 base pairs and also without exhibiting a periodic pattern of strand cleavage as observed with hydroxyl radicals.20, 50 Bjorklund and Davis have also explored the distributions of guanine alkali-labile lesions generated by photoexcitation of anthraquinone linked to the 5′-terminus of nucleosomal DNA and they concluded that the extent of damage at guanine sites decreased more sharply in nucleosomal than in free DNA with increasing distance from the site of hole injection.53, 54 Based on quantum-chemical calculations, it was concluded that structural distortions of DNA in nucleosomes at the kinks at the histone octamer contact points, could influence the stability of a guanine radical cation, and therefore might affect the efficiency of hole migration along the DNA helix.55
In our experiments, the 2AP photosensitizer was positioned at nucleotide 27 counted from the dyad axis of the nucleosome particle and the three different [GGG]-sites. The “in” and “out”-type sequences are defined in Figure 7.
Figure 7.

Structural features of nucleosomes viewed from the top of their disc-like shapes. Figure contributed by Y. Cai and S. Broyde, adapted from PDB:2NZD.56
Kinetic considerations
Based on the previous direct spectroscopic observations of electron/hole transfer reactions in the 2AP-containing DNA duplexes, in which guanine bases are separated from the 2AP residue by one to three base pairs, the kinetics of reaction and formation of covalent products are consistent with the scheme shown in Figure 8.22, 23, 35–37, 57, 58
Figure 8.

Mechanisms of oxidatively generated guanine products.22, 23, 35–37, 57, 58 The laser pulse-induced two-photon ionization of 2AP in DNA generates 2AP•+/2AP(−H)• radicals and hydrated electrons. The 2AP•+/2AP(−H)• radicals oxidize a nearby guanine base within the same oligonucleotide strand by a one-electron oxidation mechanism to form G(−H)• radicals. The latter combines with O2•− radicals to form chemical end-products.
The selective photoionization of 2AP residues in double-stranded oligonucleotides induced by 308 nm laser pulses generates first 2AP radical cations injected into DNA by the tandem two-photon ionization mechanism (hole injection, Figure 8).35, 36 The consecutive two-photon absorption of 2AP occurs within the ~ 12 ns characterizing the duration of the laser pulse (full width at half-maximun ~12 ns).22, 37 Measurements of transient absorption spectra of different radical intermediates23 show that the one-electron oxidation of guanines by 2AP•+ occurs within 100 ns after the actinic pulse, thus leading to the formation of guanine radical cations, G•+.36, 37 All radical cations rapidly deprotonate to form neutral radicals (Figure 8).36, 57, 58 Pulse radiolysis experiments published by the Kobayashi group have shown that in double-stranded DNA, depending on the position of G-bases within a DNA strand, the characteristic times of deprotonation of G• + are less than 300 ns.59, 60 A second kinetic component observable on microsecond time scales (k) was time-resolved and assigned to the one-electron oxidation of G-bases by the 2AP neutral radical, 2AP(−H)• that arises from the deprotonation of 2AP•+ cation radicals. Based on the observation of deuterium solvent isotope effects on the rate constant of the microsecond component, we demonstrated that this reaction occurs via a proton-coupled hole transfer mechanism in which electron transfer from G to 2AP(−H)• is coupled with a simultaneous protonation of the 2AP(−H)− and G•+ virtual intermediates.57
We have shown earlier that in the case of the [2AP]TnGGT12-n 15-mer duplexes with complementary strands and T opposite 2AP, the formation of G•+/G(−H)• radicals is biphasic as determined from their transient absorption spectra that exhibit maxima at 315 nm.36, 37 The fast component was attributed to the formation of G•+ radicals that decay within ~ 100 ns after the termination of the actinic laser pulse. The slower component was attributed to proton-coupled electron transfer from guanine to 2AP(−H)• radicals that generates neutral guanine radicals G(−H)•. The prompt fractional yields (≤ 100 ns) of G(−H)• radicals (ΦG [G•+]) depend strongly on the number of T residues between 2AP and G:37 ΦG [G•+] = 0.8 in [2AP]TGGT…, 0.25 in [2AP]TTGGT… and the prompt G•+ component is no longer detectable in the case of the [2AP]TTTGGT… duplexes (Table 1). The latter can be explained in terms of a competition between the deprotonation of 2AP•+ and electron transfer from the closest G in the sequence. The rate of electron transfer is too slow to compete with deprotonation in the case of [2AP]TTTGGT… with three bridging T, and only the slow phase corresponding to the proton coupled electron transfer step 2AP(−H)• → G(−H)• (rate constant k) is observable.37 Once the G(−H)• radicals are formed, proton-coupled hole (electron) transfer can occur to other nearby guanine residues as in the case of our DNA damage reporter sequence.
Table 1.
Prompt (≤100 ns) yields of guanine radical cations (ΦG [G•+]), and rate constants of proton-coupled hole transfer reactions 2AP(−H)• → G(−H)• (rate constant k) that generate neutral guanine radicals via the oxidation of guanine bases by neutral 2AP radicals in DNA duplexes.36, 37
| Sequencea) | ΦG[G• +] | k, s−1 |
|---|---|---|
| [2AP]TGGTTTTTTTTTTT | 0.8±0.1 | (5.0±0.5)×105b) |
| [2AP]TTGGTTTTTTTTTT | 0.25±0.05 | (1.0±0.1)×104 |
| [2AP]TTTGGTTTTTTTTT | ~ 0 | (3.3±0.3)×103 |
| [2AP]TTGGGTTTTTTTTT | n.d. c) | (1.8±0.2)×104 |
Oligodeoxyribonucleotide sequences are written in the 5′ to 3′ direction. The complementary strands with T opposite the 2AP residue are not shown.
The uncertainties are given as standard errors of the best least-squares fits of the appropriate fits of kinetic equations to the transient absorption profiles of the G(−H)• decay curves recorded at 315 nm.
n.d. = not determined.
While analogous measurements for nucleosomes do not exist, we expect that in nucleosomal DNA the most efficient hole transfer occurs between 2AP•+ and [GGG]1 separated by only one T base (Figure 8). The subsequent hole transfer from [GGG1]•+ to [GGG]2 and then to [GGG]3 should be less efficient since these groups of triple G are separated by two and three T bases, respectively. Since deprotonation limits the lifetime of G• + cations,59, 60 the migration of these cation radical states through the TT and TTT bridges is unlikely (Figure 8) and cannot account for the observed damaged patterns at the [GGG]2,3 sites (Figure 5). However, oxidation of the remote [GGG]2,3 sites can occur via the proton-coupled hole transfer that is terminated by the relatively slow chemical reactions that lead to Iz and 8-oxoG formation (Figure 8).
In the [2AP]TGGT… sequence context the proton-coupled hole transfer (microsecond kinetic component) occurs within a time scale of ~ 2 μs (estimated from the k value, Table 1). In the case of two thymines separating the 2AP(−H)• radical and ..GG.. (as in the [2AP]TGGT… sequence context), the value of k decreases by a factor of ~50, but is not significantly different in the case of the …[2AP]TTGGT… and …[2AP]TTGGGT… sequences. When the number of thymines is increased from two to three ([2AP]TTGGT… vs. [2AP]TTTGGGT…), the proton-coupled hole transfer rate constant decreases by a factor of 3. While analogous measurements for nucleosomal DNA do not exist, the parameters provided in Table 1 provide a crude estimate of ~2 μs for the characteristic time of oxidation of guanines by 2AP(−H)• radicals in the 5′-....[2AP]TGGGT… sequence context (proton-coupled hole hopping, Figure 8).
The partial reversibility of one-electron transfer events between 2AP(−H)• and G(−H)• radicals is evident from the significant oxidatively generated damage of 2AP that is observed in addition to damage at every GGG-site in both free and nucleosomal DNA (Figure 5). The rate constants of the oxidation of G by 2AP(−H)• radicals in 5′-2APTTGGT… and 5′-2APTTTGGT… sequences can be considered as a crude estimate of the rate constants of these reactions. Our laser flash photolysis experiments provide lower limits of 60–100 μs for proton-coupled hole hopping over two T-bridging bases and ~300 μs for three T bridging bases (Figure 8, based on the k values summarized in Table 1). Since the piperidine DNA cleavage patterns after irradiation are similar in free DNA and in nucleosomal DNA, the efficiencies of hole transfer in free and nucleosomal DNA appear to be roughly similar. Once the holes become localized on a guanine residue, subsequent reaction of the guanine radical lead to the formation of oxidatively generated end-products (Figure 8). The extent of cleavage is the highest in the first GGG-triplet and then gradually decreases, [GGG]1 > [GGG]2 > [GGG]3 (Figure 5). These results suggest that hole hopping competes with the trapping of guanine radicals at particular DNA sites, followed by their chemical transformation (kchem) to stable end-products (Figure 8).
Formation of guanine oxidation products
It is widely accepted that the guanine radicals can undergo reactions by either of two competitive pathways: (1) reactions with nucleophiles, and (2) combination with oxyl radicals, if available.61, 62 The well-known example of nucleophilic addition to guanine radical is hydration of G•+ radicals.61 The experimental evidence for this reaction was provided by the insertion of isotopic 18O in 8-oxo-7,8-dihydroguanine (8-oxoG) during the riboflavin photosensitized oxidation of calf thymus DNA in H218O solutions.63 Mechanistically, formation of 8-oxoG occurs via the addition of H2O to G•+ to give rise to the reducing 8-hydroxy-7,8-dihydroguanyl radical followed by fast oxidation of this radical by oxygen to form 8-oxoG.61 However, the efficiency of this pathway is expected to be very low, because the hydration rate constant of 6×104 s−1 estimated by Giese and Spichty in double-stranded DNA,64 is by two orders of magnitude smaller than the rate constant of ≥ 3×106 s−1 for deprotonation of G•+ in DNA reported by the Kobayashi group.59, 60 Nevertheless, Douki and Cadet have shown that the oxidation of guanine in calf thymus DNA photosensitized by riboflavin leads predominantly to the formation of 8-oxoG rather than Iz/Oz lesions; this result is consistent with the formation of G•+ radical cations that undergo hydration to yield 8-oxoG.65 However, in the case of our experiments, the initial one- electron oxidation product is the 2AP•+ radical cation that, after rapid deprotonation to the 2AP(−H)• neutral radical, results in the formation of the G(−H)• neutral radical by a proton-coupled electron transfer mechanism (Figure 8).22, 23, 35–37, 57, 58 Thus, in our experiments on the initial photoionization of 2AP, the formation of end products of guanine involves the neutral guanine radicals as precursors of the observed end-products. The ab initio molecular orbital calculation by Saito and co-workers showed that the relative stabilities of G(−H)• radicals decreased according to the order 5′- TG1[G(−H)•]2G3 > 5′- T[G(−H)•]1G2G3, > 5′- TG1G2[G(−H)•]3.40 The distribution of spin densities in these neutral radicals is correlated with the hole distributions in the same 5′-TG1G2G3 sequences which thus accounts for the distributions of the guanine lesions observed in our GGG sequences in free DNA (Figure 5).
Our experiments have shown that the major pathways of guanine lesion formation in 2AP-modified DNA duplexes exposed to 308 nm laser pulse two-photon excitation involve combination of guanine radicals with superoxide radical anions, O2•− derived from the trapping of hydrated electrons (generated by the photoionization of 2AP) by O2 in air-saturated or oxygenated solutions.23 The end-products isolated from the irradiated fully complementary duplexes that include the sequence 5′-d(CC[2AP]TCGCTACC) and its complementary strand include predominantly Iz lesions and their hydrolysis product 2,2,4-triamino-5-(2H)-oxazolone (Oz) lesions with minor amounts of 8-oxoG lesions. Mechanistically, the formation of these lesions occurs via the addition of O2•− radicals to the C5 and C8 positions of O2•− radicals (Figure 9).
Figure 9.
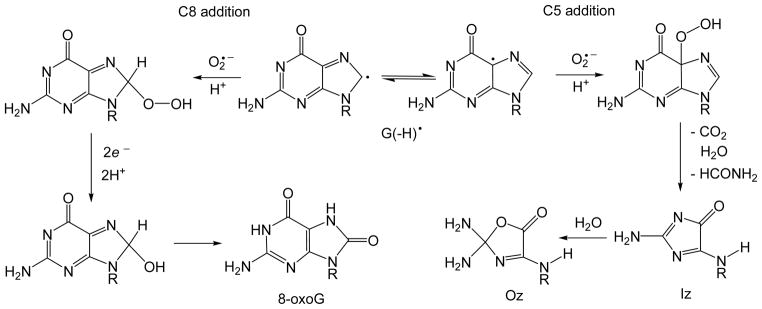
Formation of 8-oxoG and Iz lesions via the addition of O2•− radicals to the C5 and C8 positions of G(−H)• radicals in DNA.23, 62
In agreement with this mechanism, the experiments in air-equilibrated or oxygen-saturated H218O solutions showed that O-atoms in Iz and 8-oxoG lesions originate from 16O2 (not from H218O).23 The appearance of the 18O in 8-oxoG has been detected during the oxidation of human A549 lung epithelial cells by H218O2,66 which can generate 18O2•− radicals in the presence of traces of metal ions (Fe3+, Cu2+).67 Although, the Iz and 8-oxoG lesions were not isolated from nucleosomal DNA we expect that the mechanisms shown in Figure 9 could be responsible for the end-product formation in nucleosomes. In our experiments, one actinic laser flash generates ~1 μM concentrations of G(−H)• and O2•− radicals. The latter can combine with the characteristic time of ~2 ms estimated from the rate constant of 4.7×108 M−1s−1 for the combination of these radicals in DNA duplexes.23 The characteristic times of the proton-coupled hole hopping between the GGG-sites are in the range of 0.1–0.3 ms (Figure 8). However, the actual characteristic times can be longer than the values estimated on the basis of reactions between 2AP(−H)• and GG (Table 1), and thus radical combination can complete with proton-coupled hole hopping.
The hot piperidine method is known to convert Iz and Oz to single-strand breaks,42 while lesions such as 8-oxoG41 are resistant. On the other hand, all of these lesions are substrates of Fpg. Both types of treatments lead to strand cleavage when resolved on denaturing polyacrylamide gels.41–43 The results shown in Figure 6 indicate that the distribution of guanine oxidation products is different in the “out” than the two “in” positions in nucleosomal DNA and all three positions in free DNA. In all positions except the “out” positions in nucleosomes, the identical proportions of hot piperidine and Fpg-sensitive guanine lesions are observed. However, at the [GGG]2 “out” position, there is a greater proportion of Fpg-sensitive adducts. This suggests that at the “out” position a greater proportion of lesions other than Iz and Oz are formed. These lesions are most probably 8-oxoG.
The formation of 8-oxoG is widely believed to occur via the nucleophilic addition of water to C8 of guanine radical cations, G•+ ; however, it is unclear how such a hydration mechanism might apply to guanine radicals, G(−H)•. We propose that the addition of O2•− can occur not only to the C8 position of guanine, but also to the C5 position, and that the relative importance of these two pathways depends on the secondary structure of DNA, as we have shown for other oxyl radicals, e.g., •NO2.68 We found that the ratio of 5-guanidino-4-nitroimidazole (derived from a C5 addition) to 8-nitro-G (C8 addition) gradually decreases from 3.4 in the model compound, 2′,3′,5′-tri-O-acetylguanosine, to 2.1 – 2.6 in single-stranded oligodeoxynucleotides, and to 0.8 – 1.1 in double-stranded DNA. This effect is accounted for in terms of the relative accessibilities of small reactive intermediates and their further transformation to end-products such as Iz (C5 addition) or 8-oxoG (C8 addition) (Figure 9). The relative proportions of Iz and 8-oxoG lesions thus depend on the competition for O2•− by the C5 and C8 positions of guanine radicals, respectively. We surmise that the greater proportions of 8-oxoG adducts and Fpg sensitivity at the nucleosomal “out” position relative to the two “in” positions may be due to a greater accessibility, or greater reactivity of the C8 position of G(−H)• radicals with O2•−.
While nucleosomal DNA is of the B-form, there are significant differences that distinguish it from the normal protein-free B-DNA in aqueous environments.69 Overall, the nucleosomal DNA is somewhat overtwisted and the curvature is significantly greater than in free DNA (but base-pair step-dependent) in order to accommodate the wrapping of the superhelix around the histone octamer. The differences in the proportions of hot piperidine and Fpg-sensitive guanine lesions in the different “in” and “out” rotational settings suggest that these structural, position-dependent factors govern the reactivities of guanine radicals by nucleophilic and radical reaction pathways.
Conclusion
By site-selectively incorporating 2-aminopurine into nucleosomal DNA, a base analog that does not significantly perturb DNA structure, photoinduced hole injection can be triggered at selected rotational settings in nucleosomal DNA. This approach is useful for detailed electron transfer studies and formation of oxidatively damaged guanine bases as a function of rotational setting in nucleosomes. The yields of photoinduced guanine damage diminishes by 70 – 75% for GGG sequences that are separated by 12 base pairs from the 2AP residue as compared to the GGG sequence separated by only one thymine residue. This distance dependence of hole migration is only moderately smaller (by ~ 20–30%) than in the identical histone-free DNA 147-mer DNA sequence. The competition of hole hopping with hole trapping, and thus the distance of hole hopping in free DNA, is widely assumed to be limited by the hydration of the guanine radical cation G•+. However, we have shown previously that the dominant trapping reaction when hole injection is achieved by the 2AP photoionization mechanism occurs on time scales of μs to ms and involves the combination of superoxide anions with neutral guanine radicals that are formed by the rapid deprotonation of G•+.23 Hot piperidine and Fpg protein strand cleavage assays indicate that the patterns of DNA damage in nucleosomes are similar to free DNA. In free DNA, the major end-products are piperidine-sensitive imidazolone, and minor proportions of 8-oxoG are also formed. At the “in” rotational settings in nucleosomes the patterns of piperidine-induced strand cleavage are similar, suggesting a predominance of Iz lesions. However, a greater proportion of Fpg-sensitive lesions, attributed to 8-oxoG, are found at the “out” settings in nucleosomal DNA. These results, together with earlier findings,23 suggest that the ratio of Iz/8-oxoG depends on the reaction of O2•− with either the C5 or C8 position of G(−H)• with the C5 reaction dominating in free DNA and at the [GGG]1,3-sites “in” nucleosomal rotational settings. We conclude that the Iz/8-oxoG ratio depends on the reactivities and accessibility of the C5 and C8 positions of guanine radicals to O2•− that depends on the site-specific base pair structural parameters that are known to vary significantly as a function of position in nucleosomal DNA.
Acknowledgments
This work was supported by the National Institute of Environmental Health and Sciences Grant 5 R01 ES 011589-10. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health and Sciences or the National Institutes of Health. Components of this work were conducted in the Shared Instrumentation Facility at NYU that was constructed with support from a Research Facilities Improvement Grant (C06 RR-16572) from the National Center for Research Resources, National Institutes of Health. The acquisition of the MALDI-TOF mass spectrometer was supported by the National Science Foundation (CHE-0958457).
References
- 1.Coussens LM, Werb Z. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dedon PC, Tannenbaum SR. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 3.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 4.Cadet J, Douki T, Ravanat JL. Nat Chem Biol. 2006;2:348–349. doi: 10.1038/nchembio0706-348. [DOI] [PubMed] [Google Scholar]
- 5.Saito I, Takayama M, Sugiyama H, Nakatani K, Tsuchida A, Yamamoto M. J Am Chem Soc. 1995;117:6406–6407. [Google Scholar]
- 6.Hall DB, Holmlin RE, Barton JK. Nature. 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 7.Ly D, Kan Y, Armitage B, Schuster GB. J Am Chem Soc. 1996;118:8747–8748. [Google Scholar]
- 8.Giese B, Amaudrut J, Kohler AK, Spormann M, Wessely S. Nature. 2001;412:318–320. doi: 10.1038/35085542. [DOI] [PubMed] [Google Scholar]
- 9.Saito I, Nakamura T, Nakatani K, Yoshioka Y, Yamaguchi K, Sugiyama H. J Am Chem Soc. 1998;120:12686–12687. [Google Scholar]
- 10.Kurnikov IV, Tong GSM, Madrid M, Beratan DN. J Phys Chem B. 2002;106:7–10. [Google Scholar]
- 11.Yokojima S, Yoshiki N, Yanoi W, Okada A. J Phys Chem B. 2009;113:16384–16392. doi: 10.1021/jp9054582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nunez ME, Hall DB, Barton JK. Chem Biol. 1999;6:85–97. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 13.Schuster GB. Acc Chem Res. 2000;33:253–260. doi: 10.1021/ar980059z. [DOI] [PubMed] [Google Scholar]
- 14.Giese B. Acc Chem Res. 2000;33:631–636. doi: 10.1021/ar990040b. [DOI] [PubMed] [Google Scholar]
- 15.Yoshioka Y, Kawai H, Sato T, Yamaguchi K, Saito I. J Am Chem Soc. 2003;125:1968–1974. doi: 10.1021/ja028039m. [DOI] [PubMed] [Google Scholar]
- 16.Senthilkumar K, Grozema FC, Guerra CF, Bickelhaupt FM, Siebbeles LDA. J Am Chem Soc. 2003;125:13658–13659. doi: 10.1021/ja037027d. [DOI] [PubMed] [Google Scholar]
- 17.Senthilkumar K, Grozema FC, Guerra CF, Bickelhaupt FM, Lewis FD, Berlin YA, Ratner MA, Siebbeles LDA. J Am Chem Soc. 2005;127:14894–14903. doi: 10.1021/ja054257e. [DOI] [PubMed] [Google Scholar]
- 18.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 19.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 20.Hayes JJ, Tullius TD, Wolffe AP. Proc Natl Acad Sci U S A. 1990;87:7405–7409. doi: 10.1073/pnas.87.19.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowary PT, Widom J. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 22.Shafirovich V, Dourandin A, Huang W, Luneva NP, Geacintov NE. J Phys Chem B. 1999;103:10924–10933. [Google Scholar]
- 23.Misiaszek R, Crean C, Joffe A, Geacintov NE, Shafirovich V. J Biol Chem. 2004;279:32106–32115. doi: 10.1074/jbc.M313904200. [DOI] [PubMed] [Google Scholar]
- 24.Burrows CJ, Muller JG. Chem Rev. 1998;98:1109–1151. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 25.Luger K, Rechsteiner TJ, Richmond TJ. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 26.Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 27.Simpson RJ. Cold Spring Harb Protoc. 2006 doi: 10.1101/pdb.prot4313. [DOI] [Google Scholar]
- 28.Jain SS, Tullius TD. Nat Protoc. 2008;3:1092–1100. doi: 10.1038/nprot.2008.72. [DOI] [PubMed] [Google Scholar]
- 29.Margolin Y, Shafirovich V, Geacintov NE, DeMott MS, Dedon PC. J Biol Chem. 2008;283:35569–35578. doi: 10.1074/jbc.M806809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu CS, Hernandez R, Schuster GB. J Am Chem Soc. 2004;126:2877–2884. doi: 10.1021/ja0378254. [DOI] [PubMed] [Google Scholar]
- 31.Pogozelski WK, Tullius TD. Chem Rev. 1998;98:1089–1107. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 32.Lee YA, Yun BH, Kim SK, Margolin Y, Dedon PC, Geacintov NE, Shafirovich V. Chem Eur J. 2007;13:4571–4581. doi: 10.1002/chem.200601434. [DOI] [PubMed] [Google Scholar]
- 33.Lee YA, Durandin A, Dedon PC, Geacintov NE, Shafirovich V. J Phys Chem B. 2008;112:1834–1844. doi: 10.1021/jp076777x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee YA, Liu Z, Dedon PC, Geacintov NE, Shafirovich V. ChemBioChem. 2011;12:1731–1739. doi: 10.1002/cbic.201100140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shafirovich V, Geacintov NE. Top Curr Chem. 2004;237:129–157. [Google Scholar]
- 36.Shafirovich V, Geacintov NE. In: Charge Transfer in DNA. Wagenknecht HA, editor. Wiley-VCH; Weinheim: 2005. pp. 175–196. [Google Scholar]
- 37.Shafirovich V, Dourandin A, Huang W, Luneva NP, Geacintov NE. Phys Chem Chem Phys. 2000;2:4399–4408. [Google Scholar]
- 38.Ito K, Inoue S, Yamamoto K, Kawanishi S. J Biol Chem. 1993;268:13221–13227. [PubMed] [Google Scholar]
- 39.Spassky A, Angelov D. Biochemistry. 1997;36:6571–6576. doi: 10.1021/bi962761d. [DOI] [PubMed] [Google Scholar]
- 40.Yoshioka Y, Kitagawa Y, Takano Y, Yamaguchi K, Nakamura T, Saito I. J Am Chem Soc. 1999;121:8712–8719. [Google Scholar]
- 41.Cullis PM, Malone ME, Merson-Davies LA. J Am Chem Soc. 1996;118:2775–2781. [Google Scholar]
- 42.Gasparutto D, Ravanat JL, Gerot O, Cadet J. J Am Chem Soc. 1998;120:10283–10286. [Google Scholar]
- 43.Duarte V, Gasparutto D, Jaquinod M, Cadet J. Nucleic Acids Res. 2000;28:1555–1563. doi: 10.1093/nar/28.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cadet J, Douki T, Ravanat JL. Free Radic Biol Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 45.Pacher P, Beckman JS, Liaudet L. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Breen AP, Murphy JA. Free Radic Biol Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- 47.Tullius TD, Dombroski BA. Proc Natl Acad Sci U S A. 1986;83:5469–5473. doi: 10.1073/pnas.83.15.5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luger K. Curr Opin Genet Dev. 2003;13:127–135. doi: 10.1016/s0959-437x(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 49.Luger K. Chromosome Res. 2006;14:5–16. doi: 10.1007/s10577-005-1026-1. [DOI] [PubMed] [Google Scholar]
- 50.Chafin DR, Hayes JJ. Methods Mol Biol. 2009;543:121–138. doi: 10.1007/978-1-60327-015-1_10. [DOI] [PubMed] [Google Scholar]
- 51.Huie RE, Clifton CL, Neta P. Radiat Phys Chem. 1991;38:477–481. [Google Scholar]
- 52.Nunez ME, Noyes KT, Barton JK. Chem Biol. 2002;9:403–415. doi: 10.1016/s1074-5521(02)00121-7. [DOI] [PubMed] [Google Scholar]
- 53.Bjorklund CC, Davis WB. Nucleic Acids Res. 2006;34:1836–1846. doi: 10.1093/nar/gkl030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bjorklund CC, Davis WB. Biochemistry. 2007;46:10745–10755. doi: 10.1021/bi700475b. [DOI] [PubMed] [Google Scholar]
- 55.Voityuk AA, Davis WB. J Phys Chem B. 2007;111:2976–1985. doi: 10.1021/jp066470i. [DOI] [PubMed] [Google Scholar]
- 56.Ong MS, Richmond TJ, Davey CA. J Mol Biol. 2007;368:1067–1074. doi: 10.1016/j.jmb.2007.02.062. [DOI] [PubMed] [Google Scholar]
- 57.Shafirovich V, Dourandin A, Luneva NP, Geacintov NE. J Phys Chem B. 2000;104:137–139. [Google Scholar]
- 58.Shafirovich V, Dourandin A, Luneva NP, Geacintov NE. J Chem Soc Perkin Trans. 2000;2:271–275. [Google Scholar]
- 59.Kobayashi K, Tagawa S. J Am Chem Soc. 2003;125:10213–10218. doi: 10.1021/ja036211w. [DOI] [PubMed] [Google Scholar]
- 60.Kobayashi K, Yamagami R, Tagawa S. J Phys Chem B. 2008;112:10752–10757. doi: 10.1021/jp804005t. [DOI] [PubMed] [Google Scholar]
- 61.Cadet J, Douki T, Ravanat JL. Acc Chem Res. 2008;41:1075–1083. doi: 10.1021/ar700245e. [DOI] [PubMed] [Google Scholar]
- 62.Shafirovich V, Geacintov NE. In: Radical and radical ion reactivity in nucleic acid chemistry. Greenberg M, editor. John Willey&Sons, Inc; Hoboken, New Jersey: 2009. pp. 325–355. [Google Scholar]
- 63.Kasai H, Yamaizumi Z, Berger M, Cadet J. J Am Chem Soc. 1992;114:9692–9694. [Google Scholar]
- 64.Giese B, Spichty M. Chem Phys Chem. 2000;1:195–198. doi: 10.1002/1439-7641(20001215)1:4<195::AID-CPHC195>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 65.Douki T, Cadet J. Int J Radiat Biol. 1999;75:571–581. doi: 10.1080/095530099140212. [DOI] [PubMed] [Google Scholar]
- 66.Hofer T, Badouard C, Bajak E, Ravanat JL, Mattsson A, Cotgreave IA. Biol Chem. 2005;386:333–337. doi: 10.1515/BC.2005.040. [DOI] [PubMed] [Google Scholar]
- 67.Wardman P, Candeias LP. Radiat Res. 1996:145. [PubMed] [Google Scholar]
- 68.Joffe A, Mock S, Yun BH, Kolbanovskiy A, Geacintov NE, Shafirovich V. Chem Res Toxicol. 2003;16:966–973. doi: 10.1021/tx025578w. [DOI] [PubMed] [Google Scholar]
- 69.Richmond TJ, Davey CA. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]