

Abstract
Alexander disease (AxD) is a usually fatal astrogliopathy primarily caused by mutations in the gene encoding GFAP, an intermediate filament protein expressed in astrocytes. We describe three patients with unique characteristics, and whose mutations have implications for AxD diagnosis and studies of intermediate filaments. Patient 1 is the first reported case with a non-coding mutation. The patient has a splice site change producing an in-frame deletion of exon 4 in about 10% of the transcripts. Patient 2 has an insertion and deletion at the extreme end of the coding region, resulting in a short frameshift. In addition, the mutation was found in buccal DNA but not in blood DNA, making this patient the first reported chimera. Patient 3 has a single base deletion near the C-terminal end of the protein, producing a short frameshift. These findings recommend inclusion of intronic splice site regions in genetic testing for AxD, indicate that alteration of only a small fraction of GFAP can produce disease, and provide caution against tagging intermediate filaments at their C-terminal end for cell biological investigations.
Keywords: Alexander disease, GFAP, chimera, astrocyte, leukodystrophy, aggregate
Introduction
Alexander disease (AxD; MIM# 203450) is a devastating neurological disorder that has highly variable ages of onset and clinical presentation (reviewed in Li et al., 2002; Brenner et al., 2009; Prust et al., 2011). The infantile form, typically presenting before two years of age, often displays megalencephaly, seizures, and impaired physical and mental development; death usually ensues within a few years. Later onset cases present with quite different clinical symptoms and have slower disease progression. They typically have bulbar signs, which include difficulties with coordination, speech and swallowing. The two forms are unified by the pathological hallmark of AxD--widespread and abundant Rosenthal fibers, which are astrocytic protein aggregates that mainly contain GFAP (MIM# 137780) and small stress proteins. Heterozygous mutations in GFAP, an intermediate filament protein expressed abundantly in astrocytes, account for most instances of both forms of AxD (Brenner et al., 2001; Li et al., 2005). At present, 86 different mutations have been described that affect 63 different amino acid positions in the protein (Brenner et al., 2009; Flint and Brenner, 2011). All these previously identified GFAP mutations in AxD patients are either missense coding changes or additions and/or deletions of one or a few amino acids. In this study we report two novel types of mutations associated with AxD, an intronic mutation that results in the in-frame deletion of exon 4, and an insertion/deletion at the end of the coding region which results in a frameshift. Also described is a case with a C-terminal frameshift resulting from the deletion of a single nucleotide.
Materials and Methods
Mutation analysis
This investigation was performed under institutional review board approval from the University of Alabama at Birmingham and the University of Wisconsin-Madison. DNA for initial mutation detection was isolated from frozen brain tissue for patient 1, from blood and buccal samples for patient 2 and from blood for patient 3. DNA for parental testing was isolated from blood. GFAP exons and portions of the flanking introns were amplified by PCR and sequenced as previously described (Brenner et al., 2001). To investigate exon skipping in patient 1, RNA was extracted from this patient’s frozen brain tissue, as well as from a normal control and two other AxD cases, patients 5 and 10 of Brenner, et al., (2001), reverse transcribed, and amplified with an upstream primer located in exon 1 (5′-CGATCAACTCACCGCCAACA) and a downstream primer located in either exon 6 (5′-AGGTCCTGGTACTCCTGCAA) or in the 3′ untranslated region (5′-GGGAAATGTGCCAGCAGAG). After agarose gel electrophoresis, the presumptive PCR product from the exon 4-skipped transcript was excised and sequenced. The Dor Yeshorim Institute (New York, NY) provided ethnically matched (Ashkenazi Jewish) DNA samples for determining whether the mutation present in patient 1 was generally present in this population. A 202 bp DNA fragment spanning the mutation site was amplified using primers 5′AACCCCAGCTGTGGCCTGTA and 5′-CTTCGGCTTCATGCATGTTG, digested with MslI (which specifically recognizes the mutant sequence), and analyzed on an agarose gel. DNA from the patient was processed with each set of samples as a positive control. To investigate ectodermal mosaicism of patient 2, DNA was isolated from nail clippings from each hand and foot using the method of Cline et al. (2003). The nail DNAs, and for certain experiments also the blood and buccal DNAs, were analyzed for the presence of the mutant allele by PCR with a common 5′ primer (5′-CCAGGCATGACGCTAAATGC-3′) and a 3′ primer specific for the mutant allele (5′-AGGTGGGTCCTGCCGATCA-3′) or the wild type allele (5′-GTGGGTCCTGCCTCACATCA-3′). The DNA obtained from the blood, buccal and nail clipping samples of patient 2 were confirmed to originate from the same individual by genetic testing by the UAB Medical Genomics Lab. GenBank references sequences NG_008401.1 and NM_002055.4 were used to describe the observed mutations.
Functional analysis of mutant GFAP proteins
Plasmid pcDNA3.1-hGF(Δex4), which expresses human GFAP lacking exon 4, was constructed by excising the exon 4-containing XcmI/PmlI fragment from the corresponding wild type vector (Li et al., 2005) and replacing it with the corresponding fragment obtained by RT-PCR of RNA isolated from patient 1. Plasmid pcDNA3.1-hGF(I/D/F094), which expresses the human GFAP with the insertion/deletion/frameshift mutation found in patient 2, was constructed by replacing a PmlI/EcoRI fragment that spans the 3′ end of the GFAP coding region with one obtained by PmlI/EcoRI digestion of a PCR product generated using a 5′ primer upstream of the PmlI site and a 3′ primer that included the EcoRI site and the mutant sequence. For both constructs, the entire PCR-generated insert was sequenced to confirm it was correct. To evaluate functionality, the expression vectors were transfected into SW13vim− cells plated onto glass coverslips in 24-well plates and immunostained 2 days later as previously described (Li et al., 2005). The transfection efficiency was approximately 20%.
Statistical analysis
The Chi square test was employed to compare the cell numbers under each category among different sets of transfection studies.
Results
Table 1 lists the three novel GFAP mutations found in the patients described below.
Table 1.
Patients with GFAP Mutations Identified in This Study
| Patient | Nucleotide Change | Protein Change |
|---|---|---|
| 1 | c.(619-3C>G) | p.Glu207_Lys260del1 |
| 2 | c.1292_1299delinsATC | p.Val431Aspfs*14 |
| 3 | c.1249delG | p.Asp417Metfs*15 |
The reference sequence is NG_008401.1 (genomic) for the patient 1 nucleotide change; and NM_002055.4 (cDNA) for the nucleotide changes for patients 2 and 3 and for all three protein changes, wherein the “A” of the initiating ATG is taken as position +1 in accordance with journal guidelines (www.hgvs.org/mutnomen); and the initiating codon is codon 1. Variants were submitted to the Human Intermediate Filament Database (http://www.interfil.org).
About 10% of the GFAP transcripts splice out exon 4, resulting in the indicated protein change
Patient 1 clinical history
Patient 1 was a 39 year old woman with paraplegia, autonomic and bulbar dysfunction with dysphagia and a neurogenic bladder, who died of multiple systems failure. She first presented at 14 years with difficulties in speech, walking and enuresis, although her voice had a nasal quality from childhood. Intermittent episodes of tightening of the left hand and subsequently a tense feeling at the left angle of the mouth developed at age 23. An EEG, brain CT scan and CSF examination were normal and the paroxysmal episodes were controlled with 100 mg daily of carbamazepine. Two years later leg weakness began, much more in the left than the right. Cranial MRI at age 29 was normal, and no follow-up MRI was performed. Visual and auditory evoked responses were also normal at 29 years, but somatosensory evoked potentials were abnormal. Nine years later abnormal pattern-shift visual evoked responses were noted. Cognition appeared normal at age 29. Family history of degenerative neurological disease was negative. She had difficulty walking, with weakness greater in the left leg than the right. Muscle stretch reflexes were lively but ankle jerks were absent and the plantar response was extensor bilaterally. Vibratory sensation was mildly decreased in the legs. For many years she had difficulty swallowing, with frequent episodes of choking, and during her last seven years she required ephedrine for orthostatic hypotension. In her final three years she remained wheelchair-bound. She died suddenly in a long term care facility.
Neuropathological examination at autopsy showed marked atrophy of the medulla and all levels of the spinal cord. Microscopic examination revealed Rosenthal fibers in perivascular locations and diffusely underlying the ependyma of the lateral and third ventricles, and the most severe myelin loss in the periventricular and deep white matter. Myelin loss and Rosenthal fibers were also observed in the deep cerebellar white matter, dentate nuclei and in the medullary pyramids. In the spinal cord there was moderate to severe loss of myelinated fibers in the gracile tract, in the lateral and anterior corticospinal tracts bilaterally, and in the spino-thalamic and spino-cerebellar tracts with variable deposition of Rosenthal fibers
Patient 1 DNA sequencing
DNA sequencing for patient 1 revealed no coding mutation within the GFAP gene. However, a heterozygous C to G change was present in intron 3, three bp before the beginning of exon 4 (designated as c.(619-3C>G)) (Supp. Figure S1). The presence of a G 3 bp before an exon (IVS-3) is highly unusual. Among 542 primate introns surveyed, 72% have a C at this position, and fewer than 1% have a G (Shapiro and Senapathy, 1987). This raised the possibility that the nucleotide change may affect splicing efficiency. A literature search indeed provided precedent for an IVS-3C>G change causing exon skipping (Ieiri et al, 1991; Mayer et al., 2000).
Identification of the mutant GFAP splice variant
To determine if the c.(619-3C>G) results in skipping of one or more exons of the GFAP gene, RT-PCR was performed on RNA isolated from the patient’s brain using an upstream primer in exon 1 and a downstream primer either in exon 6 or the 3′ untranslated region (3′ UTR). Data obtained using the exon 6 primer are shown in Supp. Figure S2. In addition to a product of the size expected for wild type GFAP (675 bp), a minor band was observed of the size predicted if exon 4 were skipped (513 bp). This smaller band was obtained using patient RNA, but not from RNA isolated from a neurologically normal control, or from AxD patients with either an R239C or R416W coding mutation. Excision and sequencing of the smaller PCR product showed it indeed arose from an exon 4-skipped transcript--it contained wild type cDNA sequences through the end of exon 3 joined directly to wild type sequences starting at the beginning of exon 5 (data not shown). No smaller products were observed for the exon 6 primer (Supp. Figure S2) or when the 3′ UTR primer was used (data not shown), indicating that missplicing was largely limited to skipping of exon 4.
Quantitation of WT/mutant transcripts ratio
Supp. Figure S2 indicates that the exon 4-skipped transcript is present at a significantly lower level than the wild type. To estimate the ratio of wild type to mutant transcript present in the patient RNA, we compared the relative intensities of their respective RT-PCR products to those obtained by PCR of different ratios of plasmids encoding the wild type and mutant GFAPs. The data shown in Fig. 1 indicate that the ratio of wild type to mutant transcripts in the patient RNA is in the range of 12:1. Control experiments showed that the ratio of the two PCR products did not change appreciably when the number of PCR cycles was varied from 10 to 35 (data not shown).
Figure 1.
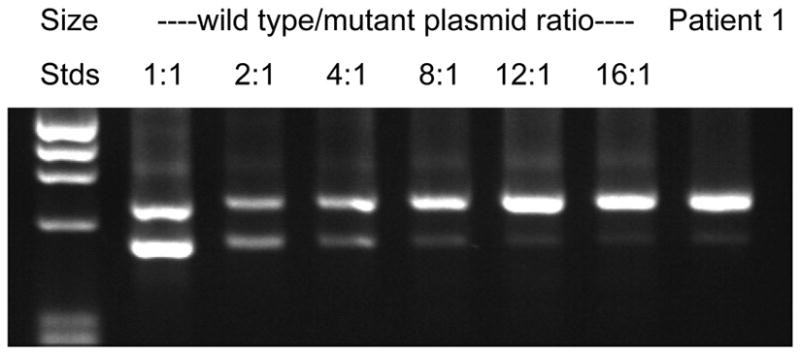
Relative amount of mutant transcript. The ratio of wild type to mutant transcripts was determined by comparing the band intensities obtained by PCR of patient cDNA (far right lane) with those amplified from wild type and mutant plasmids mixed at different ratios (lanes 2–7). The results indicate that the level of the truncated transcript is about 12-fold lower than the wild type transcript.
Functional analysis of Patient 1 mutant GFAP
Many of the GFAP mutations associated with AxD have been shown to arise de novo, providing strong evidence for causality. The mother of Patient 1 tested negative for the nucleotide change present in her daughter, but the father was deceased. As an alternative, DNA from 525 ethnically matched (Ashkenazi Jewish) individuals was tested for the presence of the nucleotide change, with negative results for all 1050 chromosomes. To further investigate the possibility of causality we examined the functional consequences of the mutation. An expression plasmid for the mutant protein (p.Glu207_Lys260del) was constructed by producing an in frame deletion of the 54 amino acids encoded by exon 4. Different ratios of the wild type and mutant expression vectors were then transfected into SW13vim− cells, which lack endogenous cytoplasmic intermediate filaments (Hedberg & Chen, 1986). As has been previously observed (Li et al., 2005), the wild type protein nearly always formed a filamentous network (Fig. 2A), whereas the mutant GFAP produced a completely different pattern characterized by short needle-like protein aggregates (Fig. 2B). Co-transfection of wild type and mutant expression vectors at a 1:1 ratio usually yielded aggregates (Fig. 2C), consistent with a dominant effect of the mutant protein. Importantly, a dominant effect was still discernible with a wild type to mutant ratio as high as 40:1 (Fig. 2D, Fig. 3). Control experiments verified that the relative amounts of wild type and mutant GFAP protein produced were proportional to the ratio of plasmids transfected (Supp. Figure S3).
Figure 2.

Polymerization properties of wild type and mutant proteins. Expression plasmids encoding wild type (WT) GFAP and/or mutant GFAPs predicted for Patient 1 (Δex4) or Patient 2 (I/D/F) were transiently transfected into SW13vim− cells and the cells stained two days later for GFAP as described in Materials and Methods. A, WT; B, Δex4; C, 1:1 WT:Δex4; D, 8:1 WT:Δex4; E, I/D/F.
Figure 3.
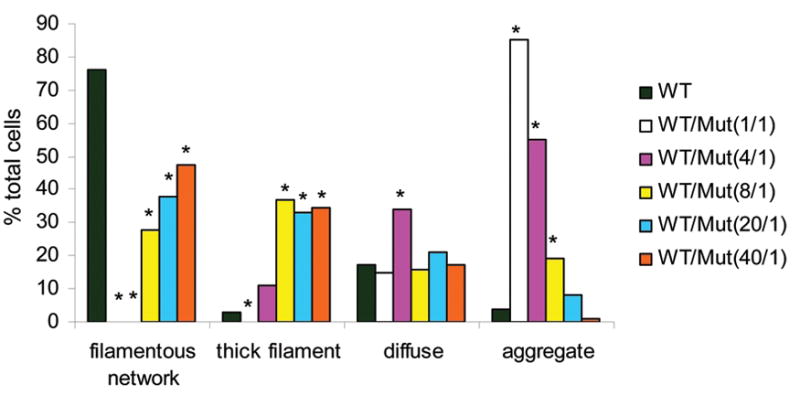
Effect of the ratio of wild type GFAP (WT) to Patient 1 mutant GFAP (Δex4) on filament assembly in transfected SW13vim− cells. SW13vim− cells were transfected with different ratios of WT to Δex4 expression plasmids, while the total amount of plasmid was held constant. Cells were then stained for GFAP, and the patterns classified as a normal filamentous network (cf Fig. 3A), thick filaments (Fig. 3D & E), diffuse staining (background in 3C) or aggregates (3B & C). The percentages of each pattern for a minimum of 150 cells are shown for the different WT to Δex4 ratios. *The difference compared with WT is significant (p<0.01) by Chi Square test.
Patient 2 clinical history
Patient 2 was macrocephalic at birth (98%), but his father’s head circumference was 61 cms (well above the 98%) and his mother’s was 57.5 cm (just below the 98%). Apgar scores were normal. At 1 year of age he was evaluated for developmental delay and intermittent ataxia. An MRI at 17 months of age showed moderate ventriculomegaly, but normal signal intensity of the gray and white matter and normal diffusion imaging. Subsequent MRIs at 3, 5 and 6 years of age were normal except for the ventriculomegaly and a small, stable T1 and T2 hypoenhancing region in the middle of the pituitary, thought unrelated to AxD. He walked at 23 months, but stumbled when walking or turning. This has not been progressive. At 33 months he was hypotonic but not ataxic, and was verbal and followed commands. At 4.5 years he could feed himself, say the alphabet, count to 10 and use 3 word phrases. He was seizure free until 4 years 9 months, when he manifested tonic clonic seizures and staring episodes that were controlled with valproic acid. An EEG was epileptiform. His neurological examination was unremarkable without focal deficits except for cognitive delays. He had a clumsy gait, but no cerebellar deficits. At the age of 6 yrs and 7 months he had slight dysarthria and gait was slightly clumsy. He was socially age appropriate and interactive, answering and following instructions.
Patient 2 DNA sequencing
Buccal DNA of Patient 2 was analyzed for GFAP mutations by GeneDx (Gaithersburg, MD) due to several clinical signs consistent with AxD (macrocephaly, delayed development, clumsy gait, dysarthria, seizures) and negative tests for multiple other disorders. An 8 bp deletion accompanied by a 3 bp insertion (c.1292_1299delinsATC) was detected, which results in a frame-shift at the extreme C-terminus of the protein. This is predicted to alter the identity of the last two amino acids of the wild type protein and to elongate the tail by an additional 11 amino acids (p.Val431Aspfs*14) (Fig. 4). The case was brought to our attention because of the unique nature of the insertion/deletion/frameshift (I/D/F) mutation, and the willingness of the parents to be tested to determine if the mutation arose de novo in their child and was thus likely to be causative for the disease. Surprisingly, the blood DNAs from both the parents and the patient proved to be wild type. We then sequenced the patient’s buccal DNA sample provided by GeneDx, and confirmed their finding of the I/D/F mutation (Fig. 4). The possibility of a sample mix-up was eliminated by DNA analysis by the UAB Medical Genomics Lab using a panel of markers used for paternity testing. These tests documented that the two DNA samples came from the same individual. Thus this patient is the first documented example of a mosaic for a GFAP mutation in AxD.
Figure 4.
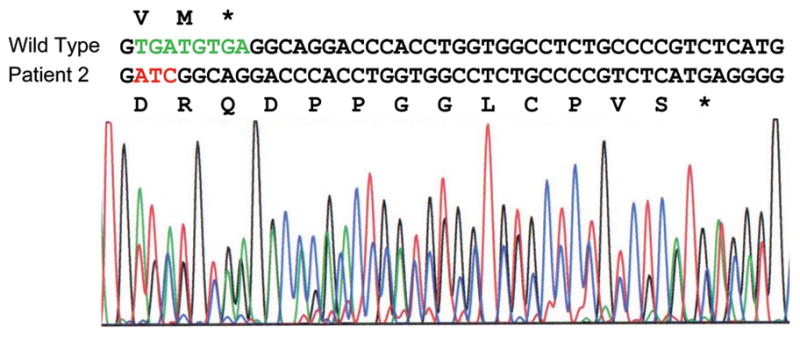
Mutation present in Patient 2. The mutation involves deletion of 8 bp (green) and insertion of 3 bp (red), resulting in a frameshift that changes the last two amino acids of wild type GFAP from VM to DR and adds 11 additional residues before encountering a stop codon.
The procedure used to sequence the blood DNA would not have detected the mutant sequence were it present in fewer than about 25% of the cells. Since the mutation involved a multi-nucleotide insertion and deletion, allele-specific PCR primers could be designed to permit a more sensitive assay for the presence of the mutant allele in the blood DNA. Reconstruction experiments in which blood DNA from a control was mixed with decreasing amounts of buccal DNA from the patient indicated that the allele-specific PCR could detect as little as 1 part in 1,000 of mutant DNA (Supp. Figure S4). Nevertheless, the mutant allele remained undetectable in the patient blood DNA sample. In contrast, the relative peak heights for the mutant and wild type nucleotides in the buccal DNA sequence suggest that 100% of these cells carry the mutation (Fig. 4). Current understanding of mammalian development indicates that mesoderm (blood) and ectoderm (buccal and brain) arise from a common multicellular precursor pool (Soriano and Jaenisch, 1986); our finding thus suggests that the mutation arose following specification of the germ layers, and that accordingly, the patient would also be mosaic in his ectoderm. To test this in a non-invasive manner, we isolated DNA from nail clippings from each hand and foot, and used the allele-specific PCR protocol to test for the presence of the mutant and wild type alleles. Only the wild type allele was detected (Supp. Figure S5). Forensic DNA testing showed that each of the nail DNA samples arose from the same individual as the blood and buccal DNA samples, ruling out the possibility of contamination. Thus Patient 2 appears to be an ectodermal mosaic for the GFAP mutation.
Functional analysis of Patient 2 mutant GFAP
Since the I/D/F mutation in Patient 2 is mosaic, it clearly arose de novo (as expected, it was not found in the blood DNA of either parent). This provides very strong evidence that the mutation is disease causing. To investigate its effect on GFAP polymerization, SW13vim− cells were transiently transfected with a construct that expressed the mutant protein, as was done above for the Δex4 mutation of Patient 1. The mutant protein did assemble into filaments, but unlike the fine filamentous network that coursed throughout the cell that is typically produced by wild type GFAP (Fig. 2A), the I/D/F mutant GFAP consistently produced thick bundles that were primarily perinuclear (Fig. 2E). Co-transfection of a 1:1 mixture of mutant and wild type expression vectors produced an assembly pattern identical to that of the mutant alone, except for a general increase in diffuse background staining (data not shown). This is consistent with the dominant nature of the heterozygous GFAP mutations associated with AxD.
Patient 3 clinical history
At four months Patient 3 had not yet rolled over and had poor head control and diffusely low muscle tone. Feeding was associated with recurrent vomiting, and weight and linear growth slowed from the 50th to the 3rd percentiles. An upper GI study revealed moderate gastroesophageal reflux, and a gastrostomy tube was placed. He began reaching for toys between five and six months, but showed no bimanual grasping or transferring. He was never able to sit without full support of his trunk. At age six months he was evaluated by an ophthalmologist for intermittent strabismus and was noted to have vertical nystagmus, prompting a cranial MRI which is described below. On examination at age seven months, he was alert and interactive with fairly good head control. Extraocular movements were full and nystagmus was present, but not prominent. Muscle tone was reduced generally. Muscle stretch reflexes were normal and clonus at the ankles was absent. He picked up objects with a raking grasp bilaterally and still showed no bimanual grasping or transferring. He had prominent dystonia of the left arm. Between seven and nine months his height and weight growth stabilized and progressed along the 3rd percentile, and head growth decelerated from the 50th to the 25th percentile, stabilizing at that level. He was able to reach for objects, roll from front to back, and stand with support. No regression was evident. Although nystagmus was no longer observed, strabismus became virtually continuous. Muscle tone of his extremities was increased on the left side, while axial tone remained low. He tended to reach for objects preferentially on the right. Oral feeding improved between nine and eleven months, but he was then admitted to the hospital with respiratory failure secondary to aspiration from gastroesophageal reflux. He was slowly weaned from the ventilator and then underwent fundoplication. Following this surgery he never truly regained awareness and could not be weaned from the ventilator a second time. He expired just before one year of age.
This patient had two sets of cranial MRIs, the first at six months, prompted by the ophthalmology findings of nystagmus, and the second at one week before death. The MRI at age 6 months revealed atrophy of the frontal lobes (Fig. 5A), and an abnormally high T2 signal in the frontal white matter, putamen, globus pallidus, dentate nucleus and the head of the caudate nucleus (Fig. 5A,B). Contrast enhancement was present in the dentate nucleus (Fig. 5C), frontal white matter (Fig. 5D, E), head of the caudate nucleus (Fig. 5D), putamen (Fig. 5D) and the hypothalamus (Fig. 5E). Follow-up MRI at the age of 12 months showed extensive cystic degeneration of the frontal white matter (Fig. 5F).
Figure 5.

MRI for Patient 3. The MRI at 6 months (A–E) shows an abnormally high signal in the frontal white matter, the head of the caudate nucleus, the putamen, globus pallidus (B) and dentate nucleus (A). The frontal lobes are atrophic (A). Contrast enhancement of the dentate nucleus (C), frontal white matter (D, E), head of the caudate nucleus (D), the putamen (D), and the hypothalamus (E) is shown. Follow-up MRI at the age of 12 months (F) shows extensive cystic degeneration of the frontal white matter.
Patient 3 DNA sequencing
Gene testing for AxD was performed by GeneDx following the suggestive findings of the first set of MRIs. A c.1249delG was observed, which predicted a frameshift commencing with a change of Asp417 to methionine followed by an additional 13 altered amino acids before encountering a stop codon (p.Asp417Metfs*15) (Fig. 6). The resultant GFAP protein is just two amino acids shorter than the wild type, but the last sixteen amino acids are abnormal. We subsequently tested both parents and the child, confirming its presence in the child and finding it absent in both parents.
Figure 6.

Mutation present in Patient 3. The predicted coding change resulting from the c.1249delG mutation is shown. The G that is deleted is shown in the black box in the wild type sequence.
Discussion
In this study we identified two new classes of GFAP mutations associated with AxD; aberrant splicing and an insertion/deletion that produces a frameshift. The patient with the intronic splicing mutation had adult onset of clinical signs, and autopsy revealed a distribution of Rosenthal fibers that are diagnostic of this disease. No coding mutation was found for this patient, but a heterozygous C>G change was present just before the consensus AG splice acceptor sequence at the end of intron 3. The IVS-3 position is a C in 72% of primate introns, a T in 23%, an A in 4% and a G in less than 1% (Shapiro and Senapathy, 1987) (in the human GFAP gene, 7 of the introns have a C, and the other has a T). The rarity of a G at the IVS-3 position led us to suspect that it may reduce splicing efficiency, and this possibility was supported by reports that other IVS-3C>G mutations are involved with defective splicing and produce human diseases, including congenital goiter (Ieiri et al., 1991), tuberous sclerosis (Mayer et al., 2000), mucopolysaccharidosis type I (Teng et al., 2000) and Glanzmann thrombasthenia (Rosenberg et al., 2005). Semiquantitative RT-PCR revealed that an exon 4-skipped transcript was present in patient brain RNA, but at a level about 12-fold lower than the wild type. Nevertheless, a ratio of wild type to mutant GFAP of 40:1 was sufficient to disturb the GFAP assembly pattern in transfected cells (Fig. 3). It has previously been shown for other intermediate filaments that relatively low levels of truncated protein, even as little as one percent, can disrupt filament polymerization and lead to formation of aberrant aggregates (Christian et al., 1990; Wong and Cleveland, 1990; Chin et al., 1991; Cary and Klymkowsky, 1995; Lin and Szaro, 1996).
The approximately 12:1 ratio for wild type to mutant transcripts observed for Patient 1 differs from the 1:1 ratio of the alleles. The simplest explanation for this discrepancy is that the GFAP c.(619-3C>G) mutation only occasionally causes missplicing. In several of the human disease cases in which the effect on an IVS-3C/G mutation has been evaluated, essentially complete exon skipping has been observed (Ieiri et al., 1991; Mayer et al., 2000; Teng et al., 2000); however, the presence of an IVS-3G in some wild type sequences suggests the effect of this nucleotide is context dependent. Alternatively, the relatively low level of mutant GFAP transcript could result from it being less stable than the wild type.
Whenever possible, the parents of patients with novel GFAP mutations are tested to determine if the mutation arose de novo, as such a finding provides powerful evidence for its disease causation. The mother of this patient tested negative for the DNA change, but her father was deceased and no DNA sample was available for testing. However, the c.(619-3C>G) mutation has not been reported in any of over 100 AxD patient and control DNAs that have been sequenced (Quinlan et al., 2007; Brenner et al., 2009), is not recorded in the NCBI dbSNP database, and was not found in 525 ethnically matched controls.
Skipping of exon 4 results in an in-frame deletion of 54 of the 432 GFAP amino acids. This deletion occurs within the central rod domain, removing the last 7 amino acids of coil 1B, all of coil 2A and the first 4 amino acids of coil 2B. Expression of this mutant protein in SW13vim− cells results in formation of unusual needle-like aggregates rather than the filamentous structures formed by the wild type (Fig. 2). Interestingly, this rare needle-like pattern has previously been reported only for the GFAP E210K mutation (Li et al., 2005), which resides in the part of coil 1B contributed by exon 4, suggesting that perturbation of the coil 1B structure leads to their formation.
That the deletion is in frame and produces a protein close to normal size is consistent with the hypothesis that AxD results from a toxic gain of function rather than a loss of function (Li et al., 2002). Multiple different point mutations have been identified in the region covered by the deletion, with several producing a severe infantile onset form of AxD. The relatively mild (albeit fatal) adult form of AxD of our patient could reflect the nature of the deletion, or more likely, the limited amount of mutant transcript present.
Patient 2 provides a striking exception to the general rule that most de novo AxD mutations arise in the parental germ cells, particularly that of the father (Li et al., 2006). For this patient the GFAP mutation is mosaic, and thus clearly arose in the embryo rather than in a parent. Moreover, the highly regional nature of its presence indicates that the mutation arose relatively late in development; it was detected in buccal DNA (and is presumably present in brain to produce symptoms), but not in blood DNA or DNA isolated from nail clippings analyzed separately from each hand and foot. Diagnosis of AxD by brain biopsy has been largely supplanted by genetic testing given that approximately 95% of cases display GFAP mutations. The findings for Patient 2 suggest that buccal DNA may be preferable to blood DNA for such tests. If buccal DNA analysis is negative, brain biopsy may still have a role when a patient is suspected of having AxD and other possibilities have been eliminated. In such rare instances it would be of interest to analyze the biopsy sample for GFAP mutations as well as the presence of Rosenthal fibers, although mosaicism could still be a confound.
The mutation itself is highly unusual. The deletion of 8 bp and the insertion of 3 bp is the greatest change in GFAP gene structure so far reported. It is also the most C-terminal of any mutation yet described, changing only the last 2 amino acids and adding an additional 11 amino acids before encountering a stop codon (Fig. 4). Although this mutation technically produces a frameshift, its effect is essentially the same as an insertion since it occurs at the very end of the protein. The de novo nature of the mutation in our patient is strong evidence that it is disease-causing, and we have also found that the mutant GFAP fails to polymerize normally in transiently transfected cells, and is dominant to the wild type. These polymerization properties of the extended, mutant GFAP add further caution to the use of C-terminal tags of intermediate filament proteins (Haubold et al., 2003).
Patient 2 is also remarkable for displaying essentially normal MRI features. Five MRI characteristics for AxD have been described, with the presence of four being considered diagnostic (van der Knaap et al., 2001). Briefly, these are 1) white matter abnormalities with a frontal preponderance, 2) a periventricular rim of decreased T2 signal and elevated T1 signal, 3) abnormalities of the basal ganglia and thalami, 4) brain stem abnormalities and 5) contrast enhancement. Exceptions to these findings are common among later onset AxD patients, but are nearly always met for infantile patients. Patient 2 presented in infancy, yet meets none of these criteria. Possible explanations include the nature of the mutation or its limited presence in the CNS due to mosaicism. Another is that the patient has a disorder other than AxD, although AxD seems the simplest explanation given the presence of a de novo GFAP mutation, and that the same mutation was found by GeneDx for another suspected AxD patient (no clinical information is available for that patient; Sherri Bale, GeneDx, personal communication).
The c.1249delG mutation found for our third patient was recently reported in a Japanese patient (Murakami et al., 2008), also presenting in infancy, but was included here for several reasons: the parents in this prior case were not tested, leaving it unclear whether the predicted C-terminal frameshift was actually disease causing; the MRI findings for our patient are striking, and the clinical progression for our patient differed significantly from that of this prior case.
The finding that neither parent has the c.1249delG coding change found in their child is strong evidence that this mutation causes AxD. Genetic testing of this patient for AxD was prompted by the MRI findings made at 6 months of age, although they did not fully meet the usual diagnostic criteria summarized above. Neither criterion 2 nor 4 were met at 6 months, although criterion 4 was met in the MRI taken shortly before his death near 1 year of age. The MRI findings for this patient are also remarkable in that the signal enhancement of the frontal white matter, hypothalamus and dentate nucleus is unusually extensive. The atrophy of the frontal white matter is in contrast with the swelling of the abnormal white matter usually seen. The atrophy and cystic degeneration of the frontal white matter is similar to that reported for the Murakami et al. patient, but the contrast enhancement of our patient is much more extensive.
The clinical courses of our patient and Murakami’s patient were quite different. The Japanese child had no reported feeding problems or GE reflux despite being <25th percentile in weight, and had a head circumference that was >50th percentile without evidence of a decelerating pattern like the infant reported here. Neither child had megalencephaly, which is often observed in infantile cases of AxD. Unlike our child, the Japanese child made slow developmental progress and had acquired a few meaningful words. He was regarded by his physicians as exhibiting relatively mild clinical involvement (N. Murakami, personal communication). It appears that the findings on cranial imaging were also different; while the forebrain and deep gray matter findings were similar in both children, involvement of the pons and medulla was not described in the Japanese child. This may explain the significant differences in GE reflux and the terminal breathing pattern in our child.
In summary, in addition to revealing novel classes of GFAP mutations that can produce AxD, the findings reported here raise the possibility that mutations might be missed because they reside in non-coding regions of the gene, or because the patient might be mosaic for a mutation. They also suggest that buccal DNA may be preferable to blood DNA for clinical testing, and recommend including intronic splice site regions in the sequence analysis. The findings also show that even alterations at the extreme C-terminal end of GFAP can have fatal consequences.
Supplementary Material
Acknowledgments
Contract grant sponsors: Funding was provided by NINDS P01NS42803; D.F. was supported by NIH training grant 2T32GM008111, and technical assistance was provided by NIH IDDRC grant P30HD38985.
We are grateful to the patients and their families for agreeing to participate in this study. We thank L. Messiaen and the staff of the UAB Medical Genomics Lab for verifying the identity of the DNA samples for Patient 2 and Sherri Bale of GeneDx for bringing this patient to our attention. None of the authors has any financial interests in the information in this report.
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Brenner M, Goldman JE, Quinlan RA, Messing A. Alexander disease: a genetic disorder of astrocytes. In: Parpura V, Haydon P, editors. Astrocytes in (patho)physiology of the nervous system. New York: Springer; 2009. pp. 591–648. [Google Scholar]
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nature Genetics. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- Cary RB, Klymkowsky MW. Disruption of intermediate filament organization leads to structural defects at the intersomite junction in Xenopus myotomal muscle. Development. 1995;121:1041–1052. doi: 10.1242/dev.121.4.1041. [DOI] [PubMed] [Google Scholar]
- Chin SS, Macioce P, Liem RK. Effects of truncated neurofilament proteins on the endogenous intermediate filaments in transfected fibroblasts. J Cell Sci. 1991;99 (Pt 2):335–350. doi: 10.1242/jcs.99.2.335. [DOI] [PubMed] [Google Scholar]
- Christian JL, Edelstein NG, Moon RT. Overexpression of wild-type and dominant negative mutant vimentin subunits in developing Xenopus embryos. New Biol. 1990;2:700–711. [PubMed] [Google Scholar]
- Cline RE, Laurent NM, Foran DR. The fingernails of Mary Sullivan: developing reliable methods for selectively isolating endogenous and exogenous DNA from evidence. J Forensic Sci. 2003;48:328–333. [PubMed] [Google Scholar]
- Flint D, Brenner M. Alexander disease. In: Raymond GV, Eichler F, Fatemi A, Naidu S, editors. Leukodystrophies. London: Mac Keith Press; 2011. pp. 106–129. [Google Scholar]
- Haubold K, Herrmann H, Langer SJ, Evans RM, Leinwand LA, Klymkowsky MW. Acute effects of desmin mutations on cytoskeletal and cellular integrity in cardiac myocytes. Cell Motil Cytoskeleton. 2003;54:105–121. doi: 10.1002/cm.10090. [DOI] [PubMed] [Google Scholar]
- Hedberg KK, Chen LB. Absence of intermediate filaments in a human adrenal cortex carcinoma-derived cell line. Exp Cell Res. 1986;163:509–517. doi: 10.1016/0014-4827(86)90081-9. [DOI] [PubMed] [Google Scholar]
- Ieiri T, Cochaux P, Targovnik HM, Suzuki M, Shimoda S, Perret J, Vassart G. A 3′ splice site mutation in the thyroglobulin gene responsible for congenital goiter with hypothyroidism. J Clin Invest. 1991;88:1901–1905. doi: 10.1172/JCI115513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Messing A, Goldman JE, Brenner M. GFAP Mutations in Alexander Disease. Int J Dev Neurosci. 2002;20:259–268. doi: 10.1016/s0736-5748(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Li R, Johnson AB, Salomons GS, Goldman JE, Naidu S, Quinlan R, Cree B, Ruyle SZ, Banwell B, D’Hooghe M, Siebert JR, Rolf CM, Cox H, Reddy A, Gutiérrez-Solana LG, Collins A, Weller RO, Jakobs C, Messing A, Van der Knaap MS, Brenner M. GFAP mutations in infantile, juvenile and adult forms of Alexander disease. Annals Neurol. 2005;57:310–326. doi: 10.1002/ana.20406. [DOI] [PubMed] [Google Scholar]
- Li R, Johnson AB, Salomons GS, van der Knaap MS, Rodriguez D, Boespflug-Tanguy O, Gorospe JR, Goldman JE, Messing A, Brenner M. Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum Genet. 2006;119:137–144. doi: 10.1007/s00439-005-0116-7. [DOI] [PubMed] [Google Scholar]
- Lin W, Szaro BG. Effects of intermediate filament disruption on the early development of the peripheral nervous system of Xenopus laevis. Dev Biol. 1996;179:197–211. doi: 10.1006/dbio.1996.0251. [DOI] [PubMed] [Google Scholar]
- Mayer K, Ballhausen W, Leistner W, Rott H. Three novel types of splicing aberrations in the tuberous sclerosis TSC2 gene caused by mutations apart from splice consensus sequences. Biochim Biophys Acta. 2000;1502:495–507. doi: 10.1016/s0925-4439(00)00072-7. [DOI] [PubMed] [Google Scholar]
- Murakami N, Tsuchiya T, Kanazawa N, Tsujino S, Nagai T. Novel deletion mutation in GFAP gene in an infantile form of Alexander disease. Pediatr Neurol. 2008;38:50–52. doi: 10.1016/j.pediatrneurol.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Prust MJ, Wang J, Morizono H, Messing A, Brenner M, Gordon ES, Hartka T, Sokohl A, Schiffmann R, Gordish-Dressman H, Albin R, Amartino H, Brockman K, Dinopoulos A, Dotti MT, Fain D, Fernandez R, Ferreira J, Fleming J, Gill D, Griebel M, Heilstedt H, Kaplan P, Lewis D, Nakagawa M, Pederson R, Reddy A, Sawaishi Y, Schneider M, Sherr E, Takiyama Y, Wakabayashi K, Gorospe R, Vanderver A. GFAP mutations, age of onset and clinical subtypes in Alexander disease. Neurology. 2011 doi: 10.1212/WNL.0b013e3182309f72. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan R, Brenner M, Goldman JE, Messing A. GFAP mutations in Alexander disease. Exp Cell Res. 2007;313:2077–2087. doi: 10.1016/j.yexcr.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N, Hauschner H, Peretz H, Mor-Cohen R, Landau M, Shenkman B, Kenet G, Coller BS, Awidi AA, Seligsohn U. A 13-bp deletion in alpha(IIb) gene is a founder mutation that predominates in Palestinian-Arab patients with Glanzmann thrombasthenia. J Thromb Haemost. 2005;3:2764–2772. doi: 10.1111/j.1538-7836.2005.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–7174. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P, Jaenisch R. Retroviruses as probes for mammalian development: allocation of cells to the somatic and germ cell lineages. Cell. 1986;46:19–29. doi: 10.1016/0092-8674(86)90856-1. [DOI] [PubMed] [Google Scholar]
- Teng YN, Wang TR, Hwu WL, Lin SP, Lee-Chen GJ. Identification and characterization of -3c-g acceptor splice site mutation in human alpha-L-iduronidase associated with mucopolysaccharidosis type IH/S. Clin Genet. 2000;57:131–136. doi: 10.1034/j.1399-0004.2000.570207.x. [DOI] [PubMed] [Google Scholar]
- van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Begeer JC, van Coster R, Barth PG, Thomas NH, Valk J, Powers JM. Alexander disease: diagnosis with mr imaging. Am J Neuroradiol. 2001;22:541–552. [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Cleveland DW. Characterization of dominant and recessive assembly-defective mutations in mouse neurofilament NF-M. J Cell Biol. 1990;111:1987–2003. doi: 10.1083/jcb.111.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.