

Abstract
Adiponectin, an adipose derived hormone with pleiotropic functions, binds to several proteins, including T-cadherin. We have previously reported that adiponectin deficient (Adipo−/−) mice have increased IL-17A-dependent neutrophil accumulation in their lungs after subacute exposure to ozone (0.3 ppm for 72 hrs). The purpose of this study was to determine whether this anti-inflammatory effect of adiponectin required adiponectin binding to T-cadherin. Wildtype, Adipo−/−, T-cadherin deficient (T-cad−/−), and bideficient (Adipo−/−/T-cad−/−) mice were exposed to subacute ozone or air. Compared to wildtype mice, ozone-induced increases in pulmonary IL-17A mRNA expression were augmented in T-cad−/− and Adipo−/− mice. Compared to T-cad−/− mice, there was no further increase in IL-17A in Adipo−/−/T-cad−/− mice, indicating that adiponectin binding to T-cadherin is required for suppression of ozone-induced IL-17A expression. Similar results were obtained for pulmonary mRNA expression of saa3, an acute phase protein capable of inducing IL-17A expression. Comparison of lung histological sections across genotypes also indicated that adiponectin attenuation of ozone-induced inflammatory lesions at bronchiolar branch points required T-cadherin. BAL neutrophils and G-CSF were augmented in T-cad−/− mice and further augmented in Adipo−/−/T-cad−/− mice. Taken together with previous observations indicating that augmentation of these moieties in ozone exposed Adipo−/− mice is partially IL-17A dependent, the results indicate that effects of T-cadherin deficiency on BAL neutrophils and G-CSF are likely secondary to changes in IL-17A, but that adiponectin also acts via T-cadherin independent pathways. Our results indicate that T-cadherin is required for the ability of adiponectin to suppress some but not all aspects of ozone-induced pulmonary inflammation.
Introduction
Ozone (O3) is an environmental pollutant generated by chemical reactions of automobile emissions (NO and hydrocarbons) with sunlight. O3 acts as oxidizing agent on cell membranes and on proteins and lipids in the lung and airway lining fluid, leading to epithelial injury and an inflammatory response that includes induction of acute phase cytokines and chemokines, and neutrophil influx [1], [2], [3].
Adiponectin, an adipose-derived hormone that decreases in obesity [4], has important anti-inflammatory effects. For example, adiponectin treatment decreases endotoxin-induced pro-inflammatory cytokine expression and augments anti-inflammatory IL-10 expression in monocytes and macrophages [5], [6]. Exogenous administration of adiponectin also decreases allergic airways inflammation in mice [7]. In addition, we have previously reported that compared to wildtype (WT) mice, adiponectin deficient (Adipo−/−) mice exposed to subacute O3 (0.3 ppm for 24 to 72 h) have increased neutrophilic inflammation, and increased pulmonary expression of certain cytokines and chemokines, including IL-17A and G-CSF [8].
Several adiponectin binding proteins have been cloned including AdipoR1, AdipoR2, and T-cadherin (T-cad) [9], [10], all of which are expressed in the lungs [11], [12]. T-cad (cdh13 or H-cadherin) is a 95 kd glycoprotein which differs from other cadherin proteins by lacking both transmembrane and cytoplasmatic domains. Instead, T-cadherin is anchored, mainly on the apical surface of cells [13], via a glycosylphosphatidylinositol (GPI) linkage [14]. Importantly, T-cadherin primarily binds the hexameric and high molecular weight isoforms of adiponectin [10]. These are also the isoforms that dominate in the lung lining fluid [15]. In the heart, T-cadherin appears to mediate the beneficial effects of adiponectin. Following pressure overload, mice deficient in Tcadherin (T-cad−/− mice), exhibit increased cardiac hypertrophy compared to WT mice [16], similar to Adipo−/− mice. Similarly, the size of infarctions in hearts of mice subjected to ischemia-reperfusion is greater in T-cad−/− than WT mice [16]. Furthermore, the ameliorative effects of adiponectin in these models are not observed in T-cad−/− mice [16].
The purpose of this study was to examine the hypothesis that T-cadherin is required for the anti-inflammatory effects of adiponectin that limit the pulmonary inflammation induced by subacute O3. To address this hypothesis, we assessed pulmonary inflammation in T-cad−/− mice and their WT controls exposed to either air or O3 (0.3 ppm) for 72 hours. For comparison we also examined Adipo−/− mice and their WT controls. T-cadherin functions not only as an adiponectin binding protein, but also as a cell-cell adhesion molecule that can impact cell polarization, migration, adhesion, and survival [14]. Hence, effects of T-cadherin deficiency on responses to O3 may be the result of the cell-adhesion rather than the adiponectin-binding properties of T-cadherin. To address this issue, we also examined mice deficient in both T-cadherin and adiponectin (Adipo−/−/T-cad−/− mice). We reasoned if effects of T-cadherin deficiency were a reflection of adipnectin binding to T-cadherin, then we would not see any difference between T-cadherin deficient mice and mice deficient in both adiponectin and T-cadherin.
Methods
Animals
This study was approved by the Harvard Medical Area Standing Committee on Animals under protocol number 03078 and carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals from the National Institute of Health. All efforts were made to minimize suffering. Adipo−/− and T-cad−/− mice were obtained from Dr. Matsuzaka (Osaka, Japan). T-cad−/− mice [17] and Adipo−/− were bred together to obtain Adipo−/−/T-cad−/− mice as previously described [18]. Others have reported small but potentially significant genetic differences in C57BL/6 mice from different vendors [19]. Differences in the microbiome between C57BL/6 mice from Jackson Laboratories and Taconic Farms have also been reported [20]. Hence, as controls for the Adipo−/− mice, we used C57BL/6 from Jackson Laboratories (WT-jax) because this was the genetic background for the Adipo−/− mice. As controls for the T-cad−/− and Adipo−/−/T-cad−/− mice, we used C57BL/6 mice from Taconic Farms (WT-tac) because this was the background for the T-cad −/− mice. Note that we generated the Adipo−/−/T-cad−/− mice by backcrossing offspring from Adipo−/−×T-cad−/− matings onto T-cad−/− mice [18].
Protocol
Age and gender matched Adipo−/−, T-cad−/−, Adipo−/−T-cad−/−, and control mice were exposed to either air or O3 (0.3 ppm, 72 hours) as previously described [8]. Our experience is that neutrophil recruitment plateaus after 48 h of exposure, but significant changes in BAL macrophages do not occur in wildtype mice until 72 h of exposure [8]. Our previous data also indicate that statistically significant changes in BAL macrophages do not occur in wildtype mice until 72 h of exposure [8]. Immediately after the exposure, mice were euthanized by i.p. overdose of sodium pentobarbital. Two cohorts of mice were used. In the first, blood was drawn, the trachea was cannulated to perform bronchoalveolar lavage (BAL), and lungs were stored at −80°C for extraction of total RNA. In the second cohort, lungs were fixed for histological assessment of O3-induced pulmonary lesions [21].
Ozone exposure
Mice were exposed to either O3 (0.3 ppm) or ambient air for 72 hours as previously described [8]. Briefly, cages without the microisolator cover were placed in a steel and plexiglass chamber, and supplied with a mixture of O3 and air. O3 was produced by passing medical grade oxygen through a high voltage ozonizer and bled into the chamber. O3 concentration in the chamber was controlled by regulating the amount of ambient air flowing into the chamber. Mice were supplied with normal chow and water ad libitum.
Bronchoalveolar lavage and serum
BAL was performed using two instillations of 1 mL of cold PBS. BAL samples were centrifuged, supernatants were assessed for inflammatory cytokines, and BAL cells were resuspended and counted using a hemocytometer. Cytospin was performed for differential cell analysis. Blood was drawn by cardiac puncture to obtain serum.
Cytokines and Chemokines
A panel of 32 cytokines, chemokines and growth factors (eotaxin, G-CSF, GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-15, IL-17, IP-10, KC, LIF, LIX, MCP-1, M-CSF, MIG, MIP-1α, MIP-1β, MIP-2, RANTES, TNF-α, and VEGF) were quantified in BAL by a multiplex assay (Eve Technologies, Alberta, Canada) as previously described [22] and a commercial ELISA was used for quantification of sTNFR1 and adiponectin (R&D Systems, MN).
RNA generation and qRT-PCR
Total lung RNA was extracted using the protocol provided in the RNAeasy kit (Qiagen, MD), and quantified by Nanodrop (ThermoScientific, NJ). cDNA was synthesized from RNA by using a commercial kit [8]. Real time PCR (RT-PCR) was used to assess the mRNA expression of IL-17A, serum amyloid A3 (saa3), and Ki67 (a marker of cell proliferation), by the SYBR green method. Primer sequences are provided in Table 1. Gene expression was normalized to 18S (internal control) followed by analysis by the ΔΔCT method.
Table 1. Primers sequence used for qRT-PCR.
| Gene | Sense | Anti-sense |
| 18S | 5′-GTAACCCGTTGAACCCCATT-3′ | 5′-CCATCCAATCGGTAGTAGCG-3′ |
| IL-17A | 5′-CCAGGGAGAGCTTCATCTGT-3′ | 5′-AGGAAGTCCTTGGCCTCAGT-3′ |
| ki67 | 5′-AGGGTAACTCGTGGAACCAA-3′ | 5′-GGAGGTGAAAACCACACTGG-3′ |
| saa3 | 5′-CCTGGGCTGCTAAAGTCATC-3′ | 5′-CACTCATTGGCAAACTGGTC-3′ |
Histology
Mice were euthanized and lungs inflated to 20 cmH2O with 4% PBS buffered paraformaldehyde (pH = 7.4) overnight. Lungs were washed with PBS and transferred to 70% ethanol, and dehydrated. The left lung was sectioned, embedded in paraffin, and stained with hematoxylin and eosin. The severity of lesions located at terminal bronchioles was assessed by scoring the number of cellular layers below the epithelium as follows: 0 for no lesions, 1: 1–2 cellular layers, 2: for 3 cellular layers, 3: for 4 cellular layers, and 4: for 5 cells or more layers. Total score for each mouse was computed by averaging the scores of all terminal bronchioles present on all sections of left lung.
Statistical Analysis
The significance of differences between groups was assessed using Factorial ANOVA combined with LSD Fisher as post-hoc analysis (Statistica Software, Statsoft, OK). Data that were not normally distributed were log transformed before statistical analysis. Means and standard errors from log values were retrocalculated with appropriate error propagations. P<0.05 (two tail) was considered significant. Because the controls for the Adipo−/− and the T-cad−/− and Adipo−/−/T-cad−/− mice were different, the data from these mice were analyzed separately. All values are expressed as mean±standard mean of error.
Results
BAL and serum adiponectin
Serum adiponectin was substantially higher in T-cad−/− than WT mice (Fig. 1A), consistent with previous observations [15], [17], [18]: adiponectin bound to T-cadherin on the endothelium serves as a repository for adiponectin that is delivered back to the circulation in T-cad−/− mice [16], [17], [23]. There was no effect of O3 exposure on serum adiponectin (Fig. 1A). In contrast, O3 exposure caused a marked increase in BAL adiponectin in T-cad−/− mice (Fig. 1B), likely as a result of increased transit from the blood into the lung consequent to O3-induced increases in the permeability of the alveolar/capillary barrier, as previously discussed [8]. BAL adiponectin was slightly lower in air exposed T-cad−/− versus WT mice and slightly higher in O3 exposed T-cad−/− versus WT mice although these changes were not significant (Fig. 1B).
Figure 1. Serum and BAL adiponectin.

Total adiponectin was measured by ELISA in serum (A) and bronchoalveolar lavage (BAL) fluid (B) of wildtype (Taconics) and T-cadherin deficient (T-cad−/−) mice exposed to air and ozone (O3, 0.3 ppm) for 72 h. * p<0.05 versus genotype matched air exposed mice; #p0.05 versus wildtype mice with the same exposure. Results of adiponectin in BAL are expressed as mean ± SEM of data from 5 mice exposed to air per group and 7 ozone exposed mice per group. The number of mice used for measurements of serum adiponectin was 4 mice per group.
Effect of T-cadherin deficiency on O3-induced pulmonary inflammation
Compared to air, factorial ANOVA demonstrated that O3 increased BAL neutrophils, macrophages, and protein (Fig. 2), consistent with previous reports by ourselves and others using this type of O3 exposure [8], [24]. BAL neutrophils and protein were higher in O3-exposed Adipo−/− versus WT mice, consistent with our previous observations [8]. Note that BAL neutrophils are also significant greater in Adipo−/− versus WT mice after 24 or 48 h of exposure [8]. We also observed significantly more BAL neutrophils in O3-exposed T-cad−/− mice versus their corresponding WT controls. BAL neutrophils were also higher in O3-exposed Adipo−/−/T-cad−/− than T-cad−/− mice. No significant change in BAL protein was observed in T-cad−/− mice exposed to O3 compared to WT mice under the same exposure (p = 0.06), despite a trend towards an increase in T-cad−/− mice. Interestingly, BAL protein was significantly greater in O3-exposed Adipo−/−/T-cad−/− versus WT mice (p = 0.005). There was no significant effect of genotype on ozone induced changes in BAL macrophages (Fig. 2B). Note that it is not possible to directly compare Adipo−/−/T-cad−/− and Adipo−/− mice due to differences in the background strain (see methods).
Figure 2. Lung inflammation and injury.
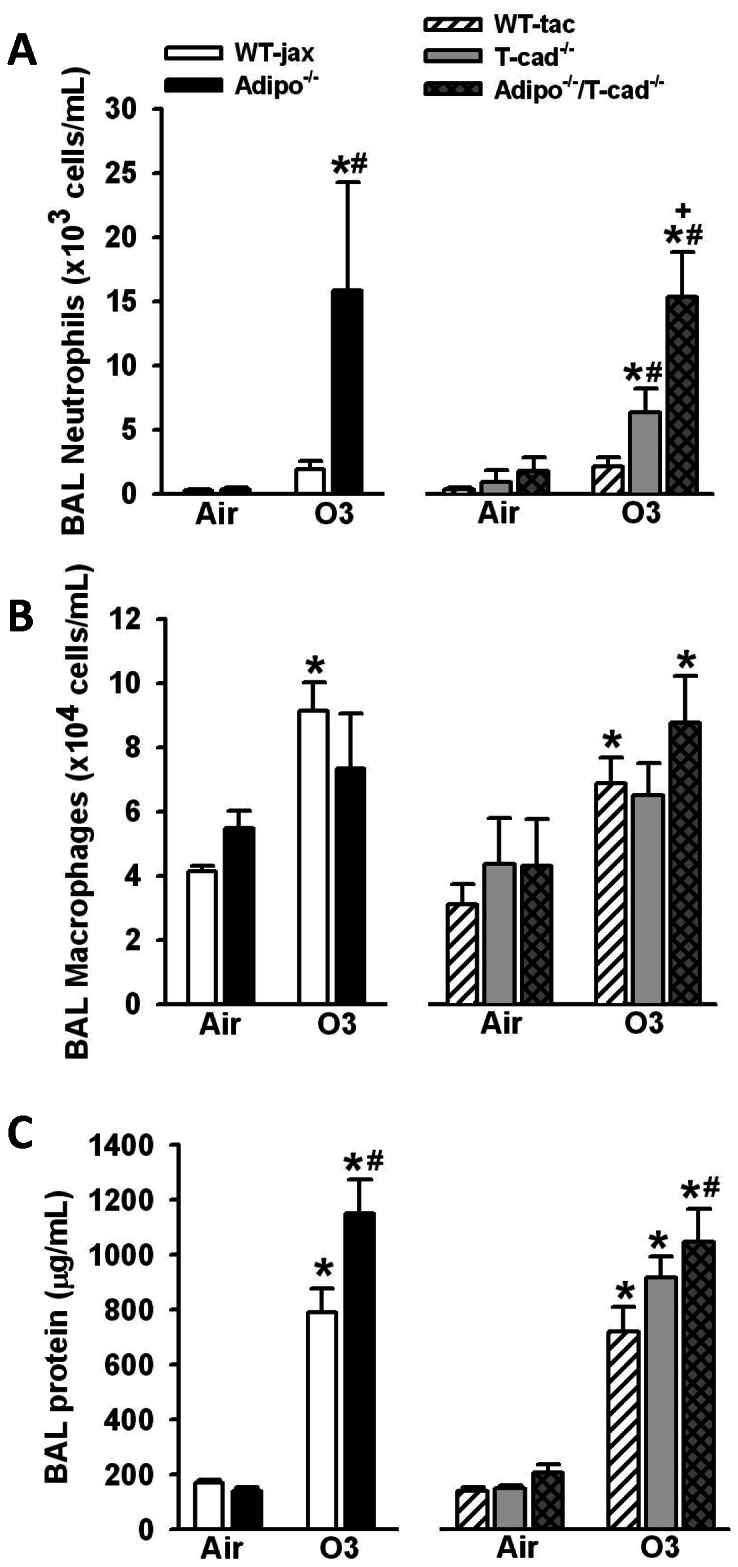
BAL neutrophils (A), macrophages (B) and protein (C) in mice exposed to room air or O3 (0.3 ppm) for 72 hours. *p<0.05 versus air exposed mice of the same genotype, #p<0.05 versus wildtype mice with the same exposure, + p<0.05 versus T-cadherin deficient mice with the same exposure. Results are mean ± SEM of data from 4–7 air exposed mice and 6–10 ozone exposed mice.
To further evaluate the impact of T-cadherin deficiency on O3 induced inflammation, we performed a multiplex assay of cytokines and chemokines. Of the factors assayed by multiplex, factorial ANOVA indicated a significant effect of O3 exposure in both cohorts of mice (WT/Adipo−/− and WT/T-cad−/−/Adipo−/−/Tcad−/−) for G-CSF, IL-5, IL-6, LIF, KC, and eotaxin. BAL IL-6 and G-CSF were significantly higher in Adipo−/− versus WT mice exposed to O3 (Fig. 3A, B), consistent with our previous observations [8]. We also observed significantly higher BAL LIF and IL-5 in O3-exposed Adipo−/− versus WT mice (Fig. 3C, D). BAL G-CSF was also significantly higher in T-cad−/− versus WT mice exposed to O3, and higher still in Adipo−/−/T-cad−/− versus T-cad−/− mice (Fig. 3B). Surprisingly, O3-induced changes in BAL IL-5 were significantly reduced by T-cadherin deficiency, and this change was reversed by combined adiponectin and T-cadherin deficiency. Neither BAL IL-6 nor LIF was significantly affected by T-cadherin deficiency, although there was significantly greater BAL LIF in O3-exposed Adipo−/−/T-cad−/− versus WT mice and a similar trend for IL-6. There was no genotype effect for either eotaxin or KC (data not shown). IL-17A was below the limit of detection of the Bioplex assay, but IL-17A mRNA expression was induced by subacute O3 exposure. O3-induced increases in IL-17A were significantly greater in Adipo−/− than WT mice (Fig. 3E), consistent with our previous observations [8]. O3-induced increases in IL-17A were also significantly greater in T-cad−/− versus WT mice, and there was no further increase in Adipo−/−/T-cad−/− versus T-cad−/− mice.
Figure 3. Cytokine and chemokine expression.

BAL IL-6 (A), G-CSF (B), LIF (C), IL-5 (D), pulmonary IL-17A mRNA expression (E), and soluble TNFR1 (sTNFR1) (F) in mice exposed to room air or O3 (0.3 ppm) for 72 hours. *p<0.05 versus air exposed mice of the same genotype, #p<0.05 versus wildtype mice with the same exposure, + p<0.05 versus T-cadherin deficient mice with the same exposure. Results are mean ± SEM of data from 3–5 air exposed mice and 3–7 ozone exposed mice for IL-6, G-CSF, LIF, and IL-5; 4–9 air exposed and 4–9 for ozone exposed mice for IL-17A mRNA; and 4–7 air exposed and 6–10 ozone exposed mice for sTNFR1;.
We also measured BAL concentrations of sTNFR1 (soluble TNFα receptor 1), the extracellular domain of TNFR1 (Fig. 3F). sTNFR1 is cleaved from cell surfaces by the enzyme TACE (TNFα converting enzyme). TACE activity is increased by conditions associated with oxidative stress [25]. Compared to air, O3 exposure resulted in a marked increase in BAL sTNFR1 in all mouse genotypes used in this study (Fig. 3F). BAL sTNFR1 was significantly greater in O3-exposed Adipo−/− versus WT mice, consistent with previous observations [8]. There was no significant difference in BAL sTNFR1 in O3-exposed T-cad−/− versus WT mice. However, BAL sTNFR1 was significantly higher in O3-exposed Adipo−/−/T-cad−/− mice than in either WT or T-cad−/− mice. The results are consistent with the hypothesis that adiponectin deficiency results in increased oxidative stress leading to greater TACE activation, and that T-cadherin is not involved in this effect of adiponectin.
Histology
In O3-exposed mice, histopathologic examination revealed inflammatory lesions localized at bronchiolar branch points. These lesions were characterized by focal interstitial expansion by mononuclear cells and reactive hyperplasia of epithelial cells (see Fig. 4B arrow, and a higher magnification of a different airway in Fig. 4C). The severity of the lesions varied, and was quantified as described in Methods. Compared to air, O3 exposure resulted in a significant increase in terminal bronchiolar lesions (Fig. 4D). O3-induced lesions were significantly greater in Adipo−/− versus WT mice. O3-induced lesions were also significantly greater in T-cad−/− versus WT mice, but there was no further augmentation in Adipo−/−/T-cad−/− versus T-cad−/− mice (Fig. 4D).
Figure 4. Terminal bronchiolar lesions.

H&E stained histological sections of lungs of wildtype mice exposed to air (A) or ozone (B) showing bronchiolar/central acinar lesions in ozone exposed mice (magnification 100×). Arrow in red is pointing a terminal bronchiole with lesion. (C) A detail of these lesions in an ozone exposed mouse (magnification 400×). (D) An average lesion score was calculated for each mouse as described in Methods and these were averaged across mice from each genotype. * p<0.05 versus air exposed mice of the same genotype, # p<0.05 versus wildtype mice with the same exposure. Results are mean ± SEM of data from 6–12 air exposed mice and 8–16 ozone exposed mice.
qRT-PCR
To further evaluate the role of T-cadherin in adiponectin dependent effects on O3-induced inflammation, we examined the mRNA expression of Ki67 and saa3. We chose to examine Ki67 because it is a well-established marker of cell proliferation [26]. Others have reported BrdU labeling within the terminal bronchiolar epithelium after O3 exposure [27] consistent with epithelial repair following injury, and the lesions we observed in mice exposed to O3 included epithelial cell hyperplasia (Fig. 4). We chose to examine saa3 because others have reported that it is increased by O3 to a greater extent in lungs of other types of O3-sensitive versus O3-resistant mice [28], [29] qRT-PCR data indicated a robust induction of Ki67 and saa3 by O3 (Fig. 5). O3-induced increases in Ki67 were not affected by either adiponectin or T-cadherin deficiency (Fig. 5A). O3-induced expression of saa3 was higher in Adipo−/− versus WT mice (Fig. 5B). O3-induced expression of saa3 was also significantly greater in T-cad−/− than WT mice, and there was no further increase in Adipo−/−/T-cad−/− versus T-cad−/− mice.
Figure 5. Real time PCR.

Pulmonary mRNA expression of ki67 (A) and saa3 (B) in lungs of mice exposed to room air or O3 (0.3 ppm ) for 72 h. Expression was normalized to 18 s and expressed relative to wildtype mice exposed to O3. *p<0.05 versus air exposed mice of the same genotype, #p<0.05 versus wildtype mice with the same exposure. Results are mean ± SEM of data from 4–7 air exposed mice and 6–10 ozone exposed mice.
Discussion
Adiponectin deficiency augments the pulmonary inflammation induced by subacute O3 exposure in mice [8], indicating an anti-inflammatory role for adiponectin. Results from the current study show that T-cadherin, an adiponectin binding protein, is required for aspects of this anti-inflammatory effect of adiponectin.
We have previously reported that BAL concentrations of adiponectin increase following subacute O3 exposure and that O3-induced neutrophilic influx into the lungs is augmented in mice deficient in adiponectin [8]. These results indicate an anti-inflammatory role for adiponectin during subacute ozone exposure. We have also demonstrated that the augmented neutrophilia observed in Adipo−/− mice is the result of increased IL-17A expression and consequent G-CSF production [8]. The goal of this study was to determine whether T-cadherin, an adiponectin binding protein, contributes to these anti-inflammatory effects of adiponectin. Although several other adiponectin binding proteins have been described [9], [30], we chose to examine T-cadherin because we have shown, using CHO cells overexpressing T-cadherin, that T-cadherin primarily binds the hexameric and high molecular weight (HMW) isoforms of adiponectin [10], the isoforms most abundant in the lung lining fluid [15]. T-cadherin has also been shown to bind adiponectin in vivo: T-cadherin is prominently expressed on the apical surface of endothelial cells and immunohistochemistry indicates strong binding of adiponectin to these cells in wildtype mice. In contrast, this binding is lost in T-cad−/− mice [17]. In addition, T-cadherin is required for the protective effects of adiponectin against cardiac hypertrophy induced by pressure overload and against cardiac injury induced by ischemia-reperfusion [31]. Our results indicate that T-cadherin deficiency mimics aspects of the effects of adiponectin deficiency. After subacute O3, T-cad−/− mice, like Adipo−/− mice, had increased BAL neutrophils and G-CSF, increased pulmonary IL-17A mRNA expression, as well as increased terminal bronchiolar lesions compared to wildtype mice (Fig. 2A,3B,3E, and 4D), suggesting that binding to T-cadherin is required for effects of adiponectin on these outcomes. In contrast, O3-induced increases in BAL protein, a marker of lung injury, O3-induced increases in sTNFR1, a marker of oxidative stress, and O3-induced increases in BAL IL-6 and LIF were augmented in Adipo−/− versus wildtype mice, but not in T-cad−/− versus wildtype mice (Figs. 2,3). IL-6 is required for recruitment of neutrophils after subacute ozone [2], [32]. The role of LIF has not been established, but may be similar to IL-6 since it is a member of the same family of cytokines and shares signal transduction pathways with IL-6 [33]. The results indicate that adiponectin-dependent changes in sTNF1, IL-6, and LIF involve adiponectin acting through other adiponectin binding proteins such as AdipoR1, AdipoR2, and calreticulin [9], [30], or through non receptor mediated effects of adiponectin [34].
In addition to its role as an adiponectin binding protein, T-cadherin has other functions. It acts as a binding protein for lipoproteins [10], [35], [36], [37] and also plays an important role in neuron growth [38] and in polarization and migration of endothelial cells [14]. T-cadherin also reduces surfactant protein D secretion in a pulmonary epithelial cell line [11]. It is possible that loss of such functions, rather than loss of the ability of T-cadherin to bind adiponectin, accounts for the observed effects of T-cadherin deficiency in O3 exposed mice. To test this hypothesis, we also examined Adipo−/−/T-cad−/− mice. Although we did not assess adiponectin binding to T-cadherin in this study, our results suggest that adiponectin binding to T-cadherin is required for the ability of adiponectin to inhibit O3-induced IL-17A and saa3 expression, as well as the development of terminal bronchiolar lesions. O3-induced IL-17A mRNA expression, terminal bronchiolar lesions, and saa3 expression, were augmented in T-cad−/− mice, but no further augmentation was observed in mice with combined adiponectin and T-cadherin deficiency mice (Fig. 3E,4D,5B), indicating that the presence of T-cadherin was necessary for the ability of adiponectin to inhibit these outcomes. In contrast, compared to mice deficient in T-cadherin alone, combined adiponectin and T-cadherin deficiency further augmented BAL neutrophils and G-CSF (Figs. 2A, 3B). Augmented ozone-induced increases in BAL neutrophils and G-CSF are partially dependent on IL-17A [8]. Taken together with the observation that the increased IL-17A expression in Adipo−/− mice requires T-cadherin (Fig. 3E), the results are consistent with the hypothesis that the augmented O3-induced increases in BAL neutrophils and G-CSF in T-cad−/− versus wildtype mice derive from increases in IL-17A, whereas the additional effects of combined adiponectin and T-cadherin deficiency are the result of adiponectin acting through non-IL-17A dependent pathways (Figure 6).
Figure 6. Schematic representation of T-cadherin dependent and independent effects of adiponectin that act to inhibit ozone induced neutrophil influx into the lungs.

In contrast to the increased inflammatory responses observed in T-cad−/− versus wildtype mice after subacute O3 exposure reported here, we have previously reported reduced airway inflammation in T-cad−/− versus wildtype mice after allergen sensitization and challenge [18]. In that study, we concluded that T-cadherin does not mediate the effects of adiponectin. Instead, we suggested that the effects of T-cadherin deficiency may result from augmented circulating adiponectin in T-cad−/− mice (Fig. 1A) acting on other adiponectin binding proteins on circulating or lymphoid tissue Th2 lymphocytes, a key effector cell for allergic airway responses. As discussed above, T-cadherin is highly expressed in endothelial cells [14], where it appears to act as a repository for adiponectin. In the absence of T-cadherin, this adiponectin is delivered back to the blood resulting in increased circulating concentrations [15], [16], [17], [18], [23], as observed (Fig. 1A). One explanation for the divergent effects of T-cadherin deficiency in that study [18] versus this one is as follows. As discussed above, T-cadherin does appear to at least partially mediate the effects of adiponectin that reduce inflammation induced by subacute O3. In contrast, other adiponectin binding proteins appear to mediate the anti-inflammatory effects of adiponectin after allergen challenge [18]. The difference in adiponectin binding proteins employed by adiponectin in the two models is likely related to the cell types involved in the two different types of pulmonary inflammation. Whereas CD4+ lymphocytes are critical for allergic airways inflammation, the response to subacute O3 mainly involves the innate immune response, particularly epithelial cells, macrophages, and pulmonary γδ T-cells [8].
In wildtype mice, we observed an increase in BAL concentrations of the Th2 cytokine, IL-5 after subacute O3 exposure (Fig. 3D). Others have also reported an increase in BAL IL-5 after O3 exposure in mice, albeit using a different ozone exposure protocol [39]. The role of IL-5 in mediating responses to O3 has not been established. In contrast to the effects of T-cadherin deficiency on other O3-induced changes in the lung, BAL IL-5 was reduced in T-cadherin deficient mice and restored in adiponectin/T-cadherin bideficient mice. This response is similar to the effects of T-cadherin deficiency on Th2 cytokines in allergen sensitized and challenged mice described above [18], suggesting that the source of IL-5 after O3 may be Th2 cells.
Despite the increased circulating concentrations of adiponectin in T-cad−/− mice (Fig. 1A), we and others [15], [23] have reported reduced BAL adiponectin in naïve T-cad−/− versus wildtype mice, similar to the results of this study (Fig. 1B, air exposed mice). Such observations indicate that in unchallenged mice, adiponectin is not transported into the lung via simple diffusion: if accumulation of BAL adiponectin relied solely on diffusion, it would be greater in T-cad−/− versus WT mice, since these mice have greater serum adiponectin (Fig. 1A). Instead, we suggested that T-cadherin may serve to transport adiponectin across the alveolar capillary barrier [15]. Such a hypothesis is consistent with the observation that HMW adiponectin, the adiponectin isoform most readily bound to T-cadherin [10], is also the major isoform present in BAL fluid of naïve mice, whereas the trimeric isoform, which should diffuse most easily, is barely detectable [15], [23]. Loss of such a transport function for T-cadherin is unlikely to explain the effects of T-cad−/− observed in this study since BAL concentrations of adiponectin were actually greater in O3 exposed T-cad−/− versus wildtype mice (Fig. 1B). Subacute O3 exposure results in a marked increase in the permeability of the lungs (Fig. 2C), consistent with lung injury. Based on previous observations indicating that trimeric adiponectin accounted for the majority of the increased BAL adiponectin after ozone [8], we reasoned that in the setting of increased lung permeability, diffusion rather than T-cadherin-mediated transport begins to dominate movement of adiponectin from the blood into the lungs.
Others have demonstrated epithelial injury in mice exposed to O3 in this manner, especially in the terminal bronchioles and central acinus, as evidenced by increased BrdU incorporation into these cells, likely reflecting cell proliferation after injury [27], [40], [41]. Consistent with these observations, RT-PCR confirmed increased O3-induced expression of one of these genes, Ki67 (Fig. 5A), a common marker of cell proliferation [26]. However, neither adiponectin deficiency nor T-cadherin deficiency had any effect on Ki67 mRNA expression (Fig. 5A), suggesting that adiponectin does not regulate cell proliferation in this model. In contrast, we observed effects of both adiponectin deficiency and T-cadherin deficiency on the extent of terminal bronchiolar lesions observed after subacute O3 (Fig. 4). Furthermore, adiponectin/T-cadherin bideficiency did not further augment the effects of T-cadherin deficiency on these lesions (Fig. 4), indicating that the effects of T-cadherin deficiency were the result of loss of adiponectin binding to this receptor. Taken together, the results suggest that the impact of adiponectin on terminal bronchiolar lesions is the result of more inflammatory cell recruitment to these sites of injury rather than more epithelial proliferation.
Saa3 is an acute phase protein capable of recruiting monocytes and macrophages to sites of inflammation, perhaps by forming a complex with the extracellular matrix protein, hyaluronan [42]. O3-induced increases in saa3 mRNA expression were substantially (almost 6 fold) greater in Adipo−/− versus WT mice (Fig. 5B). T-cadherin deficiency also resulted in a marked increase in saa3 expression in O3-exposed mice, and no further augmentation was observed in mice with combined adiponectin and T-cadherin deficiency (Fig. 5B), indicating that the presence of T-cadherin was necessary for adiponectin to suppress saa3 mRNA expression, similar to our observations with IL-17A (Fig. 3E). We have previously reported that interstitial macrophages and γδ T cells are the source of the augmented IL-17A produced in the lungs following subacute O3 exposure in Adipo−/− mice [8]. Saa3 has the capacity to induce IL-17A expression in T cells [43] and it is possible that it also regulates IL-17A expression in lung after subacute O3 exposure.
One technical issue requires discussion. We exposed mice to O3 for 72 hours. Others have shown gene specific differences in the kinetics of gene expression following O3 [28], [29]. Hence, it is likely that cytokines and chemokines other than those we identified in Fig. 3 as being impacted by O3 were induced at earlier times in the exposure and then declined. Moreover, it is possible that the earlier expression of these moieties contributes to responses observed after 72 h exposure. However, we have examined the time course of key outcomes described here that differ in WT versus Adipo−/− mice (BAL neutrophils, IL-17 mRNA expression) and have found that these genotype-related differences exist throughout the 72 h exposure period described here[8].
In summary, our results confirm an anti-inflammatory role for adiponectin in pulmonary responses to subacute O3 and indicate that adiponectin binding to T-cadherin is required for aspects of this response, including the induction of IL-17A and consequent recruitment of neutrophils to the lungs.
Acknowledgments
The authors wish to thank Dr. Roderick Bronson at the Harvard Medical School Rodent Histopathology Laboratory for initial assessment of histological sections. The authors would also like to thank Dr. Huiqing Si for her valuable help in breeding mice used in this manuscript.
Funding Statement
This study was supported by the U.S. National Institute of Health [HL-084044, ES-013307, and ES-00002]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cho HY, Zhang LY, Kleeberger SR (2001) Ozone-induced lung inflammation and hyperreactivity are mediated via tumor necrosis factor-alpha receptors. Am J Physiol Lung Cell Mol Physiol 280: L537–546. [DOI] [PubMed] [Google Scholar]
- 2. Johnston RA, Schwartzman IN, Flynt L, Shore SA (2005) Role of interleukin-6 in murine airway responses to ozone. Am J Physiol Lung Cell Mol Physiol 288: L390–397. [DOI] [PubMed] [Google Scholar]
- 3. Zhao Q, Simpson LG, Driscoll KE, Leikauf GD (1998) Chemokine regulation of ozone-induced neutrophil and monocyte inflammation. Am J Physiol 274: L39–46. [DOI] [PubMed] [Google Scholar]
- 4. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, et al. (1999) Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257: 79–83. [DOI] [PubMed] [Google Scholar]
- 5. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, et al. (2010) Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem 285: 6153–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H (2004) Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun 323: 630–635. [DOI] [PubMed] [Google Scholar]
- 7. Shore SA, Terry RD, Flynt L, Xu A, Hug C (2006) Adiponectin attenuates allergen-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol 118: 389–395. [DOI] [PubMed] [Google Scholar]
- 8. Kasahara DI, Kim HY, Williams AS, Verbout NG, Tran J, et al. (2012) Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: role of IL-17A. J Immunol 188: 4558–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, et al. (2003) Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423: 762–769. [DOI] [PubMed] [Google Scholar]
- 10. Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, et al. (2004) T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A 101: 10308–10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takeuchi T, Misaki A, Fujita J, Sonobe H, Ohtsuki Y (2001) T-cadherin (CDH13, H-cadherin) expression downregulated surfactant protein D in bronchioloalveolar cells. Virchows Arch 438: 370–375. [DOI] [PubMed] [Google Scholar]
- 12. Nakanishi K, Takeda Y, Tetsumoto S, Iwasaki T, Tsujino K, et al. (2011) Involvement of endothelial apoptosis underlying chronic obstructive pulmonary disease-like phenotype in adiponectin-null mice: implications for therapy. Am J Respir Crit Care Med 183: 1164–1175. [DOI] [PubMed] [Google Scholar]
- 13. Koller E, Ranscht B (1996) Differential targeting of T- and N-cadherin in polarized epithelial cells. J Biol Chem 271: 30061–30067. [DOI] [PubMed] [Google Scholar]
- 14. Philippova M, Joshi MB, Kyriakakis E, Pfaff D, Erne P, et al. (2009) A guide and guard: The many faces of T-cadherin. Cell Signal 21: 1035–1044. [DOI] [PubMed] [Google Scholar]
- 15. Zhu M, Hug C, Kasahara DI, Johnston RA, Williams AS, et al. (2010) Impact of adiponectin deficiency on pulmonary responses to acute ozone exposure in mice. Am J Respir Cell Mol Biol 43: 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, et al. (2010) T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest 120: 4342–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hebbard LW, Garlatti M, Young LJ, Cardiff RD, Oshima RG, et al. (2008) T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. Cancer Res 68: 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Williams AS, Kasahara DI, Verbout NG, Fedulov AV, Zhu M, et al. (2012) Role of the adiponectin binding protein, T-cadherin (cdh13), in allergic airways responses in mice. PLoS One 7: e41088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bothe GW, Bolivar VJ, Vedder MJ, Geistfeld JG (2004) Genetic and behavioral differences among five inbred mouse strains commonly used in the production of transgenic and knockout mice. Genes Brain Behav 3: 149–157. [DOI] [PubMed] [Google Scholar]
- 20. Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, et al. (2008) Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4: 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herbert RA, Hailey JR, Grumbein S, Chou BJ, Sills RC, et al. (1996) Two-year and lifetime toxicity and carcinogenicity studies of ozone in B6C3F1 mice. Toxicol Pathol 24: 539–548. [DOI] [PubMed] [Google Scholar]
- 22. Williams AS, Chen L, Kasahara DI, Si H, Wurmbrand AP, et al. (2012) Obesity and airway responsiveness: Role of TNFR2. Pulm Pharmacol Ther [Epub ahead of print]: PMID 22584291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konter JM, Parker JL, Baez E, Li SZ, Ranscht B, et al. (2012) Adiponectin Attenuates Lipopolysaccharide-Induced Acute Lung Injury through Suppression of Endothelial Cell Activation. J Immunol 188: 854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kleeberger SR, Levitt RC, Zhang LY (1993) Susceptibility to ozone-induced inflammation. I. Genetic control of the response to subacute exposure. Am J Physiol 264: L15–20. [DOI] [PubMed] [Google Scholar]
- 25. Serino M, Menghini R, Fiorentino L, Amoruso R, Mauriello A, et al. (2007) Mice heterozygous for tumor necrosis factor-alpha converting enzyme are protected from obesity-induced insulin resistance and diabetes. Diabetes 56: 2541–2546. [DOI] [PubMed] [Google Scholar]
- 26. Gerdes J, Schwab U, Lemke H, Stein H (1983) Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer 31: 13–20. [DOI] [PubMed] [Google Scholar]
- 27. Yu M, Pinkerton KE, Witschi H (2002) Short-term exposure to aged and diluted sidestream cigarette smoke enhances ozone-induced lung injury in B6C3F1 mice. Toxicol Sci 65: 99–106. [DOI] [PubMed] [Google Scholar]
- 28. Backus GS, Howden R, Fostel J, Bauer AK, Cho HY, et al. (2010) Protective role of interleukin-10 in ozone-induced pulmonary inflammation. Environ Health Perspect 118: 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bauer AK, Rondini EA, Hummel KA, Degraff LM, Walker C, et al. (2011) Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: a role for heat-shock protein 70. Environ Health Perspect 119: 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takemura Y, Ouchi N, Shibata R, Aprahamian T, Kirber MT, et al. (2007) Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest 117: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Denzel MS, Scimia M-C, Zumstein PM, Walsh K, Ruiz-Lozano P, et al. (2010) T-cadherin is critical for adiponectin-mediated cardioprotection in mice. The Journal of Clinical Investigation 120: 4342–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shore SA, Johnston RA, Schwartzman IN, Chism D, Krishna Murthy GG (2002) Ozone-induced airway hyperresponsiveness is reduced in immature mice. J Appl Physiol 92: 1019–1028. [DOI] [PubMed] [Google Scholar]
- 33. Gearing DP, Comeau MR, Friend DJ, Gimpel SD, Thut CJ, et al. (1992) The IL-6 signal transducer, gp130: an oncostatin M receptor and affinity converter for the LIF receptor. Science 255: 1434–1437. [DOI] [PubMed] [Google Scholar]
- 34. Wang Y, Lam KS, Xu JY, Lu G, Xu LY, et al. (2005) Adiponectin inhibits cell proliferation by interacting with several growth factors in an oligomerization-dependent manner. J Biol Chem 280: 18341–18347. [DOI] [PubMed] [Google Scholar]
- 35. Kuzmenko YS, Kern F, Bochkov VN, Tkachuk VA, Resink TJ (1998) Density- and proliferation status-dependent expression of T-cadherin, a novel lipoprotein-binding glycoprotein: a function in negative regulation of smooth muscle cell growth? FEBS Lett 434: 183–187. [DOI] [PubMed] [Google Scholar]
- 36. Niermann T, Kern F, Erne P, Resink T (2000) The glycosyl phosphatidylinositol anchor of human T-cadherin binds lipoproteins. Biochem Biophys Res Commun 276: 1240–1247. [DOI] [PubMed] [Google Scholar]
- 37. Resink TJ, Kuzmenko YS, Kern F, Stambolsky D, Bochkov VN, et al. (1999) LDL binds to surface-expressed human T-cadherin in transfected HEK293 cells and influences homophilic adhesive interactions. FEBS Lett 463: 29–34. [DOI] [PubMed] [Google Scholar]
- 38. Ranscht B, Dours-Zimmermann MT (1991) T-cadherin, a novel cadherin cell adhesion molecule in the nervous system lacks the conserved cytoplasmic region. Neuron 7: 391–402. [DOI] [PubMed] [Google Scholar]
- 39. Kierstein S, Krytska K, Sharma S, Amrani Y, Salmon M, et al. (2008) Ozone inhalation induces exacerbation of eosinophilic airway inflammation and hyperresponsiveness in allergen-sensitized mice. Allergy 63: 438–446. [DOI] [PubMed] [Google Scholar]
- 40. Mautz WJ, Kleinman MT, Bhalla DK, Phalen RF (2001) Respiratory tract responses to repeated inhalation of an oxidant and acid gas-particle air pollutant mixture. Toxicol Sci 61: 331–341. [DOI] [PubMed] [Google Scholar]
- 41. Kleeberger SR, Levitt RC, Zhang LY, Longphre M, Harkema J, et al. (1997) Linkage analysis of susceptibility to ozone-induced lung inflammation in inbred mice. Nat Genet 17: 475–478. [DOI] [PubMed] [Google Scholar]
- 42. Han CY, Subramanian S, Chan CK, Omer M, Chiba T, et al. (2007) Adipocyte-derived serum amyloid A3 and hyaluronan play a role in monocyte recruitment and adhesion. Diabetes 56: 2260–2273. [DOI] [PubMed] [Google Scholar]
- 43. Ather JL, Ckless K, Martin R, Foley KL, Suratt BT, et al. (2011) Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol 187: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]