

Abstract
Objectives
Low levels of high-density lipoprotein (HDL) cholesterol are associated with an increased risk of acute myocardial infarction possibly through impaired endothelial atheroprotection and decreased nitric oxide (NO) bioavailability. Asymmetric dimethylarginine (ADMA) mediates endothelial function by inhibiting nitric oxide synthase activity. In patients with acute myocardial infarction, we investigated the relationship between serum levels of HDL and ADMA.
Approach and Results
Blood samples from 612 consecutive patients hospitalized for acute MI <24 hours after symptom onset were taken on admission. Serum levels of ADMA, its stereoisomer, symmetric dimethylarginine (SDMA) and L-arginine were determined using high-performance liquid chromatography. Patients with low HDL (<40 mg/dL for men and <50 mg/dL for women) were compared with patients with higher HDL. Most patients (59%) had low HDL levels. Median ADMA levels were markedly higher in the low HDL group (0.69 vs. 0.50 µmole/L, p<0.001). In contrast, SDMA and L-arginine levels were similar for the two groups (p = 0.120 and p = 0.064). Notably, ADMA, but not SDMA or L-arginine, was inversely correlated with HDL (r = −0.311, p<0.001). In stratified analysis, this relationship was only found for low HDL levels (r = −0.265, p<0.001), but not when HDL levels were higher (r = −0.077, p = 0.225). By multivariate logistic regression analysis, ADMA level was strongly associated with low HDL levels (OR(95%CI):6.06(3.48–10.53), p<0.001), beyond traditional confounding factors.
Conclusions
Our large population-based study showed for the first time a strong inverse relationship between HDL and ADMA in myocardial infarction patients, suggesting a functional interaction between HDL and endothelium, beyond metabolic conditions associated with low HDL levels.
Introduction
Reduced endothelial nitric oxide (NO) bioavailability contributes to the development and progression of atherosclerosis, leading to coronary artery disease (CAD). Asymmetric dimethylarginine (ADMA), an endogenous competitive inhibitor of all isoforms of nitric oxide synthases (NOS), may compete with L-arginine as the substrate for the enzyme or inhibit NOS phosphorylation and thus decrease NO bioavailability [1], [2]. ADMA and its stereoisomer, symmetric dimethylarginine (SDMA) are endogenously produced by the methylation of arginine residues from nuclear proteins and are released after proteolysis. Unlike ADMA, which is a primary factor in the control of NOS activity, SDMA has insignificant inhibitory effects on the enzyme. High ADMA concentrations have been associated with most cardiovascular risk factors, including dyslipidemia [3], [4]. Moreover, several lines of evidence have demonstrated that elevated ADMA levels are associated with endothelial dysfunction in healthy subjects and in patients with CAD or hypercholesterolemia, [3], [5].
Epidemiological studies and randomized clinical trials have showed that low plasma levels of high-density lipoprotein (HDL) are associated with an increased risk of cardiovascular events in CAD patients, suggesting that HDL-C (HDL cholesterol) has cardioprotective effects [6]. This strong relationship has stimulated interest in determining mechanisms and optimal management of low levels of HDL-C. Beyond reverse cholesterol transport, the molecular mechanisms underlying the anti-atherogenic properties of HDL-C include antioxidant, anti-inflammatory and vasculo-protective effects [7]. Moreover, experimental studies have identified various direct endothelial protective effects of HDL-C, including the stimulation of endothelial NO production [8]. HDL-C restores endothelial function by increasing NO bioavailability [9]. Decreased HDL-C levels are frequently associated with the defective functionality of HDL-C particles, which have abnormal metabolism and chemical composition, reflecting in part their attenuated cholesterol efflux capacity but also their decreased anti-inflammatory and antioxidative activities [10]. However, there is little information on the association between ADMA and HDL-C, limited to an in vitro study using human umbilical vein endothelial cells (HUVECs) [11]. From a large prospective population of MI patients, we hypothesized that elevated circulating levels of ADMA, as a surrogate marker of endothelial health, could be associated with low HDL-C levels.
Patients and Methods
Study Subjects
All the consecutive patients aged >18 y and hospitalized <24 hours after symptom onset for acute MI in the coronary care unit of Dijon University Hospital between 1st January 2011 and 30th June 2012 were included. Patients with relevant co-morbidities (infection, autoimmune disorders and cancers) were excluded from the study. MI was defined by an increase in serum troponin I (2X> upper limit of the hospital normal (ULN) range) associated with symptoms of ischemia and/or characteristic ECG signs (ST-segment-T wave changes, left bundle branch block, or development of pathological Q waves) [12]. STEMI was defined by new ST-segment elevation >1 mm or left bundle branch block on the qualifying ECG.
The study was approved by the Consultative Committee of Protection of Persons in Biomedical Research of Burgundy and conducted in accordance with Declaration of Helsinki. All subjects gave their written consent to participate in the study.
Group Definition
Patients were analyzed according to their HDL-C levels. Low HDL-C was defined by <40 mg/dL in men and <50 mg/dL in women [13].
Data Collection
Data on demographics, risk factors [history of hypertension, diabetes, hyperlipidemia, body mass index (BMI)], chronic treatments and prior MI were prospectively collected. Echocardiography was performed at 2±1 days and the Simpson method was used to assess left ventricular ejection fraction (LVEF).
Biological Data
Blood samples were drawn on admission (Median time from symptom onset to blood sampling: 16(8–30) hours). Homocysteine concentrations were determined using a chemiluminescence method on an Immulite 2000 analyzer (Diagnostic Products Corporation, Los Angeles, USA). C-reactive protein (CRP), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C) and triglyceride (TG) concentrations were measured on a Dimension analyzer (Dade Behring, Newark, NE). The level of low-density lipoprotein cholesterol (LDL-C) was calculated using the Friedewald formula [14]. Plasma glucose concentrations (enzymatic method (glucose oxidase)) and creatinine levels were measured on a Vitros 950 analyzer (Ortho Clinical Diagnostics, Rochester, NY) and creatinine clearance was calculated with the Cockcroft formula. Glycated hemoglobin A1c (HbA1c) was measured with ion exchange HPLC (Bio-Rad Laboratories, Richmond, CA).
Dimethylarginines and L-arginine Analysis
Samples were allowed to clot at room temperature for 30 minutes and centrifuged at 2500 rpm for 10 minutes at 4°C. The serum was kept frozen at −80°C until analysis. L-arginine, ADMA, and SDMA, were measured by high performance liquid chromatography (HPLC) [15]. Before the analysis, serum was added with N-monomethyl L-arginine (NMMA) as the internal standard and phosphate–buffered saline (PBS). This mixture was extracted on solid-phase extraction (SPE) cartridges (Phenomenex Strata X-C, Torrance, CA, USA). The cartridges were first conditioned with elution buffer (10/0.5/40/50; NH3 concentrated/1 M NaOH/bidistilled water/CH3OH; v/v/v/v) followed by bidistilled water before being loaded with the diluted sample. The SPE cartridge was consecutively washed with HCl (100 mmol/L) and methanol (1∶1; v:v).ADMA and SDMA were eluted with elution buffer. All conditioning, washing and elution steps were achieved by vacuum suction. The eluate was dried under nitrogen, derivatized with ortho-phthaldialdehyde (OPA) reagent (1∶1; v:v) and injected into the HPLC system, with a fluorescent detector Finingan Surveyor (Thermo Fisher (λexc:340 nm, λem:455 nm) and Chromolith® RP-18E column (100×4.6 mm) including a guard cartridge (10×4.6 mm) supplied by Merck (Darmstadt, Germany). Chromatographic separation, at room temperature, was performed isocratically at 100% mobile phase A, with 25 mmol/L phosphate buffer (pH 6.8) containing 6.5% CH3CN, at a flow rate of 1.1 mL/min. After SDMA elution, mobile phase was switched to 100% mobile phase B, with ultrapure water:CH3CN (50∶50, v:v), and the flow rate was increased to 3.0 mL/min to elute strongly retained compounds. Assays were performed in duplicate.
The detection limits were 0.05 and 1.19 µmol/L and interday variabilities were 5.7 and 4.6% for ADMA and L-arginine, respectively.
Statistical Analysis
All the analyses have been made on anonymous de-identified data that were available to others. Data are presented as median (interquartile range [IQR]), or proportion (n(%)). For continuous variables, normality was checked by the Kolmogorov-Smirnov test. The Student t test or Mann-Whitney test was used to test for continuous data in the two groups. Categorical variables were compared by the chi-square test or Fisher’s exact test. Spearman’s correlation was applied to test for associations between dimethylarginines, L-arginine and biological variables. Mann-Whitney test was used to test L-arginine and dimethylarginines concentrations in patients according to their HDL levels.
In order to further investigate whether the relationship between HDL-C and ADMA was influenced by metabolic or pharmacological conditions, stratified correlation analysis was performed on subgroups of patients based on median LDL or CRP values (inframedian or supramedian), on the chronic use of statin or fibrate therapy (with or without treatment) and on levels of HDL-C (low or high HDL-C). Factors associated with low HDL-C were studied by backward logistic regression analysis. Variables entered into the multivariate model were chosen according to their relationship (cut-off at 10%) in univariate analysis (i.e. age, BMI, diabetes, prior MI, fibrate, CRP, HbA1c, triglycerides, ADMA, SDMA, L-arginine and smoking), with an exclusion cut-off at 1%. All the analyses were performed using the SPSS 13.0 software package (IBM Inc, USA).
Results
Baseline Characteristics
Patients’ characteristics are shown in Table 1. Most patients (362/612 (59%)) had low HDL-C levels. Patients with low HDL-C were younger, more frequently smokers, with prior MI, had a higher body mass index, and showed a trend towards a greater rate of diabetes than patients with high HDL-C. Clinical data and current medications were similar for the two groups, except for fibrate therapy, which was more frequent in the low HDL-C group. Biological data are shown in Table 2. Higher CRP and triglycerides and a trend toward higher HbA1c levels were found in patients with low HDL-C. No difference was observed between the two groups for LDL-C, glucose, creatinine clearance and homocysteine. Interestingly, median ADMA concentrations were markedly higher (∼ 40%, i.e. 0.69 vs. 0.50 µmole/L, p<0.001) in patients with low HDL-C than in those with higher HDL-C levels (Figure 1). In contrast, there was no or only weak difference between the two groups for SDMA and L-arginine levels (p = 0.120 and p = 0.064, respectively).
Table 1. Patients’ characteristics (n(%) or median (IQR)).
| High HDL | Low HDL | p | |
| N = 250 | N = 362 | ||
| Risk factors | |||
| Age, years | 68(57–79) | 65(52–77) | 0.007 |
| Female | 60(24) | 99(27) | 0.353 |
| BMI, kg/m2 | 26(24–29) | 27(25–30) | 0.002 |
| Hypertension | 136(55) | 200(55) | 0.934 |
| Hypercholesterolemia | 111(44) | 155(43) | 0.658 |
| Family history of CAD | 65(26) | 100(28) | 0.521 |
| Diabetes | 45(18) | 86(24) | 0.085 |
| Smoking | 62(25) | 130(36) | 0.002 |
| Prior myocardial infarction | 27(11) | 65(18) | 0.015 |
| Clinical data | |||
| HR, beats/min | 77(67–90) | 76(66–88) | 0.542 |
| SBP, mmHg | 140(121–163) | 137(120–160) | 0.308 |
| DBP, mmHg | 80(70–96) | 80(69–90) | 0.093 |
| STEMI | 147(59) | 188(52) | 0.093 |
| LVEF, % | 55(45–60) | 55(45–60) | 0.670 |
| Current medications | |||
| Statin | 66(26) | 92(25) | 0.784 |
| Fibrate | 7(3) | 23(6) | 0.045 |
| ACE inhibitor | 42(17) | 74(20) | 0.258 |
| Aspirin | 56(22) | 78(21) | 0.802 |
Low HDL was defined as <40 mg/dL in men and <50 mg/dL in women.
ACE: Angiotensin converting enzyme; BMI: body mass index; CAD: Coronary artery disease; DBP: Diastolic blood pressure; HR: heart rate; LVEF: Left ventricular ejection fraction; SBP: Systolic blood pressure; STEMI: ST segment elevation myocardial infarction.
Table 2. Biological data (Median (IQR)).
| High HDL | Low HDL | p | |
| N = 250 | N = 362 | ||
| CRP, mg/L | 4.0(3.0–10.7) | 6.5(3.0–18.0) | <0.001 |
| Homocysteine, µmol/L | 13(10–17 | 12(10–17 | 0.239 |
| Creatinine clearance, ml/min | 75(54–92) | 80(52–107) | 0.311 |
| Glucose, mmol/L | 6.84(5.72–8.37) | 6.93(5.77–9.32) | 0.211 |
| HbA1c, % | 5.8(5.5–6.3) | 5.9(5.6–6.7) | 0.082 |
| HDL-cholest, mg/dL | 54(47–64) | 33(27–38) | <0.001 |
| LDL-cholest, mg/dL | 121(94–152) | 125(94–153) | 0.390 |
| Total-cholest, mg/dL | 199(168–227) | 191(158–224) | 0.023 |
| Triglycerides, mg/dL | 93(68–135) | 138(95–199) | <0.001 |
| ADMA, µmol/L | 0.50(0.41–0.73) | 0.69(0.47–1.02) | <0.001 |
| SDMA, µmol/L | 0.52(0.42–0.70) | 0.51(0.38–0.71) | 0.120 |
| L-arginine, µmol/L | 83.9(68.7–114.4) | 91.1(72.5–120.9) | 0.064 |
| L-arg/ADMA ratio | 163(118–225) | 132(92–199) | <0.001 |
Low HDL was defined as HDL <40 mg/dL in men and <50 mg/dL in women.
ADMA: Asymmetric dimethylarginine; CRP: C-reactive protein; HbA1c: glycated hemoglobin; HDL-C: high-density lipoprotein; LDL-C: low-density lipoprotein; SDMA: Symmetric dimethylarginine.
Figure 1. L-arginine and dimethylarginines concentrations in patients according to their HDL levels.
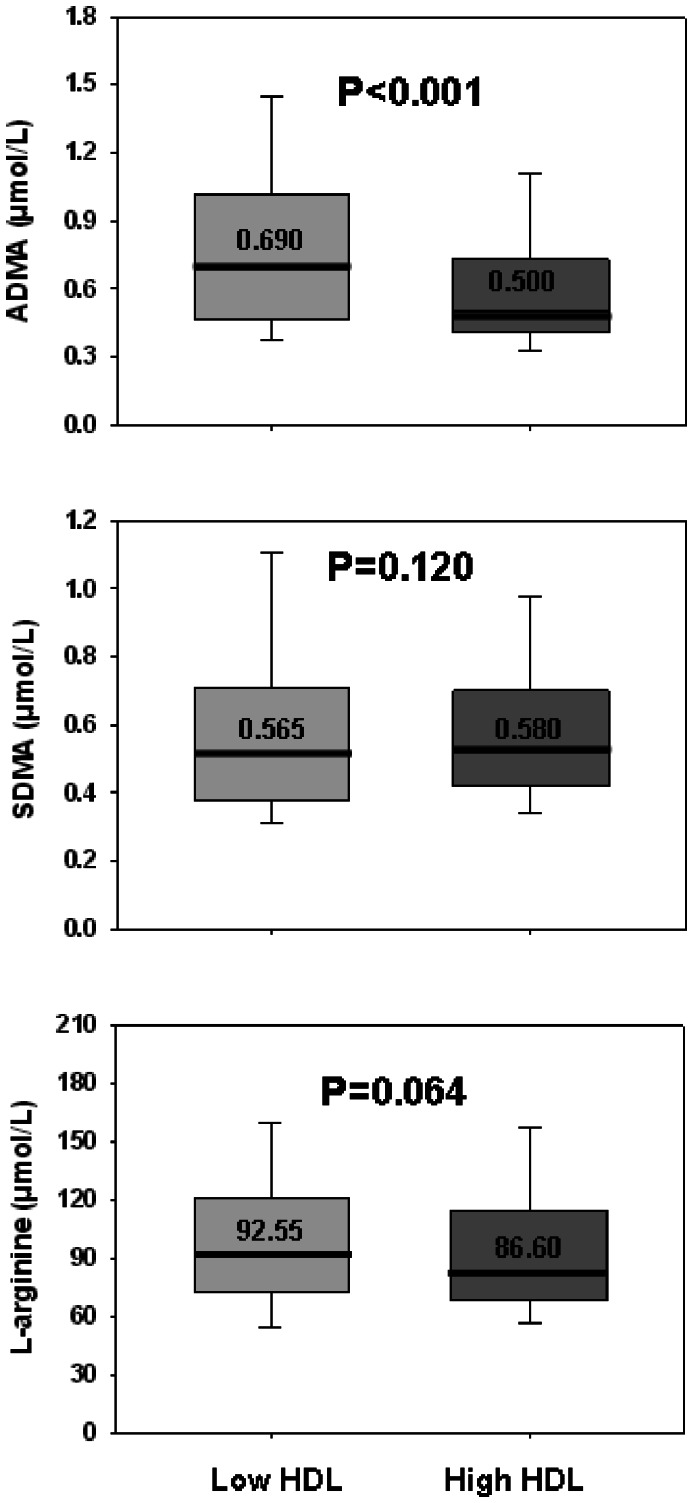
The horizontal line is the median, box is the IQR (interquartile range) and whiskers are the 95% range. P values are from Mann-Whitney tests.
ADMA and HDL-C
Spearman correlation analyses for ADMA, SDMA, L-arginine and biological parameters are reported in Table 3. ADMA was positively related to SDMA and L-arginine, with a trend towards a negative relationship with homocysteine (p = 0.067). Moreover, ADMA showed a strong negative correlation with HDL-C (r = −0.311, p<0.001) (Figure 2). SDMA had only a weak and positive association with HDL-C (p = 0.040), but a strong correlation with both LDL and homocysteine (p<0.001). L-arginine was associated positively with LDL and triglyceride levels, but negatively with HDL-C (p = 0.006). Subgroup correlation analysis showed that whatever the LDL and CRP levels (i.e. stratified by median values) and treatments (stratified by statin or fibrate use), the significant relationship between ADMA and HDL-C was maintained in all subgroups (p<0.001). In contrast, when patients were classified according to HDL-C levels (stratified by low/high HDL-C) (Table 4), only patients with low HDL-C levels showed a strong relationship between HDL-C and ADMA (r = −0.265, p<0.001). In contrast, there was no significant correlation between these two parameters in patients with higher HDL-C (p = 0.225).
Table 3. Correlations between dimethylarginines, L-arginine and biological data (N = 612).
| ADMA | SDMA | L-arginine | ||||
| r | p | r | p | r | p | |
| ADMA | – | – | 0.248 | <0.001 | 0.260 | <0.001 |
| SDMA | 0.248 | <0.001 | – | – | 0.057 | 0.162 |
| L-arginine | 0.260 | <0.001 | 0.057 | 0.162 | – | – |
| HDL-Cholest | −0.311 | <0.001 | 0.083 | 0.040 | −0.111 | 0.006 |
| LDL-Cholest | 0.070 | 0.088 | −0.193 | <0.001 | 0.169 | <0.001 |
| Total-Cholest | −0.041 | 0.309 | −0.163 | <0.001 | 0.144 | <0.001 |
| Triglycerides | −0.009 | 0.825 | −0.074 | 0.068 | 0.106 | 0.009 |
| Creatinine clearance | −0.078 | 0.061 | −0.448 | <0.001 | 0.107 | 0.010 |
| Homocysteine | −0.077 | 0.067 | 0.386 | <0.001 | −0.042 | 0.321 |
| HbA1c | −0.072 | 0.087 | 0.029 | 0.495 | −0.038 | 0.368 |
| CRP | 0.051 | 0.212 | 0.145 | <0.001 | −0.025 | 0.546 |
| Glucose | 0.014 | 0.737 | 0.009 | 0.821 | −0.060 | 0.142 |
ADMA: Asymmetric dimethylarginine; CRP: C-reactive protein; HbA1c: glycated hemoglobin; HDL-C: high-density lipoprotein; LDL-C: low-density lipoprotein; SDMA: Symmetric dimethylarginine.
Figure 2. Correlations between ADMA and HDL circulating levels (r = −0.311, p<0.001).

Table 4. Correlations between dimethylarginines, and Larginine in the high and low HDL groups.
| ADMA | ||||
| HighHDL | LowHDL | |||
| N = 250 | N = 362 | |||
| r | p | r | p | |
| HDL-Cholest | −0.077 | 0.225 | −0.265 | <0.001 |
| SDMA | 0.319 | <0.001 | 0.253 | <0.001 |
| L-arginine | 0.171 | 0.007 | 0.311 | <0.001 |
Low HDL was defined as <40 mg/dL in men and <50 mg/dL in women.
ADMA: Asymmetric dimethylarginine; HDL-C: high-density lipoprotein; SDMA: Symmetric dimethylarginine.
Backward logistic regression analysis with low HDL-C as a dependent variable showed that the level of ADMA was strongly associated with the HDL-C levels (OR(95%CI): 6.06(3.48–10.53), p<0.001), beyond confounding factors such as triglycerides (OR(95%CI):3.16(2.30–4.33), p<0.001) and smoking (OR(95%CI): 1.80(1.20–2.71),p = 0.005) (Figure 3). SDMA was not independently associated with HDL-C in the final multivariate model. The Quality Index validated the ability of the model to correctly predict HDL-C (−2LL = 664.72, p (Hosmer-Lemeshow) = 0.540, with more than 75% of correct classifications in the at-risk group).
Figure 3. Multivariate backward logistic regression analysis with low HDL levels as the dependent variable.

Discussion
In recent years, it has been established that HDL-C may exert atheroprotective effects through its beneficial effect on endothelial cells. However, only few data are available in humans on the relationship between HDL-C levels and their association with ADMA, as a surrogate marker of endothelial health. Our study is the first large prospective study to show that patients with low HDL-C levels were characterized by markedly high ADMA levels, whatever the levels of lipids and inflammation markers. Given that this relationship between ADMA and HDL-C levels was only found in patients with low HDL-C levels, our work also lends further support to the concept of the functional heterogeneity of HDL-C in such high-risk patients.
ADMA and Endothelial Dysfunction
In MI patients, circulating ADMA has been independently associated with clinical outcomes probably through its deleterious impact on vascular homeostasis [16], [17]. The role of ADMA as an endogenous competitive inhibitor of eNOS is now well established [18], [19]. High plasma ADMA concentrations are associated with endothelial dysfunction in patients with CV risk factors or CAD, while this association is weak in healthy individuals[20]–[23]. Endothelial dysfunction is primarily characterized by impaired NO-induced vasodilation, and associated with traditional CV risk factors such as dyslipidemia, arterial hypertension and metabolic syndrome [3], [5], [24]. More precisely, in the vascular endothelium of CAD patients, high serum ADMA levels are associated with impaired vascular function reflected by increased vascular O2 •− production and reduced NO bioavailability, due to accelerated eNOS uncoupling [25].
Low HDL-C and Dysfunctional HDL-C
Our study, like other recent studies [26], showed that most patients with acute MI had low HDL-C levels. Low HDL-C is an independent risk factor for CAD, whatever the level of LDL-C. However, the effects of therapies to raise HDL-C, such as cholesterylester transfer protein (CETP) inhibitors, are controversial, partly because of the complex interaction of the lipoprotein particle with the enzyme, which depends on the metabolic context [27]. It is becoming increasingly evident that HDL-C particles not only promote reverse cholesterol transport from the periphery (mainly macrophages) to the liver but also exert pleiotropic effects, in particular on inflammation and vascular homeostasis. However, few studies have been carried out on the association between low HDL-C levels and markers of endothelial dysfunction.
In our study, patients with low HDL-C were more likely to have metabolic risk factors such as diabetes, obesity and high triglycerides, characteristics of the atherogenic dyslipidemia phenotype [28], [29]. Moreover, active smokers have lower levels of HDL-C and HDL particles, and both increase with smoking cessation [30], [31]. In our study population, whether low HDL could be associated with qualitative modifications in HDL-C remains speculative. However, consistent data have showed that reductions in HDL-C levels frequently coincide with HDL-C functional impairment and alterations in HDL-C metabolism and structure. In common metabolic diseases, such as type 2 diabetes and metabolic syndrome, low circulating levels of HDL-C are associated with the defective functionality of small HDL particles, which also have an abnormal structure and composition.
Moreover, in atherogenic low HDL-C dyslipidemia, circulating levels of large, cholesterol-rich HDL2 decrease in parallel with HDL-C [32], [33]. The biological activities of atheroprotective HDL-C (i.e. HDL3) are also often compromised when levels of HDL-C are low. Specifically, in CAD, alterations in HDL3 composition, which include decreases in cholesteryl esters, apoA-I, and paraoxonase 1 (PON1) together with increases in triglyceride and serum amyloid A (SAA) and covalent modifications of HDL-C by oxidation and/or glycation, cause a decrease in cellular cholesterol capacity as well as decreases in the antioxidative and anti-inflammatory activities of these particles. In addition, acute phase protein SAA, which is bound by HDL-C, particularly HDL3, has recently been shown to promote endothelial dysfunction and reduce NO bioavailability by stimulation of O2 production [34], [35]. The ability of HDL to stimulate the eNOS activating pathway was impaired in CAD patients, in part due to reduced PON1-HDL-associated activity [36]. Moreover, low HDL-C has also recently been reported as an important determinant of lipid peroxidation, irrespective of risk factors and inflammatory and/or metabolic biomarkers [37].
ADMA, Endothelial Dysfunction and HDL-C
Recent studies on endothelial cell cultures or isolated vessels showed that HDL-C can exert direct biological effects on endothelial cells, via stimulation of eNOS mediated NO production, increased eNOS expression and NO-mediated vasodilation [7]. In humans, consistent data also show that under certain favorable conditions, HDL-C could exert beneficial effects on endothelium-dependent vascular reactivity. In CAD patients, HDL-C was significantly related to the Flow Mediated Dilation (FMD) of brachial arteries (beta = 0.295, P = 0.018) [38].
The underlying mechanisms of the effects of HDL-C on eNOS stimulation remain unclear. Early studies have suggested that HDL-C can prevent the detrimental effects of LDL oxidation on eNOS. In human umbilical vein endothelial cell cultures, exposure to increasing HDL-C concentrations protected against reduced NO production induced by ox-LDL, with concomitant decreases in ADMA levels [11]. However, one emerging hypothesis, based on experimental findings, suggests an effect on apoA-I and ABC transporters (ABCG1), eNOS phosphorylation-induced activation of PI3K/Akt, MAP kinase pathways and Scavenger receptor B type 1 (SRB1), or interaction of eNOS with caveolin-1 [33], [39]. On the other hand, HDL-associated lysophospholipid, i.e. shingosine-1-phosphate (S1P), binds to the lysophospholipid receptor S1P3, thus stimulating the Akt pathway and thereby eNOS phosphorylation at Ser177. HDL-C also contains active PON1 which suppresses the formation of malondialdehyde (MDA), and thereby exerts beneficial effects on the endothelium through blunting lectin-type oxidized LDL receptor 1 (LOX-1)–induced inhibition of eNOS phosphorylation.
Our study provides important additional data that suggest a specific role of HDL-C in ADMA-related endothelial dysfunction. Our data are in agreement with the findings of recent studies to assess the impact of strategies to raise HDL-C on endothelial function. In patients with type II hyperlipidemia or CAD, a 16 to 26% increase in HDL-C plasma levels was achieved by a CETP inhibitor or niacin, and was associated with improved endothelial function, but only in the subgroup of patients with low baseline HDL-C levels [40], [41]. When HDL-C level achieved is sufficient (i.e. >40 mg/dL in men and >50 mg/dL in women), corresponding to physiological particle sizes and functionality, the lack of any significant interaction between ADMA and HDL-C strongly suggests that other pathophysiological pathways, including Ca2+-activated calmodulin, Hsp90, shear stress, oestrogen and vascular endothelial growth factor (VEGF), are involved in regulating eNOS activity [42]. Moreover, as SDMA, which is not a competitive inhibitor of NO synthase, failed to show any independent relationship with the lipoprotein, the link between ADMA and HDL-C levels may be due to the modulation of eNOS activity. The recent disappointing results from the dal-VESSEL randomized clinical trial, showing a lack of improvement in brachial artery reactivity, as a surrogate of endothelial function, and inflammatory markers after 36 weeks of treatment with dalcetrapib, also add fuel to this hypothesis of the functional heterogeneity of HDL particles in CAD patients [43].
Study Limitations
As suggested by earlier studies, lipoprotein-related parameters may vary over the few days following MI. However, previous reports have indicated that lipid changes in response to MI-related inflammation are only minor during the first 24 h of MI [44], [45]. Thus, in the present study, it can be assumed that measurements of HDL-C, LDL and triglycerides at the early phase of acute MI reflect accurately and in a retrospective manner the lipoprotein profile before the MI as well as during the acute phase. The main strength of this study is the use of large prospective population-based registry, with adjustments for a wide range of possible confounding factors. These elements limit the risk of any bias in our conclusions. This work, however, suffers from the usual limitations of observational, non-randomized studies and therefore determines correlations rather than causal relationships.
Conclusions
The adverse effects or the lack of clinical benefit of strategies to raise HDL-C suggest that better understanding of the link between HDL-C and markers of vascular homeostasis is urgently required, as are efficient methods to measure HDL functionality [46]. ADMA is now emerging as a relevant clinical tool, and a useful biomarker of endothelial dysfunction. Some authors have proposed ADMA as an in vivo assessment of endothelial health and even suggested that it could be used to monitor the vascular effects of statin therapy in a clinical setting [47]. This study points out how the implementation of such surrogate markers in clinical trials could give a measure of vascular effects of the lipoprotein and thus may allow for the shortening of clinical trials, which could in turn help to reduce the cost of drug development in the future. Our large population-based study further supports the hypothesis of the functional heterogeneity of HDL particles in CAD patients, and strongly suggests that the biological activity of on-treatment HDL-C, in particular regarding endothelial health, in addition to its plasma levels, needs to be taken into account for the development of targeted strategies to raise HDL levels or to treat HDL dysfunction.
Acknowledgments
The authors thank Mr. P. Bastable for English revision of the manuscript and Françoise Bechet for technical assistance.
Funding Statement
This work was supported by the University Hospital of Dijon, the Association de Cardiologie de Bourgogne, and by grants from the Agence Régionale de Santé (ARS) de Bourgogne, the French Ministry of Research from the Institut National de la Santé et de la Recherche Médicale (INSERM) and from the Regional Council of Burgundy. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vallance P, Leone A, Calver A, Collier J, Moncada S (1992) Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 339: 572–575. [DOI] [PubMed] [Google Scholar]
- 2. Kajimoto H, Kai H, Aoki H, Yasuoka S, Anegawa T, et al. (2012) Inhibition of eNOS phosphorylation mediates endothelial dysfunction in renal failure: new effect of asymmetric dimethylarginine. Kidney Int 81: 762–768. [DOI] [PubMed] [Google Scholar]
- 3. Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, et al. (1998) Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 98: 1842–1847. [DOI] [PubMed] [Google Scholar]
- 4. Boger RH (2003) The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc Res 59: 824–833. [DOI] [PubMed] [Google Scholar]
- 5. Juonala M, Viikari JS, Alfthan G, Marniemi J, Kahonen M, et al. (2007) Brachial artery flow-mediated dilation and asymmetrical dimethylarginine in the cardiovascular risk in young Finns study. Circulation 116: 1367–1373. [DOI] [PubMed] [Google Scholar]
- 6. Barter P, Gotto AM, LaRosa JC, Maroni J, Szarek M, et al. (2007) HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med 357: 1301–1310. [DOI] [PubMed] [Google Scholar]
- 7. Besler C, Luscher TF, Landmesser U (2012) Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease. EMBO Mol Med 4: 251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, et al. (2001) High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med 7: 853–857. [DOI] [PubMed] [Google Scholar]
- 9. Spieker LE, Sudano I, Hurlimann D, Lerch PG, Lang MG, et al. (2002) High-density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation 105: 1399–1402. [DOI] [PubMed] [Google Scholar]
- 10. Kontush A, Chapman MJ (2006) Antiatherogenic small, dense HDL-guardian angel of the arterial wall? Nat Clin Pract Cardiovasc Med 3: 144–153. [DOI] [PubMed] [Google Scholar]
- 11. Peng ZY, Zhao SP, He BM, Peng DQ, Hu M (2011) Protective effect of HDL on endothelial NO production: the role of DDAH/ADMA pathway. Mol Cell Biochem 351: 243–249. [DOI] [PubMed] [Google Scholar]
- 12. Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, et al. (2012) Third universal definition of myocardial infarction. Circulation 126: 2020–2035. [DOI] [PubMed] [Google Scholar]
- 13. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 285: 2486–2497. [DOI] [PubMed] [Google Scholar]
- 14.Warnick GR (1990) Laboratory measurement of lipid and lipoprotein risk factors. Scand J Clin Lab Invest Suppl 198: 9–19. [PubMed]
- 15. Ivanova M, Artusi C, Boffa GM, Zaninotto M, Plebani M (2010) HPLC determination of plasma dimethylarginines: method validation and preliminary clinical application. Clin Chim Acta 411: 1632–1636. [DOI] [PubMed] [Google Scholar]
- 16. Zeller M, Korandji C, Guilland JC, Sicard P, Vergely C, et al. (2008) Impact of asymmetric dimethylarginine on mortality after acute myocardial infarction. Arterioscler Thromb Vasc Biol 28: 954–960. [DOI] [PubMed] [Google Scholar]
- 17. Meinitzer A, Seelhorst U, Wellnitz B, Halwachs-Baumann G, Boehm BO, et al. (2007) Asymmetrical dimethylarginine independently predicts total and cardiovascular mortality in individuals with angiographic coronary artery disease (the Ludwigshafen Risk and Cardiovascular Health study). Clin Chem 53: 273–283. [DOI] [PubMed] [Google Scholar]
- 18. Achan V, Broadhead M, Malaki M, Whitley G, Leiper J, et al. (2003) Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol 23: 1455–1459. [DOI] [PubMed] [Google Scholar]
- 19. Cooke JP (2004) Asymmetrical dimethylarginine: the Uber marker? Circulation 109: 1813–1818. [DOI] [PubMed] [Google Scholar]
- 20. Thorne S, Mullen MJ, Clarkson P, Donald AE, Deanfield JE (1998) Early endothelial dysfunction in adults at risk from atherosclerosis: different responses to L-arginine. J Am Coll Cardiol 32: 110–116. [DOI] [PubMed] [Google Scholar]
- 21. Fard A, Tuck CH, Donis JA, Sciacca R, Di Tullio MR, et al. (2000) Acute elevations of plasma asymmetric dimethylarginine and impaired endothelial function in response to a high-fat meal in patients with type 2 diabetes. Arterioscler Thromb Vasc Biol 20: 2039–2044. [DOI] [PubMed] [Google Scholar]
- 22. Chao CL, Kuo TL, Lee YT (2000) Effects of methionine-induced hyperhomocysteinemia on endothelium-dependent vasodilation and oxidative status in healthy adults. Circulation 101: 485–490. [DOI] [PubMed] [Google Scholar]
- 23. Boger RH, Lentz SR, Bode-Boger SM, Knapp HR, Haynes WG (2001) Elevation of asymmetrical dimethylarginine may mediate endothelial dysfunction during experimental hyperhomocyst(e)inaemia in humans. Clin Sci (Lond) 100: 161–167. [PubMed] [Google Scholar]
- 24. Yasuda S, Miyazaki S, Kanda M, Goto Y, Suzuki M, et al. (2006) Intensive treatment of risk factors in patients with type-2 diabetes mellitus is associated with improvement of endothelial function coupled with a reduction in the levels of plasma asymmetric dimethylarginine and endogenous inhibitor of nitric oxide synthase. Eur Heart J 27: 1159–1165. [DOI] [PubMed] [Google Scholar]
- 25. Antoniades C, Shirodaria C, Leeson P, Antonopoulos A, Warrick N, et al. (2009) Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J 30: 1142–1150. [DOI] [PubMed] [Google Scholar]
- 26. Paneni F, Cosentino F, Marrara F, Palano F, Capretti G, et al. (2012) The clinical relevance of dysfunctional HDL in patients with coronary artery disease: a 3-year follow-up study. Int J Cardiol 158: 158–160. [DOI] [PubMed] [Google Scholar]
- 27. Zeller M, Masson D, Farnier M, Lorgis L, Deckert V, et al. (2007) High serum cholesteryl ester transfer rates and small high-density lipoproteins are associated with young age in patients with acute myocardial infarction. J Am Coll Cardiol 50: 1948–1955. [DOI] [PubMed] [Google Scholar]
- 28. Lakka HM, Laaksonen DE, Lakka TA, Niskanen LK, Kumpusalo E, et al. (2002) The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA 288: 2709–2716. [DOI] [PubMed] [Google Scholar]
- 29. Malik S, Wong ND, Franklin SS, Kamath TV, L'Italien GJ, et al. (2004) Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation 110: 1245–1250. [DOI] [PubMed] [Google Scholar]
- 30. Gossett LK, Johnson HM, Piper ME, Fiore MC, Baker TB, et al. (2009) Smoking intensity and lipoprotein abnormalities in active smokers. J Clin Lipidol 3: 372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gepner AD, Piper ME, Johnson HM, Fiore MC, Baker TB, et al. (2011) Effects of smoking and smoking cessation on lipids and lipoproteins: outcomes from a randomized clinical trial. Am Heart J 161: 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kontush A, Chapman MJ (2006) Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev 58: 342–374. [DOI] [PubMed] [Google Scholar]
- 33. Mineo C, Shaul PW (2012) Novel biological functions of high-density lipoprotein cholesterol. Circ Res 111: 1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Witting PK, Song C, Hsu K, Hua S, Parry SN, et al. (2011) The acute-phase protein serum amyloid A induces endothelial dysfunction that is inhibited by high-density lipoprotein. Free Radic Biol Med 51: 1390–1398. [DOI] [PubMed] [Google Scholar]
- 35. Litvinov D, Mahini H, Garelnabi M (2012) Antioxidant and anti-inflammatory role of paraoxonase 1: implication in arteriosclerosis diseases. N Am J Med Sci 4: 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Besler C, Heinrich K, Rohrer L, Doerries C, Riwanto M, et al. (2011) Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest 121: 2693–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zelzer S, Fuchs N, Almer G, Raggam RB, Pruller F, et al. (2011) High density lipoprotein cholesterol level is a robust predictor of lipid peroxidation irrespective of gender, age, obesity, and inflammatory or metabolic biomarkers. Clin Chim Acta 412: 1345–1349. [DOI] [PubMed] [Google Scholar]
- 38. Li XP, Zhao SP, Zhang XY, Liu L, Gao M, et al. (2000) Protective effect of high density lipoprotein on endothelium-dependent vasodilatation. Int J Cardiol 73: 231–236. [DOI] [PubMed] [Google Scholar]
- 39. Prosser HC, Ng MK, Bursill CA (2012) The role of cholesterol efflux in mechanisms of endothelial protection by HDL. Curr Opin Lipidol 23: 182–189. [DOI] [PubMed] [Google Scholar]
- 40. Hermann F, Enseleit F, Spieker LE, Periat D, Sudano I, et al. (2009) Cholesterylestertransfer protein inhibition and endothelial function in type II hyperlipidemia. Thromb Res 123: 460–465. [DOI] [PubMed] [Google Scholar]
- 41. Warnholtz A, Wild P, Ostad MA, Elsner V, Stieber F, et al. (2009) Effects of oral niacin on endothelial dysfunction in patients with coronary artery disease: results of the randomized, double-blind, placebo-controlled INEF study. Atherosclerosis 204: 216–221. [DOI] [PubMed] [Google Scholar]
- 42.Forstermann U, Sessa WC (2012) Nitric oxide synthases: regulation and function. Eur Heart J 33: 829–837, 837a–837d. [DOI] [PMC free article] [PubMed]
- 43. Luscher TF, Taddei S, Kaski JC, Jukema JW, Kallend D, et al. (2012) Vascular effects and safety of dalcetrapib in patients with or at risk of coronary heart disease: the dal-VESSEL randomized clinical trial. Eur Heart J 33: 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rosenson RS (1993) Myocardial injury: the acute phase response and lipoprotein metabolism. J Am Coll Cardiol 22: 933–940. [DOI] [PubMed] [Google Scholar]
- 45. Ryder RE, Hayes TM, Mulligan IP, Kingswood JC, Williams S, et al. (1984) How soon after myocardial infarction should plasma lipid values be assessed? Br Med J (Clin Res Ed) 289: 1651–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng C, Aikawa M (2012) High-density lipoproteins: from function to therapy. J Am Coll Cardiol 60: 2380–2383. [DOI] [PubMed] [Google Scholar]
- 47. Boger GI, Rudolph TK, Maas R, Schwedhelm E, Dumbadze E, et al. (2007) Asymmetric dimethylarginine determines the improvement of endothelium-dependent vasodilation by simvastatin: Effect of combination with oral L-arginine. J Am Coll Cardiol 49: 2274–2282. [DOI] [PubMed] [Google Scholar]