

Abstract
Arginine vasopressin (AVP) is released from the posterior pituitary and controls water homeostasis. AVP binding to vasopressin V2 receptors (V2Rs) located on kidney collecting duct epithelial cells triggers activation of Gs proteins, leading to increased cAMP levels, trafficking of aquaporin-2 water channels, and consequent increased water permeability and antidiuresis. Typically, loss-of-function V2R mutations cause nephrogenic diabetes insipidus (NDI), whereas gain-of-function mutations cause nephrogenic syndrome of inappropriate antidiuresis (NSIAD). Here we provide further characterization of two mutant V2Rs, R181C and M311V, reported to cause complete and partial NDI respectively, together with a V266A variant, in a patient diagnosed with NSIAD. Our data in HEK293FT cells revealed that for cAMP accumulation, AVP was about 500- or 30-fold less potent at the R181C and M311V mutants than at the wild-type receptor respectively (and about 4000- and 60-fold in COS7 cells respectively). However, in contrast to wild type V2R, the R181C mutant failed to increase inositol phosphate production, while with the M311V mutant, AVP exhibited only partial agonism in addition to a 37-fold potency decrease. Similar responses were detected in a BRET assay for β-arrestin recruitment, with the R181C receptor unresponsive to AVP, and partial agonism with a 23-fold decrease in potency observed with M311V in both HEK293FT and COS7 cells. Notably, the V266A V2R appeared functionally identical to the wild-type receptor in all assays tested, including cAMP and inositol phosphate accumulation, β-arrestin interaction, and in a BRET assay of receptor ubiquitination. Each receptor was expressed at comparable levels. Hence, the M311V V2R retains greater activity than the R181C mutant, consistent with the milder phenotype of NDI associated with this mutant. Notably, the R181C mutant appears to be a Gs protein-biased receptor incapable of signaling to inositol phosphate or recruiting β-arrestin. The etiology of NSIAD in the patient with V266A V2R remains unknown.
Introduction
Arginine vasopressin (AVP) is released from the posterior pituitary and controls water balance homeostasis [1]–[3]. AVP, binding to arginine vasopressin V2 receptors (V2R) located on the basolateral membrane of kidney collecting duct epithelial cells, triggers activation of Gs proteins, leading to increased cAMP levels, and activation of Protein Kinase A [1]–[3]. This in turn causes trafficking of aquaporin-2 water channels to the apical membrane of collecting duct cells, resulting in increased water permeability and antidiuresis [1]–[3].
The V2R is a 7-transmembrane spanning G protein-coupled receptor (GPCR) [4], [5]. Typically, loss-of-function (in terms of cAMP signaling) V2R mutations cause nephrogenic diabetes insipidus (NDI), characterized by an inability to concentrate urine resulting in excessive urine production, dehydration and thirst [2], [3]. Administration of exogenous AVP fails to restore normal water balance [2], [3]. In contrast, gain-of-function mutations cause nephrogenic syndrome of inappropriate antidiuresis (NSIAD), characterized by inappropriately concentrated urine resulting in excessive sodium excretion, low plasma sodium concentration, and low serum osmolality [6]. A key feature is the absence of detectable levels of AVP in serum, whereas the closely related syndrome of inappropriate antidiuretic hormone secretion (SIADH) is typically associated with measurably elevated AVP levels and consequent V2R hyperactivity [6], [7].
There are now more than 200 different V2R mutations known to cause NDI [8], but only three that cause NSIAD [6], [9]. This is not surprising considering that many changes to a protein structure can result in impaired function, whereas a single point mutation able to constitutively activate a receptor is much more rare. Interestingly, the two opposing phenotypes of NDI and NSIAD can both result from distinct mutations at R137, which is present within the conserved DRY/H motif of the V2R, a region critical for stabilizing the receptor in either inactive or active conformations [10], [11]. Substitution of R137 with H results in NDI due to an inability of this mutant receptor to stimulate cAMP production [12]. Whether the loss-of-function is primarily due to G protein uncoupling, reduced forward trafficking and/or constitutive internalization is a matter of debate, and it is not inconceivable that all of these aspects of receptor pharmacology play a role in the overall phenotype [13], [14]. In contrast, substitution of the same residue with C or L favors constitutive activation and cAMP production, causing NSIAD [6]. Notably, both R137H and R137C/L mutants recruit β-arrestin constitutively and are constitutively internalized [13]–[16]. In contrast, the NSIAD-causing F229V mutation, although resulting in constitutive cAMP activation, does not result in constitutive β-arrestin interaction. Instead, this receptor is able to recruit β-arrestin in a ligand-dependent manner similar to that observed with wild-type V2R, indicating a degree of biased coupling [9].
Correct protein folding and trafficking to the plasma membrane are critical for V2R function. Indeed, it is thought that the majority of disease-causing mutations of the V2R, especially those associated with NDI, result in significant intracellular retention of the receptor due to a failure to pass quality control systems of the endoplasmic reticulum and early secretory pathway [2], [17], [18].
In this study we further characterize two previously reported mutations of the V2R from patients with NDI, R181C [19]–[25] and M311V [25], and a third potential mutation (V266A) from a patient with a putative diagnosis of NSIAD. V266A has been previously reported in combination with a nonsense mutation (W200X/V266A) in a patient with NDI [26], but mutation of V266A alone by those authors had no effect on cAMP accumulation (Emax or EC50), ELISA studies or radioligand binding. However, inositol phosphate production, β-arrestin interaction and receptor ubiquitination were not assessed. In the more recent case of a patient with symptoms of NSIAD, including hyponatremia and low ADH levels that presented during infancy, no variation in V2R was identified other than V266A. None of the patient's immediate family was clinically affected and as they declined genetic testing, unfortunately it is not known if they also possessed the same variation in V2R.
We have found that for the R181C and M311V mutant receptors, the degree of receptor activity lost (for cAMP, inositol phosphate accumulation and β-arrestin recruitment) correlated with the severity of disease. However, with the assays we have utilized, we could find no functional difference between wild-type and V266A receptor, even for inositol phosphate accumulation, β-arrestin interaction and receptor ubiquitination. Hence the etiology of NSIAD in this patient remains uncertain. Most interestingly, our analysis also revealed that R181C acts as a Gs protein-biased receptor, unable to couple to β-arrestin or stimulate inositol phosphate production, yet retaining some ability to increase intracellular cAMP.
Materials and Methods
Ethics Statement
The R181C and M311V mutations have been published previously [19]–[25]. We consulted the Stanford University Institutional Review Board (IRB) and they stated, in accordance with U.S. Code of Federal Regulations Title 45 part 46, that the V266A case should not be part of a protocol. V2R gene sequencing for the patient with the V266A variant was performed as a diagnostic test in order to guide clinical care and was performed in a CLIA approved clinical laboratory, with the sequence result being reported from the CLIA laboratory. The clinical laboratory also obtained written consent from the patient prior to performing genetic testing. There were no patient samples used in any of the research experiments presented in this manuscript.
Materials
Wild-type and mutant HA-tagged V2R and V2R/Rluc8 cDNA constructs were generated by PCR amplification of receptor cDNA (with appropriate site-directed mutagenesis) and ligated into pcDNA3 containing the corresponding expression vectors, in a manner similar to that described previously [13]. Rluc8 cDNA was kindly provided by Andreas Loening and Sanjiv Gambhir (Stanford University, CA) [27]. Vasopressin receptor 2 (V2R)/Rluc8 was generated as described previously [13], [16]. The β-arrestin2/Venus cDNA construct was prepared previously [13] from pcC2-Venus kindly provided by Atsushi Miyawaki (RIKEN Brain Science Institute, Wako-city, Japan). Ubiquitin K48,63A/Venus was prepared as described [28]. Arginine Vasopressin (AVP) was from Sigma-Aldrich.
Cell Culture and Transfection
HEK293FT (Life Technologies) and COS7 (ATCC) cells were maintained at 37°C in 5% CO2 and Dulbecco's modified Eagle's medium (DMEM) containing 0.3 mg/ml glutamine, 100 IU/ml penicillin and 100 µg/ml streptomycin (Gibco) supplemented with 10% foetal calf serum (FCS). HEK293FT cell medium included 400 µg/ml Geneticin (Gibco). Transient transfections were carried out 24 h after seeding 550,000 HEK293FT or 75,000 COS7 cells per well of a 6-well plate. Genejuice (Novagen) transfection reagent was used according to the manufacturer's instructions. Cells were harvested 24 h later using 0.05% Trypsin-EDTA (Gibco), and re-plated into 96-well plates at a density of 100,000 cells per well (HEK293FT) or 15,000 cells per well (COS7) in phenol red free DMEM containing 5% FCS, 16–18 hours prior to assay.
BRET Assays
HEK293FT or COS7 cells were transiently transfected with cDNA encoding wild-type or mutant V2R fused to Rluc8 (V2R/Rluc8) and β-arrestin2 fused to Venus (β-arrestin2/Venus). In one set of experiments, cells were transfected with V2R/Rluc8 constructs and a mutant ubiquitin (ubiquitin K48,63A) fused to Venus (ubiquitin/Venus). This mutant is reported to have a reduced capacity for poly-ubiquitin chain formation, thereby avoiding the potential for multiple acceptor moieties causing quenching or interference phenomena [29]. HEK293FT cells were transfected with 0.1 µg/well of V2R/Rluc8 and 0.3 µg/well of β-arrestin2/Venus or ubiquitin/Venus, whereas COS7 cells were transfected with 0.2 and 0.6 µg/well, respectively. Cells were harvested as above, plated into white 96 well plates (Nunc) and incubated in serum free, phenol red free DMEM at 37°C, 5% CO2 for 2 h with 30 µM EnduRen (Promega) to ensure substrate equilibrium was reached. BRET measurements were taken at 37°C using the VICTOR Light plate reader with Wallac 1420 software (PerkinElmer). Filtered light emissions were sequentially measured at 400–475 and 520–540 nm. The BRET signal was calculated by subtracting the ratio of 520–540 nm emission over 400–475 nm emission for a vehicle-treated cell sample from the same ratio for a second aliquot of the same cells treated with agonist, as described previously [30], [31]. In this calculation, the vehicle-treated cell sample represents the background, eliminating the requirement for measuring a donor-only control sample [30], [31].
Measurement of Fluorescence
Cells were transfected as above, harvested, and replated into black 96-well plates (PerkinElmer). Fluorescence was measured on an EnVision 2102 multilabel plate reader (PerkinElmer) using a 485/14 excitation filter, 535/25 emission filter and D505 mirror.
Measurement of cyclic adenosine monophosphate (cAMP) production using homogeneous time-resolved fluorescence (HTRF)
Intracellular cAMP levels were measured using a HTRF cAMP dynamic 2 assay kit (CisBio Bioassays, Bagnol sur Ceze, France). Cells were transfected as described above and seeded into white 96-well microplates (Nunc) 24 h prior to assay. Cell media was removed by aspiration and replaced with 40 µl stimulation buffer (0.5 mM IBMX, 5 mM HEPES, 0.1% BSA in Hank's Balanced Salt Solution; Life Technologies) containing agonists as indicated. Cells were incubated for 30 min at 37°C and then lysed by addition of 12.5 µl of the supplied conjugate-lysis buffer containing d2-labeled cAMP, followed by 12.5 µl of conjugate-lysis buffer containing terbium cryptate-labeled anti-cAMP antibody, both reconstituted according to the manufacturer's instructions. Plates were incubated for 1 h at room temperature and time-resolved fluorescence signals were measured at 620 and 665 nm, respectively, 50 µs after excitation at 337 nm using an EnVision 2102 plate reader (PerkinElmer).
Measurement of D-myo-inositol-1-phosphate (IP1) production using homogeneous time-resolved fluorescence (HTRF)
Intracellular IP1 levels were measured using a HTRF IP-One Tb assay kit (CisBio Bioassays, Bagnol sur Ceze, France). Cells were transfected as described above and seeded into white 96-well microplates (Nunc) 24 h prior to assay. Cell media was removed by aspiration and replaced with 40 µl stimulation buffer (as supplied, including 50 mM LiCl) containing agonists as indicated. Cells were incubated for 30 min at 37°C and then lysed by addition of 12.5 µl of the supplied conjugate-lysis buffer containing d2-labeled IP1, followed by 12.5 µl of conjugate-lysis buffer containing terbium cryptate-labeled anti-IP1 antibody, both reconstituted according to the manufacturer's instructions. Plates were incubated for 1 h at room temperature and time-resolved fluorescence signals were measured at 620 and 665 nm, respectively, 50 µs after excitation at 337 nm using an EnVision 2102 plate reader (PerkinElmer).
Quantification of cell surface receptor expression
Cell surface HA-tagged V2Rs were quantified by fluorescence microscopy using an automated system for image acquisition (IN Cell Analyzer 1000; GE Healthcare) and validated algorithms for image analysis [32], [33]. Briefly, COS7 cells were seeded in a 6-well plate (75,000 cells/well) and transiently transfected the next day with 1 µg HA-tagged V2R cDNA. Cells were harvested 24 h later and seeded into black 96-well plates (Greiner; 8000 cells/well in triplicate wells) for 16–18 h prior to staining. Cell surface receptor staining was performed on live intact cells by incubation with primary antibody (rabbit polyclonal anti-HA (Sigma-Aldrich) diluted 1∶1000 in DMEM with 1% BSA) for 1 h at 4°C. Cells were then washed with ice-cold phosphate-buffered saline (PBS), fixed (2% paraformaldehyde in PBS for 30 min), and permeabilized (0.1% Triton X-100 in PBS for 10 min). Cells were washed three times, blocked (1 h in PBS containing 0.1% Triton X-100 and 1% BSA), and incubated for 1 h with secondary antibody (Alexa Fluor 546-conjugated goat anti-rabbit IgG at 1∶400 in PBS with 0.1% Triton X-100 and 1% BSA). Nuclei were stained using Hoechst 33258 (1 µM, 20 min). Images were acquired, collecting four fields per well with a 10× objective and triple band filter set to obtain images of 100–1000 cells (per well) in a total imaged area of 2.4 mm2. Image analysis was performed with IN Cell Workstation 3.7 software (GE Healthcare) using a Dual Area Object Analysis algorithm and a filter to define the proportion of positively stained cells (cell surface HA-V2R staining >10% above background). A cell surface expression index was then calculated by multiplying the proportion of stained cells (% positive cells) by their mean fluorescence intensity in arbitrary fluorescence units, as described [32], [33].
Data Presentation and Statistical Analysis
Data were analyzed using Prism 5 graphing software (GraphPad). Sigmoidal curves were fitted to the dose-response data using non-linear regression. Statistical analysis of pEC50 values was carried out using one-way ANOVA followed by Tukey's multiple comparison post-test.
Results
V2R signaling to cAMP and functional validation of the V2R/Rluc8 fusion protein
The V2R couples to Gs protein, thereby activating adenylate cyclase and increasing intracellular cAMP. Accordingly, we tested the ability of the three mutant receptors to increase cAMP levels using a homogenous time-resolved fluorescence (HTRF) assay (Figure 1). Treatment of transiently transfected HEK293FT cells with AVP caused a robust dose-dependent increase in cAMP (Figure 1A), with particularly high potency for the wild-type and V266A mutant receptors, the pEC50 values of which were not significantly different (Table 1). In contrast, AVP was 29- or 427-fold less potent in cells transfected with M311V or R181C HA-V2R respectively (Table 1). Next, we tested for the ability of Rluc8-tagged V2R to couple to Gs in the same cell line (Figure 1B), as it is important to check that the addition of a BRET tag does not compromise normal receptor function [34], [35]. Again, AVP caused a dose-dependent increase in cAMP, and for each receptor, potency was comparable to that obtained with the HA-tagged V2R (Table 1). Furthermore, the ligand-induced β-arrestin recruitment shown below provides additional validation of V2R/Rluc8 functionality. We also tested whether a different cellular background influenced the effect of the mutations on cAMP signaling. No significant difference in potency was observed between wild-type and V266A mutant receptors expressed in COS7 cells compared to the HEK293FT cell line. Furthermore, a significantly lower (60-fold) AVP potency was again observed with M311V and significantly lower still with R181C (3890-fold lower than wild-type). Indeed, expression of the R181C receptor in COS7 cells resulted in a significantly lower potency of AVP compared to its expression in HEK293FT cells (Table 1).
Figure 1. AVP-induced cAMP accumulation.

Transfected HEK293FT cells (A, B) or COS7 cells (C) expressing either wild-type or mutant HA-tagged V2R (A, C) or Rluc8-tagged V2R (B), were treated for 30 minutes with stimulation buffer containing 0.5 mM IBMX together with the indicated concentrations of AVP or 50 mM forskolin. Following additions, cells were lysed and cAMP accumulation measured by HTRF. Data shown are HTRF signal in arbitrary units (AU) normalized to the forskolin response. Results shown are the mean ± SEM of 3 independent experiments.
Table 1. pEC50 values and fold change in EC50 for AVP-induced cAMP and IP1 accumulation.
| cAMP | IP1 | |||||||
| HEK293FT | COS7 | HEK293FT | ||||||
| HA-V2R | V2R/Rluc8 | HA-V2R | HA-V2R | |||||
| pEC50 | Fold change | pEC50 | Fold change | pEC50 | Fold change | pEC50 | Fold change | |
| wt | 10.57±0.27 | 1.0 | 10.78±0.14 | 1.0 | 10.10±0.17 | 1.0 | 7.82±0.13 | 1.0 |
| R181C | 7.94±0.20* , ** | 427 | 7.93±0.16* , ** | 708 | 6.51±0.07* , ** , *** | 3891 | ND | ND |
| V266A | 10.76±0.06 | 0.6 | 10.84±0.20 | 0.9 | 10.31±0.15 | 0.6 | 7.97±0.13 | 0.7 |
| M311V | 9.11±0.07* | 28.8 | 9.34±0.12* | 27.5 | 8.32±0.25* | 60.3 | 6.25±0.13* | 37.2 |
Transfected HEK293FT or COS7 cells were treated as per Figure 1 or 2. Data shown are the pEC50 values for AVP treatment (30 min) and are the mean ± SEM of 3 or 5 independent experiments for cAMP and IP1 respectively. Fold change from the wild-type receptor EC50 data are given in adjacent columns.
, significantly different to the corresponding wild-type receptor in the same cell line (P<0.001);
, significantly different to M311V in the same cell line (P<0.01 for HA-V2R in HEK293FT and P<0.001 for V2R/Rluc8 in HEK293FT and HA-V2R in COS7);
, significantly different to R181C HA-V2R in HEK293FT cells. ND = Not determined.
Effect of V2R mutation on IP1 accumulation
Although the V2R predominantly signals through Gs, this receptor is also known to couple to the Gq class of G-proteins, activate phospholipase C and increase intracellular Ca2+ [36], [37]. Accordingly, we used a HTRF assay of IP1 accumulation (a downstream metabolite of inositol 1,4,5-trisphosphate; IP3) to test V2R signaling through this pathway (Figure 2). Addition of AVP to transfected HEK293FT cells caused a robust dose-dependent increase in IP1 for the wild-type receptor (Figure 2), albeit with 2–3 orders of magnitude less potency than the cAMP response (Table 1). No significant difference in IP1 accumulation was observed for the V266A receptor compared to wild-type, whereas for the M311V receptor the potency of AVP was markedly reduced (37-fold). Notably AVP failed to increase IP1 levels in cells expressing R181C V2R, even at the highest dose tested (Figure 2).
Figure 2. AVP-induced IP1 accumulation.
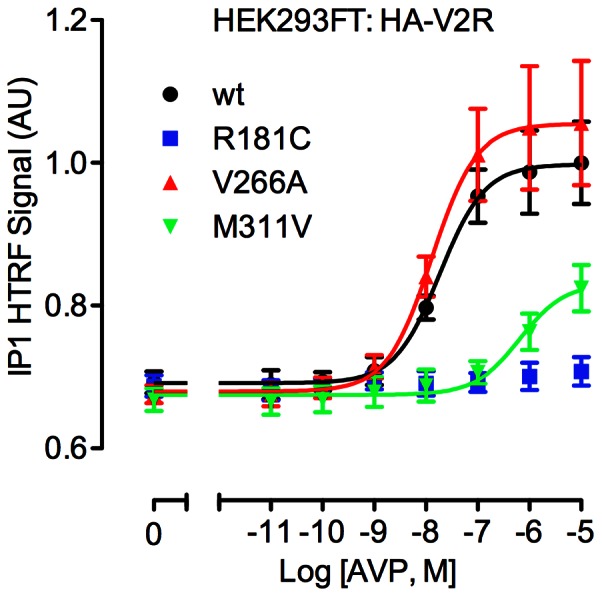
Transfected HEK293FT cells expressing either wild-type or mutant HA-tagged V2R were treated for 30 minutes with the indicated concentrations of AVP. Following additions, cells were lysed and IP1 accumulation measured by HTRF. Data shown are HTRF signal in arbitrary units (AU) normalized to the wild-type receptor response. Results shown are the mean ± SEM of 5 independent experiments.
Effect of V2R mutation on β-arrestin interaction
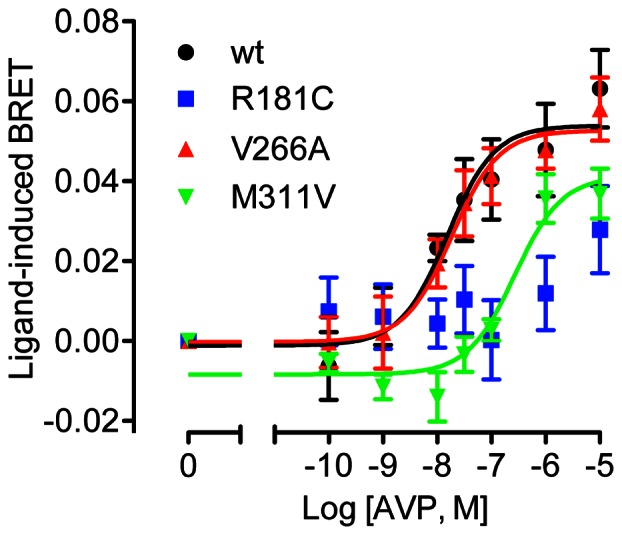
We next examined the ability of V2R/Rluc8 to interact with β-arrestin in response to AVP stimulation, using a previously validated BRET assay [13] to generate real-time kinetic profiles in both HEK293FT and COS7 cells with multiple doses of AVP (Figure 3). AVP caused a robust dose-dependent increase in BRET signal for both wild-type and V266A V2R (Figures 3 and 4, and Table 2), indicative of β-arrestin recruitment to the activated receptor. In contrast, the R181C mutant was unable to recruit β-arrestin at any dose tested, whereas the M311V receptor showed a marked reduction in both the efficacy (∼65% reduction) and potency (23-fold decrease; Table 2) of AVP. The kinetic profile with M311V V2R was also distinct from that of the wild-type receptor (Figure 3), consistent with reduced temporal stability of the receptor-arrestin complex [28]. Importantly, basal BRET ratios for wild-type and mutant V2Rs were comparable (Table 2), indicating that these mutations do not increase coupling to β-arrestin in the absence of ligand.
Figure 3. Effect of V2R mutations on the kinetics of AVP-induced β-arrestin2/Venus recruitment.

Extended BRET kinetic profiles were generated with HEK293FT cells (A, B, C, D) or COS7 cells (E, F, G, H) expressing β-arrestin2/Venus with wild-type, R181C, V266A, or M311V V2R/Rluc8, as indicated. Cells were treated with indicated concentrations of AVP and kinetic BRET responses recorded. Data shown are the mean ± SEM of 3 independent experiments.
Figure 4. Dose-response curves for the effect of V2R mutations on AVP-induced β-arrestin2/Venus recruitment.

BRET dose-response data were generated with HEK293FT cells (A) or COS7 cells (B) expressing β-arrestin2/Venus with wild-type, R181C, V266A, or M311V V2R/Rluc8. Data shown are the ligand-induced BRET response at 60 minutes post AVP treatment, and are the mean ± SEM of 3 independent experiments.
Table 2. pEC50 values, fold change in EC50, and difference in basal BRET ratio compared to wild-type for AVP-induced β-arrestin2/Venus recruitment to V2R/Rluc8.
| HEK293FT | COS7 | HEK293FT | COS7 | |||
| pEC50 | Fold change | pEC50 | Fold change | Difference in basal BRET ratio compared to wild-type | ||
| wt | 8.31±0.21 | 1.0 | 8.54±0.02 | 1.0 | 0.000±0.008 | 0.000±0.009 |
| R181C | ND | ND | ND | ND | 0.014±0.008 | 0.000±0.003 |
| V266A | 8.24±0.19 | 1.2 | 8.45±0.05 | 1.2 | 0.020±0.012 | −0.007±0.006 |
| M311V | 6.96±0.04* | 22.4 | 7.18±0.23* | 22.9 | 0.004±0.015 | 0.014±0.008 |
Transfected HEK293FT or COS7 cells were treated as per Figure 3. Data shown are the pEC50 values for AVP treatment (60 min), fold change from the wild-type EC50, and the BRET ratio for each receptor minus the mean BRET ratio for wild-type receptor (both prior to addition of agonist). Data are the mean ± SEM of 3 independent experiments.
, significantly different to the corresponding wild-type receptor in the same cell line (P<0.001). ND = Not determined.
Comparison of wild-type and mutant V2R cell surface expression
The majority of disease-causing mutations of the V2R exhibit a reduced ability to traffic to the plasma membrane [2]. To test for this possibility, we examined cell surface expression using a previously validated semi-automated imaging assay [32], [33]. COS7 cells expressing wild-type and mutant HA-tagged receptors were stained, initially as live intact cells, and cell surface expression determined by immunofluorescence (Figure 5). As shown, cell surface expression of each of the mutant V2Rs was comparable to wild-type. This, together with the comparable maximal ligand-induced cAMP responses (Figure 1), suggests that these receptors retain their ability to traffic to the plasma membrane. Furthermore, in both HEK293FT and COS7 cells, V2R/Rluc8 luminescence was not significantly different between wild-type and mutant V2Rs, indicating that these mutations also do not compromise overall receptor expression in each cell line (Table 3). Although no difference in expression was observed within each cell line, Rluc8-tagged receptor expression in COS7 cells was significantly lower than in HEK293FT cells (Table 3). This lower expression in COS7 cells is likely to explain the lower absolute cAMP accumulation (Figure 1) and BRET signals (Figures 3 and 4), and may contribute to the lower AVP potency with R181C V2R in COS7 compared to HEK293FT cells (Table 1).
Figure 5. Quantification of HA-V2R cell surface expression by automated imaging.
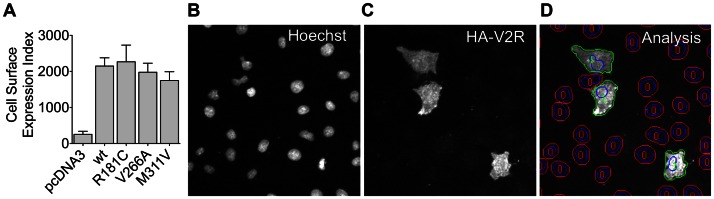
COS7 cells expressing HA-V2R constructs were stained with anti-HA antibody, initially as live intact cells, prior to image acquisition and analysis using an IN Cell Analyzer 1000. Data shown are an Expression Index defined as the percentage of positive cells (>10% above background) multiplied by their mean fluorescence intensity (A), and are the mean ± SEM of 3 experiments. Representative images of nuclei stained with Hoechst 33258 (B), cell surface HA-V2R stain (C), and automated image segmentation (D) are shown. As illustrated in (D), IN Cell Analyzer software was used to define perimeters of nuclei (blue) and cells (green or red) with a filter to distinguish cells in which staining was >10% above background (green perimeters (1)) or <10% above background (red perimeters (0)). Each of the image panels corresponds to a width of 250 µm and represents approximately 1/10th of the area captured in each field of view.
Table 3. Relative V2R/Rluc8 expression in live cells.
| HEK293FT | COS7 | |||
| RLU | % wt | RLU | % wt | |
| wt | 143394±25126 | 100±18 | 8768±3321 | 100±38 |
| R181C | 152886±19465 | 107±14 | 9380±3250 | 107±37 |
| V266A | 148174±22523 | 103±16 | 6379±1601 | 73±18 |
| M311V | 129322±17097 | 90±12 | 5057±1763 | 58±20 |
HEK293FT or COS7 cells were transfected with the indicated V2R/Rluc8 constructs together with β-arrestin2/Venus. Luminescence was measured in triplicate wells after 2 h pre-treatment with 30 mM EnduRen, as part of the extended BRET kinetic assays (Figure 3). Data shown are the mean ± SEM of 3–7 independent experiments. RLU = relative light units. Luminescence intensity in cells not expressing Rluc8 was 30–150 RLU.
Determination of receptor/ubiquitin interaction
The V2R undergoes agonist-induced conjugation of ubiquitin to K268, which regulates receptor degradation [38]. In a final series of experiments, we used a BRET assay of ubiquitin recruitment [29] to investigate the possibility that V266A mutation influences V2R ubiquitination, and thereby contributes to pathogenesis. As expected, AVP increased BRET between wild-type V2R/Rluc8 and ubiquitin/Venus in a dose-dependent manner (pEC50: 7.83±0.14) in HEK293FT cells (Figure 6). Notably, the V266A mutant displayed a very similar AVP dose-response curve (pEC50: 7.72±0.06) compared to the wild-type receptor. In contrast, the AVP potency with M311V V2R was significantly (P<0.001) lower (pEC50: 6.57±0.01) and that with R181C V2R was too low to determine, as the curve did not plateau (Figure 6).
Figure 6. Effect of V2R mutations on AVP-induced ubiquitin/Venus recruitment.
HEK293FT cells expressing ubiquitin/Venus and wild-type or mutant V2R/Rluc8 constructs were stimulated with the indicated concentrations of AVP for 120 min. Data shown are the mean ± SEM of 4 independent experiments.
Discussion
In this study we have characterized the molecular pharmacology of three V2R variants from patients with differing disease phenotypes. Two of the mutants were previously described (R181C [19]–[25], and M311V [25]), and are reported to cause complete and partial NDI respectively [25]. The third variant (V266A) was found in a patient with a diagnosis consistent with NSIAD, including chronic hyponatremia and undetectable ADH levels. Using a number of functional assays we have confirmed that the M311V mutant retains greater activity than R181C V2R, consistent with the milder phenotype of NDI associated with this mutation. In contrast, the V266A V2R appears functionally identical to the wild-type receptor in all aspects of pharmacology tested by us, and others [26]. It is possible that the V266A variation requires additional modifying events, such as compound changes in G protein, β-arrestin or other proteins important for V2R signaling or regulation, for the phenotype to become penetrant. However, we feel it is more likely that the V266A change represents a rare silent polymorphism, with the causative mutation being in another protein such as aquaporin-2. This case underscores the importance of functional testing of genetic variants discovered during clinical investigations.
Of the over 200 mutations of the V2R that have been reported, at least 100 are missense mutations [8]. The majority of these mutated receptors fail to traffic to the cell surface and are retained in the endoplasmic reticulum [2]. In this regard, treatment with membrane permeable pharmacochaperones that are thought to aid in folding and functionally rescue mutant receptors may be of therapeutic benefit [18]. However, a number of V2R mutants are properly targeted to the plasma membrane, but are unable to bind AVP or signal though Gs protein to increase cAMP [2]. In this study, it appears that all three V2R variants were able to adequately traffic to the plasma membrane, as evidenced by HA-V2R staining and maximal cAMP signaling comparable to wild-type in response to AVP treatment. As such, both R181C and M311V mutants appear to be defective in ligand binding or signaling. Indeed, although to our knowledge no high-resolution structure of the V2R exists, 3D homology modeling indicates that both R181 and M311 are key residues for AVP binding [39].
The R181C mutation is present within the second extracellular loop, proximal to the plasma membrane [25]. Prior reports indicate that cysteine residue substitutions within these regions (R181C, G185C, R202C, R203C, Y205C) cause inactivating mutations and NDI [8], [25]. The V2R contains a conserved disulfide bond between C112 of the first extracellular loop and C192 of the second extracellular loop. Hence, any additional cysteine residue may interfere with formation of the conserved disulfide bond and/or introduce different deleterious disulfide bonds [40], [41].
Previous studies also report that the R181C mutant correctly traffics to the plasma membrane but has a markedly reduced capacity for AVP binding, and reduced AVP-induced cAMP production, with an approximate 1000-fold [20], 860-fold [25], or 100-fold [22] shift in potency. We observed similarly dramatic decreases in potency for cAMP accumulation in the two cell lines tested. However, the previous reports did not investigate IP1 accumulation or β-arrestin coupling for this mutant, and although high doses of AVP caused maximal cAMP responses (Figure 1), no IP1 accumulation was measured at any dose tested in HEK293FT cells (Figure 2). Similarly, no ligand-induced interaction between R181C V2R and β-arrestin was observed at any dose in either HEK293FT or COS7 cells (Figures 3 and 4). Consequently, we suggest that R181C V2R may be a Gs protein-biased receptor, albeit one for which AVP exhibits low potency compared to wild-type V2R for Gs coupling. Furthermore, with the M311V V2R, partial agonism was apparent for IP1 accumulation and β-arrestin recruitment not just a lower potency as for cAMP signaling. Although, the wild-type receptor itself exhibits approximately 2–3 orders of magnitude lower potency for AVP when signaling to IP1 and recruiting β-arrestin (pEC50 7.82 and 8.31 respectively; Table 1 and 2) compared to cAMP signaling (pEC50 10.57), if the R181C receptor maintained the shift observed in the cAMP assay (427-fold difference from wild-type in HEK293FT cells) one would expect to detect clear IP1 and β-arrestin responses at the highest doses of AVP tested, with theoretical pEC50s of approximately 5.2 and 5.7 for IP1 and β-arrestin respectively. However, no such responses are seen, suggesting a degree of Gs protein bias. Importantly as well, the potency shift for the M311V mutant is similar for IP1 and β-arrestin compared to cAMP, indicating a lack of bias in terms of potency for M311V, in contrast to R181C.
Various GPCRs have been engineered to induce signaling bias, including the dopamine D2 receptor [42], β2 adrenergic receptor [43], and designer receptors exclusively activated by designer drugs (DREADDs) [44], [45]. However naturally occurring mutations resulting in signaling bias are rare in the literature [46], particularly when the bias is towards G protein signaling. Notably, a Gs-biased thyrotropin receptor mutant has been reported [47]. This mutant is correctly expressed at the cell surface, able to increase intracellular cAMP, but markedly defective in inositol phosphate generation, resulting in an imbalance between iodide trapping (Gs-dependent) and iodide organification (Gq-dependent) [47]. Furthermore, a comprehensive examination of signaling-biased mutants was recently completed for the calcium-sensing receptor [48]. A number of biased synthetic ligands have been reported for the V2R [49], and protean agonism [50] by non-peptide antagonists was recently revealed using two V2R mutants (Ser-333del and Y128S) [51], highlighting the diverse pharmacology arising from different conformational states of the V2R.
As a result of the likely structural changes in disulfide bond formation and therefore receptor configuration discussed above, we hypothesize that the R181C mutation stabilizes a conformation even less suitable for Gq-coupling and β-arrestin interaction than Gs-coupling. This bias in ligand-induced receptor function in the absence of notable constitutive activity differs from the bias seen by Carpentier et al. with the F229V mutation, which resulted in constitutive cAMP signaling without constitutive β-arrestin recruitment, the latter function being similar to wild-type [9]. Collectively, these findings illustrate that alterations in receptor conformation due to mutation can have different effects depending on the function assessed, thus highlighting the value of comprehensively profiling multiple functions.
Following agonist stimulation the human V2R is ubiquitinated at K268, which mediates agonist-induced receptor degradation [38]. Hence we used a BRET-based assay of receptor-ubiquitin interaction to test our mutant receptors for defects in this pathway. For ubiquitin recruitment to R181C and M311V, the rank order of potency for AVP was comparable to the data obtained for intracellular cAMP accumulation (Figure 1). Interestingly, R181C retained some ability to recruit ubiquitin to the activated receptor, despite an earlier report that β-arrestin is required for this interaction [38]. Accordingly, we suggest that alternative mechanisms may be in place, or that a very weak/transient interaction of β-arrestin may suffice. Note that as both receptor-arrestin and receptor-ubiquitin assays utilized the same assay platform, it seems unlikely that our ability to detect R181C V2R ubiquitination but not β-arrestin recruitment is due to assay sensitivity.
Notably, the V266A mutation had no effect on ubiquitin interaction, in addition to wild-type-like cAMP and β-arrestin responses. The V266A variant has been previously reported in combination with a missense mutation (W200X/V266A) in a patient with NDI [26]. Here the authors also concluded that the V266A mutation has no functional consequence (as determined by radioligand binding, cAMP assay and cell surface receptor expression by ELISA), with the W200X truncation being the primary cause of NDI in that case [26]. The lack of any observable effect, shown herein and by Schulz et al, suggests that the V266A mutation may be a genetic polymorphism; one of 21 such AVPR2 gene variants reported [8]. It is important to note that the patient herein has been diagnosed with NSIAD, not NDI as in the prior study, although presumably the W200X truncation dominates over the V266A mutation when combined. Typically, NSIAD results from constitutive V2R activity (e.g. as shown for F229V [9] and R137C/L [6], [13]). However, here we find no evidence of constitutive activity. Hence, more detailed genetic studies are required to determine the precise cause of NSIAD in this patient, a starting point being aquaporin-2.
Interestingly, Zhong and colleagues engineered a V266K mutant V2R, in order to investigate potential Gq coupling properties, as a K residue at this position in the oxytocin receptor is important for IP3 generation [52]. Again, the authors report that this mutation does not influence AVP binding affinity or cAMP production. A small change (1.5 fold) in IP3 generation was reported but the authors conclude that is negligible when compared to the large (4 fold) change observed when the V2R is engineered to include the entire oxytocin receptor third intracellular loop region [52]. Notably, we found no significant difference in IP1 accumulation for the V266A mutant (Figure 2).
In conclusion, we could find no functional difference between wild-type and V266A V2R, hence the etiology of NSIAD in this patient remains unknown. We found that the M311V mutant V2R retains greater activity than R181C V2R, with respect to β-arrestin recruitment and receptor ubiquitination as well as cAMP and IP1 production, consistent with the milder phenotype of NDI associated with this mutant. Furthermore, our analysis of the R181C V2R suggests that it is a Gs protein-biased receptor. As recently discussed by Carpentier at al. [9], distinct mutations can result in distinct effects on different aspects of receptor pharmacology, and we have illustrated this in terms of both partial (M311V) and biased (R181C) agonism. Importantly, a clearer understanding of the effects of clinical mutations at the molecular pharmacological level can inform both drug discovery and patient treatment, with the tailoring of pharmaceuticals to individuals based on their receptor signaling profiles.
Funding Statement
This research was funded by the Australian Research Council (www.arc.gov.au) Future Fellowship (FT100100271) and a Priming Grant from the Raine Medical Research Foundation (www.raine.uwa.edu.au), both awarded to KDGP. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ball SG (2007) Vasopressin and disorders of water balance: the physiology and pathophysiology of vasopressin. Ann Clin Biochem 44: 417–431. [DOI] [PubMed] [Google Scholar]
- 2. Morello J-P, Bichet DG (2001) Nephrogenic Diabetes Insipidus. Annu Rev Physiol 63: 607–630. [DOI] [PubMed] [Google Scholar]
- 3. Sands JM, Bichet DG (2006) Nephrogenic Diabetes Insipidus. Ann Intern Med 144: 186–194. [DOI] [PubMed] [Google Scholar]
- 4. Lolait SJ, O'Carroll A-M, McBride OW, Konig M, Morel A, et al. (1992) Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature 357: 336–339. [DOI] [PubMed] [Google Scholar]
- 5. Birnbaumer M, Seibold A, Gilbert S, Ishido M, Barberis C, et al. (1992) Molecular cloning of the receptor for human antidiuretic hormone. Nature 357: 333–335. [DOI] [PubMed] [Google Scholar]
- 6. Feldman BJ, Rosenthal SM, Vargas GA, Fenwick RG, Huang EA, et al. (2005) Nephrogenic syndrome of inappropriate antidiuresis. N Engl J Med 352: 1884–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baylis PH (2003) The syndrome of inappropriate antidiuretic hormone secretion. Int J Biochem Cell Biol 35: 1495–1499. [DOI] [PubMed] [Google Scholar]
- 8. Spanakis E, Milord E, Gragnoli C (2008) AVPR2 variants and mutations in nephrogenic diabetes insipidus: Review and missense mutation significance. J Cell Physiol 217: 605–617. [DOI] [PubMed] [Google Scholar]
- 9. Carpentier E, Greenbaum LA, Rochdi D, Abrol R, Goddard WA, et al. (2012) Identification and Characterization of an Activating F229V Substitution in the V2 Vasopressin Receptor in an Infant with NSIAD. J Am Soc Nephrol 23: 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rovati GE, Capra V, Neubig RR (2007) The Highly Conserved DRY Motif of Class A G Protein-Coupled Receptors: Beyond the Ground State. Mol Pharmacol 71: 959–964. [DOI] [PubMed] [Google Scholar]
- 11. Audet M, Bouvier M (2012) Restructuring G-Protein- Coupled Receptor Activation. Cell 151: 14–23. [DOI] [PubMed] [Google Scholar]
- 12. Rosenthal W, Antaramian A, Gilbert S, Birnbaumer M (1993) Nephrogenic diabetes insipidus. A V2 vasopressin receptor unable to stimulate adenylyl cyclase. J Biol Chem 268: 13030–13033. [PubMed] [Google Scholar]
- 13. Kocan M, See HB, Sampaio NG, Eidne KA, Feldman BJ, et al. (2009) Agonist-Independent Interactions between β-Arrestins and Mutant Vasopressin Type II Receptors Associated with Nephrogenic Syndrome of Inappropriate Antidiuresis. Mol Endocrinol 23: 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rochdi MD, Vargas GA, Carpentier E, Oligny-Longpré G, Chen S, et al. (2010) Functional Characterization of Vasopressin Type 2 Receptor Substitutions (R137H/C/L) Leading to Nephrogenic Diabetes Insipidus and Nephrogenic Syndrome of Inappropriate Antidiuresis: Implications for Treatments. Mol Pharmacol 77: 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tenenbaum J, Ayoub MA, Perkovska S, Adra-Delenne A-L, Mendre C, et al. (2009) The Constitutively Active V2 Receptor Mutants Conferring NSIAD Are Weakly Sensitive to Agonist and Antagonist Regulation. PLoS ONE 4: e8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kocan M, Dalrymple M, Seeber R, Feldman B, Pfleger K (2011) Enhanced BRET technology for the monitoring of agonist-induced and agonist-independent interactions between GPCRs and β-arrestins. Front Endocrinol 1: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hermosilla R, Oueslati M, Donalies U, Schönenberger E, Krause E, et al. (2004) Disease-causing V2 Vasopressin Receptors are Retained in Different Compartments of the Early Secretory Pathway. Traffic 5: 993–1005. [DOI] [PubMed] [Google Scholar]
- 18. Bernier V, Morello J-P, Zarruk A, Debrand N, Salahpour A, et al. (2006) Pharmacologic Chaperones as a Potential Treatment for X-Linked Nephrogenic Diabetes Insipidus. J Am Soc Nephrol 17: 232–243. [DOI] [PubMed] [Google Scholar]
- 19. Pan Y, Metzenberg A, Das S, Jing B, Gitschier J (1992) Mutations in the V2 vasopressin receptor gene are associated with X-linked nephrogenic diabetes insipidus. Nat Genet 2: 103–106. [DOI] [PubMed] [Google Scholar]
- 20. Pan Y, Wilson P, Gitschier J (1994) The effect of eight V2 vasopressin receptor mutations on stimulation of adenylyl cyclase and binding to vasopressin. J Biol Chem 269: 31933–31937. [PubMed] [Google Scholar]
- 21. Bichet DG, Birnbaumer M, Lonergan M, Arthus MF, Rosenthal W, et al. (1994) Nature and recurrence of AVPR2 mutations in X-linked nephrogenic diabetes insipidus. Am J Hum Genet 55: 278–286. [PMC free article] [PubMed] [Google Scholar]
- 22. Schöneberg T, Schulz A, Biebermann H, Grüters A, Grimm T, et al. (1998) V2 vasopressin receptor dysfunction in nephrogenic diabetes insipidus caused by different molecular mechanisms. Hum Mutat 12: 196–205. [DOI] [PubMed] [Google Scholar]
- 23. Wildin RS, Cogdell DE, Valadez V (1998) AVPR2 variants and V2 vasopressin receptor function in nephrogenic diabetes insipidus. Kidney Int 54: 1909–1922. [DOI] [PubMed] [Google Scholar]
- 24. Knoers NVAM, van den Ouweland AMW, Verdijk M, Monnens LAH, van Oost BA (1994) Inheritance of mutations in the V2 receptor gene in thirteen families with nephrogenic diabetes insipidus. Kidney Int 46: 170–176. [DOI] [PubMed] [Google Scholar]
- 25. Sahakitrungruang T, Tee MK, Rattanachartnarong N, Shotelersuk V, Suphapeetiporn K, et al. (2010) Functional Characterization of Vasopressin Receptor 2 Mutations Causing Partial and Complete Congenital Nephrogenic Diabetes Insipidus in Thai Families. Horm Res Paediatr 73: 349–354. [DOI] [PubMed] [Google Scholar]
- 26. Schulz A, Sangkuhl K, Lennert T, Wigger M, Price DA, et al. (2002) Aminoglycoside Pretreatment Partially Restores the Function of Truncated V2 Vasopressin Receptors Found in Patients with Nephrogenic Diabetes Insipidus. J Clin Endocrinol Metab 87: 5247–5257. [DOI] [PubMed] [Google Scholar]
- 27. Loening AM, Fenn TD, Wu AM, Gambhir SS (2006) Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng Des Sel 19: 391–400. [DOI] [PubMed] [Google Scholar]
- 28. Dalrymple MB, Jaeger WC, Eidne KA, Pfleger KDG (2011) Temporal Profiling of Orexin Receptor-Arrestin-Ubiquitin Complexes Reveals Differences between Receptor Subtypes. J Biol Chem 286: 16726–16733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perroy J, Pontier S, Charest PG, Aubry M, Bouvier M (2004) Real-time monitoring of ubiquitination in living cells by BRET. Nat Meth 1: 203–208. [DOI] [PubMed] [Google Scholar]
- 30. Mustafa S, See HB, Seeber RM, Armstrong SP, White CW, et al. (2012) Identification and profiling of a novel α1A-adrenoceptor-CXC chemokine receptor 2 heteromer. J Biol Chem 287: 12952–12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pfleger KDG, Seeber RM, Eidne KA (2006) Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat Protocols 1: 337–345. [DOI] [PubMed] [Google Scholar]
- 32. Armstrong SP, Caunt CJ, Finch AR, McArdle CA (2011) Using automated imaging to interrogate gonadotrophin-releasing hormone receptor trafficking and function. Mol Cell Endocrinol 331: 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Finch AR, Caunt CJ, Armstrong SP, McArdle CA (2010) Plasma Membrane Expression of Gonadotropin-Releasing Hormone Receptors: Regulation by Peptide and Nonpeptide Antagonists. Mol Endocrinol 24: 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pfleger KDG, Dromey JR, Dalrymple MB, Lim EML, Thomas WG, et al. (2006) Extended bioluminescence resonance energy transfer (eBRET) for monitoring prolonged protein–protein interactions in live cells. Cell Signal 18: 1664–1670. [DOI] [PubMed] [Google Scholar]
- 35. Pfleger KDG, Eidne KA (2006) Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat Meth 3: 165–174. [DOI] [PubMed] [Google Scholar]
- 36. Balasubramanian L, Sham JSK, Yip KP (2008) Calcium signaling in vasopressin-induced aquaporin-2 trafficking. Pflug Arch Eur J Phy 456: 747–754. [DOI] [PubMed] [Google Scholar]
- 37. Zhu X, Birnbaumer L (1996) G protein subunits and the stimulation of phospholipase C by Gs-and Gi-coupled receptors: Lack of receptor selectivity of Galpha(16) and evidence for a synergic interaction between Gbeta gamma and the alpha subunit of a receptor activated G protein. Proc Natl Acad Sci U S A 93: 2827–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martin NP, Lefkowitz RJ, Shenoy SK (2003) Regulation of V2 Vasopressin Receptor Degradation by Agonist-promoted Ubiquitination. J Biol Chem 278: 45954–45959. [DOI] [PubMed] [Google Scholar]
- 39. Ślusarz MJ, Giełdoń A, Ślusarz R, Ciarkowski J (2006) Analysis of interactions responsible for vasopressin binding to human neurohypophyseal hormone receptors—molecular dynamics study of the activated receptor–vasopressin–Gα systems. J Pept Sci 12: 180–189. [DOI] [PubMed] [Google Scholar]
- 40. Schülein R, Zühlke K, Krause G, Rosenthal W (2001) Functional Rescue of the Nephrogenic Diabetes Insipidus-causing Vasopressin V2 Receptor Mutants G185C and R202C by a Second Site Suppressor Mutation. J Biol Chem 276: 8384–8392. [DOI] [PubMed] [Google Scholar]
- 41. Schulz A, Grosse R, Schultz G, Gudermann T, Schöneberg T (2000) Structural Implication for Receptor Oligomerization from Functional Reconstitution Studies of Mutant V2 Vasopressin Receptors. J Biol Chem 275: 2381–2389. [DOI] [PubMed] [Google Scholar]
- 42. Lan H, Liu Y, Bell MI, Gurevich VV, Neve KA (2009) A Dopamine D2 Receptor Mutant Capable of G Protein-Mediated Signaling but Deficient in Arrestin Binding. Mol Pharmacol 75: 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, et al. (2006) β-Arrestin-dependent, G Protein-independent ERK1/2 Activation by the β2 Adrenergic Receptor. J Biol Chem 281: 1261–1273. [DOI] [PubMed] [Google Scholar]
- 44. Nakajima K, Wess J (2012) Design and Functional Characterization of a Novel, Arrestin-Biased Designer G Protein-Coupled Receptor. Mol Pharmacol 82: 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang WC, Ng JK, Nguyen T, Pellissier L, Claeysen S, et al. (2007) Modifying Ligand-Induced and Constitutive Signaling of the Human 5-HT4 Receptor. PLoS ONE 2: e1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vassart G, Costagliola S (2011) G protein-coupled receptors: mutations and endocrine diseases. Nat Rev Endocrinol 7: 362–372. [DOI] [PubMed] [Google Scholar]
- 47. Grasberger H, Van Sande J, Hag-Dahood Mahameed A, Tenenbaum-Rakover Y, Refetoff S (2007) A Familial Thyrotropin (TSH) Receptor Mutation Provides in Vivo Evidence that the Inositol Phosphates/Ca2+ Cascade Mediates TSH Action on Thyroid Hormone Synthesis. J Clin Endocrinol Metab 92: 2816–2820. [DOI] [PubMed] [Google Scholar]
- 48. Leach K, Wen A, Davey AE, Sexton PM, Conigrave AD, et al. (2012) Identification of Molecular Phenotypes and Biased Signaling Induced by Naturally Occurring Mutations of the Human Calcium-Sensing Receptor. Endocrinology 153: 4304–4316. [DOI] [PubMed] [Google Scholar]
- 49. Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, et al. (2012) Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A 109: 6733–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kenakin T (2001) Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J 15: 598–611. [DOI] [PubMed] [Google Scholar]
- 51. Takahashi K, Makita N, Manaka K, Hisano M, Akioka Y, et al. (2012) V2 Vasopressin Receptor (V2R) Mutations in Partial Nephrogenic Diabetes Insipidus Highlight Protean Agonism of V2R Antagonists. J Biol Chem 287: 2099–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhong M, Parish B, Murtazina DA, Ku C-Y, Sanborn BM (2007) Amino acids in the COOH-terminal region of the oxytocin receptor third intracellular domain are important for receptor function. Am J Physiol Endocrinol Metab 292: E977–E984. [DOI] [PubMed] [Google Scholar]