

Abstract
Selenoprotein S (SelS, VIMP) is an intrinsically disordered membrane enzyme that provides protection against reactive oxidative species. SelS is a member of the endoplasmic reticulum associated protein degradation pathway but its precise enzymatic function is unknown. Since it contains the rare amino acid selenocysteine, it belongs to the family of selenoproteins, which are typically oxidoreductases. Its exact enzymatic function is key to understanding how the cell regulates the response to oxidative stress and thus influences human health and aging. In order to identify its enzymatic function, we have isolated the selenocysteine-containing enzyme by relying on the aggregation of forms that do not have this reactive residue. That allows us to establish that SelS is primarily a thioredoxin-dependent reductase. It is capable of reducing hydrogen peroxide but is not an efficient or broad-spectrum peroxidase. Only the selenocysteine-containing enzyme is active. In addition, the reduction potential of SelS was determined to be −234 mV using electrospray ionization mass spectrometry. This value agrees with SelS being a partner of thioredoxin. Based on this information, SelS can directly combat reactive oxygen species but is also likely to participate in a signaling pathway, via a yet unidentified substrate.
INTRODUCTION
Selenoprotein S (SelS) belongs to a family of human enzymes that contains the genetically encoded amino acid selenocysteine (Sec).1, 2 Selenoproteins typically act as oxidoreductases in redox regulation and the management of oxidative species and SelS belong to a eukaryotic protein family that is related to redox stress.3 Accumulating evidence suggests that it is involved in inflammation and management of oxidative stress.4, 5 It was identified as a member of the ER-assisted protein degradation (ERAD) machinery6, 7 - a pathway responsible for transporting misfolded proteins from the ER to the cytoplasm for degradation by the proteasome.8
This work focuses on the enzymatic function of human SelS (also known by the alternative names SEPS1, Tanis, VIMP and SELENOS). SelS is a single-pass transmembrane protein9 with a short segment in the ER lumen and an extended cytoplasmic region.10 The cytoplasmic segment contains a disordered segment (residues 123–189 out of 189) that includes the Sec at position 188. The Sec forms a selenenylsulfide bond with a nearby Cys (Cys174).11 SelS was shown to dimerize through a coiled-coil region and contains a valosin-containing protein (VCP, p97) interacting motif.11, 12 It mediates the interactions of the ERAD component Derlin-1 with p97, an AAA ATPase that pulls the protein targets to the cytoplasm, where they are broken down by the proteasome.13 In addition to p97, SelS also interacts with Derlin 1 and 2, components of the putative ERAD channel6; selenoprotein K (SelK), a Sec-containing enzyme with unknown function3; the VCP accessory protein ubiquitin conjugation factor E4 A (UBE4A), a contributor to multiubiquitin chain extension14; the UBX domain–containing protein 8 (UBXD8), a sensor for unsaturated fatty acids and regulator of triglyceride synthesis15 that together with derlin-1 controls degradation of lipidated apolipoprotein B-10016; the UBX domain–containing protein 6 (UBXD6, Rep), which tethers p97 to the ER membrane17 and the kelch containing protein 2 (KLHDC2, HCLP-1), a regulator of the LZIP transcription factor.18 As mentioned above, these interactions take place in the cytoplasm, in the context of the ERAD complex. However, the actual function of SelS in the ERAD machinery and its relevance to management of oxidative stress is unknown.
Previously, the cytosolic segment of a SelS U188C mutant (cSelS U188C) was characterized by NMR spectroscopy and biochemical methods.11 It was found that SelS’ C-terminal domain is unstructured, classifying it as an intrinsically disordered protein. This is a class of proteins that are rich in charged and polar residues and typically adopt a stable tertiary structure only in the presence of their protein partner.19 The enzymatic activity of cSelS U188C, however, was not characterized. This information is necessary to tie together not only the role of SelS in the ERAD machinery but also its strong relation to redox stress. Here, we set out to characterize in vitro the enzymatic activity of human SelS that contains its native Sec.
Characterization of the Sec-containing form requires incorporation of Sec at position 188. However, the specific incorporation of selenoproteins is typically the bottleneck in their characterization. Selenoproteins are produced in vivo by synthesizing Sec on its dedicated tRNA. The Sec codon, which is shared with the opal stop codon, UGA, is recognized by the dual action of a mRNA loop termed the SEC Insertion Sequence (SECIS) and ancillary proteins.20 A major challenge for efficient production of selenoproteins is that protein translation is prematurely terminated at the UGA codon due to its misreading as a stop codon. Hence, heterologous expression contains a high ratio of truncated protein. For SelS, the separation of the truncated and full-length Sec-containing forms, which differ by only two amino acids, is complicated by the strong dimerization of truncated and full forms of the protein. To construct a purification strategy that would isolate the full-length, Sec-containing form, we developed an expression system that allows high yield preparation of both the full-length and cytoplasmic portions of SelS (cSelS).21 E. coli selenium expression machinery was employed to insert a genetically encoded Sec at the active site. We have purified the full-length cSelS by relying on the tendency of the truncated protein to aggregate in the absence of reducing agents. With a sample enriched with the native Sec-containing protein at hand, we demonstrate that only the Sec-containing enzyme has reductase and peroxidase activities in vitro.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, and Chemical Reagents
Enzymes used for molecular biology were acquired from New England Biolabs (Ipswich, MA). The pMHTDelta238 plasmid expressing Tobacco Etch Virus (TEV) protease fused to the cytoplasmic maltose binding protein (cMBP)22 was purchased from the Protein Structure Initiative: Biology Materials Repository.23 Chromatography media was supplied by GE Healthcare Bio-Sciences Corporation (Pittsburgh, PA) and New England Biolabs. The expression construct of human thioredoxin 1 (hTrx) was generously provided by Prof. Marletta.24 Human protein disulfide isomerase (hPDI) was a gift from Prof. Thorpe.25 The expression construct for human glutaredoxin 1 (hGrx) was a gift from Prof. Mieyal.26 15S- hydroperoxy- 5Z, 8Z, 11Z, 13E- eicosatetraenoic acid was from Cayman (Ann Arbor, MI). Bovine insulin was from Cell Application (San Diego, CA). Rat thioredoxin reductase 1 (rTrxR) was from Cayman (recombinant form) or Sigma (purified from rat liver). The pSUABC plasmid was generously provided by Prof. Arner from the Karolinska Institutet.27 All other chemicals and reagents were supplied by Sigma-Aldrich (St. Louis, MO), Acros Organics (Geel, Belgium) and GoldBio (St. Louis, MO). All reagents and solvents were at least analytical grade and were used as supplied.
Cloning
Homo sapiens SelS gene (GenBank accession no. GI: 45439348) was codon optimized for expression in E. coli and the gene synthesized by GeneScript (Piscataway, NJ). The cytoplasmic portion of the gene (residues 52–189, abbreviate as cSelS) was cloned into a pMAL-C5X (New England Biolabs) as a fusion with the cytoplasmic maltose binding protein (cMBP). To enable selenocysteine insertion, a non-encoding E. coli formate dehydrogenase SECIS element was inserted immediately after the stop codon.28 A short linker NSSS and a TEV protease cleavage site, ENLYFQS, was used to connect the two proteins. Following cleavage with TEV protease, no non-native residues were retained in cSelS.
Expression and Purification of cSelS
For protein expression of cSelS mutants (cSelS U188C and cSelS U188S), the plasmids were transformed into an E. coli BL21(DE3) strain. Cells were grown in LB, supplemented with 0.2% glucose at 37 °C, with good aeration and the relevant antibiotic selection (100 μg/ml). When the Optical Density (OD) at 600 nm reached 0.5, the temperature was lowered to 18 °C, and the cells were allowed to shake at the lower temperature for an additional hour. Protein expression was induced with 0.5 mM isopropyl-1-thio-b-Dgalactopyranoside (IPTG) when the optical density (OD) at 600 nm reached 0.7. The cells were harvested after 18–20 h, and the cell paste (7 g/L) was resuspended in 50 mM sodium phosphate, 200 mM sodium chloride, pH 7.5 (amylose buffer), supplemented with 0.5 mM benziamidine, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1 mM ethylenediaminetetraacetic acid (EDTA). Cells were lysed using a high-pressure homogenizer (EmulsiFlex-C5, Avestin, Ottawa, Canada) on ice, and all subsequent procedures were carried out at 4 °C. Cell debris was removed by centrifugation at 20000 g for 1 h. The supernatant was loaded on an amylose column, and the column was washed with the amylose buffer. The fusion cMBP-cSelS was eluted using an amylose buffer containing 20 mM maltose. The purity of the eluted cMBP-cSelS was about 90%. Cleavage of the fusion partner cMBP was carried out by adding a hexahistidine-tagged TEV protease to the dialysis bag for 12 h. The TEV protease was added at a molar ratio of 1:10 relative to the fusion protein. Following cleavage, the protein was then loaded on a 5-ml HiTrap SP HP and eluted with a salt gradient between 200 and 1000 mM sodium chloride, over 20 column volumes. The fractions containing SelS were concentrated to 20 mg/ml, loaded on a sephacryl S-100 column (GE Healthcare) and eluted at 0.4 ml/min with 50 mM sodium phosphate and 200 mM sodium chloride (pH 7.5). The column was calibrated using GE Healthcare Bio-Sciences gel filtration protein standards: bovine serum albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen A (25 kDa), and ribonuclease A (13.7 kDa). The void volume was measured using blue dextran 2000. Protein purity, as determined by 16% Tris-tricine SDS-PAGE gel was higher than 99%. Protein concentration was determined using an extinction coefficient of 1415 M−1 CM−1 for cSelS U188C and 13980 M−1 CM−1 for both cSelS U188S and cSelS 188Δ. The successful incorporation of selenium was confirmed by mass and inductively coupled plasma (ICP) spectroscopy.
For protein expression of the cSelS and cSelS 188Δ, which was generated by mutating UGA to UAA, the plasmid was cotransformed into E. coli BL21(DE3), along with the pSUABC plasmid expressing E. coli SelA, SelB, and SelC under the control of their endogenous promoters.27 Cells were grown in Studier’s MDAG defined media at 37 °C,29 with good aeration and the relevant antibiotic selection (100 μg/ml ampicillin + 34 μg/ml chloramphenicol). When the OD at 600 nm reached 2.4, the temperature was lowered to 18 °C, and the cells were allowed to shake at the lower temperature for an additional hour. Protein expression was induced with 0.5 mM IPTG. At induction, 5 μM Na2SeO3 and 100 μg/ml L-cysteine were added to the media. The remaining steps are identical to those described above. It was possible to distinguish two well-separated peaks on the HiTrap SP HP column, eluting at 400 mM sodium chloride and 600 mM sodium chloride. The latter contains aggregates and was not included in further purifications. The fraction eluted at lower ionic strength was concentrated to 20 mg/ml and further purified on a sephacryl S-100 column. The dimeric form of the protein was separated from other fractions and assayed for activity. Other fractions also contain full-length protein but at a much lower ratio (less than 1%) compared to the truncated form. The percent of selenium incorporation in samples was measured by ICP spectroscopy. The percent of selenium incorporation in all batches used for activity assays was at least 50%.
Inductively Coupled Plasma (ICP) Spectrometry
ICP spectrometry (Thermo IRIS Intrepid II XSP Dual View) was used to determine the ratio of sulfur and selenium in protein samples. The instrument was calibrated using customized reference standard from AccuStandard (New Haven, CT), containing 2 μg/ml of elemental sulfur and selenium in 2% nitric acid.
Electrospray Ionization Mass Spectroscopy
Mass spectra were obtained using a QTOF Ultima (Waters, MA), operating under positive electrospray ionization (+ESI) mode, connected to an LC-20AD (Shimadzu, Kyoto, Japan). Protein samples were separated from small molecules by reverse phase chromatography on a C4 column (Waters XBridge BEH300), using an acetonitrile gradient from 30–71.4%, with 0.1% formic acid as the mobile phase, in 25 minutes, at a flow rate of 0.2 ml/min at room temperature. Data were acquired from m/z 350 to 2500, at a rate of 1 sec/scan. The obtained spectra were deconvoluted using maximum entropy in MassLynx (Waters, MA).
Circular Dichroism Spectroscopy
CD spectra of SelS were measured by using a J-810 circular dichroism spectropolarimeter (Jasco, Essex, UK) that had been calibrated using camphor sulfonic acid for optical rotation and benzene vapor for wavelength. Far-UV spectra were recorded using a 1 mm path-length cell for the 190–250 nm region at 20 °C. Samples for CD spectroscopy were prepared in 10 mM potassium phosphate (pH 7.5) and 50 mM sodium sulfate. Three accumulation scans were collected for baseline, and eight accumulation scans were taken for each sample.
Measurements of cSelS and cSelS U188C Reduction Potential by ESI mass spectrometry
A 0.5 ml sample, which contained 0.2 μM fully oxidized protein, was dialyzed in buffer containing 50 mM sodium phosphate (pH 7.0), 1 mM EDTA, as well as various ratios of glutathione (GSH) and oxidized glutathione (GSSG) at a total GSH+GSSG concentration of 20 mM. The ratio of GSH/GSSG was used to poise the reduction potential of the buffer. To prevent excessive oxidation, the protein solutions were degassed and subsequently flushed with nitrogen. The samples were incubated at 25 °C for 18 hours to ensure full equilibration. The equilibrated reaction mixtures were quenched by adding ice-cold 100% W/V trichloroacetic acid (TCA) to a final concentration of 20% and were mixed immediately. The quenched reaction mixture was then spun for 10 minutes at 16110 g. The supernatant was decanted and the pellet was washed with 0.25 ml ice-cold acetone twice and spun at 16110 g for 10 minutes at 4 °C after each wash. After the final acetone wash, the pellet was dried by exposing it to air for 10 minutes and was then re-suspended in 50 μl 15 mM iodoacetamide solution. 10–20 μl of the re-suspended sample was used to acquire mass spectrum. Experiments were repeated three times, using two independent preparations, for each protein. Following deconvolution, the fractional ratio of reduced to oxidized protein was determined from the presence of mass increments of 116 Da (transfer of two acetamide groups). The percentage of reduced and alkylated to oxidized protein was determined by integrating the relevant mass spectroscopy peak area in Excel (Microsoft, WA). Identical results were obtained when using N-ethylmaleimide (NEM) for labeling.
The reaction of cSelS with the GSSG/GSH redox pair (reaction 1) and the corresponding equilibrium constant (Keq) is given in Equation 1:
| Reaction 1 |
| (Eq. 1) |
The reduction potential of cSelS at pH 7.0 and 25 °C (E0′ (cSelS)) was then calculated from the Nernst equation (Equation 2):
| (Eq. 2) |
where E0'(GSH/GSSG) = −240 mV (pH 7.0 and 25 °C) 30, R is the gas constant (8.315 J K−1 mol−1), T is the absolute temperature (298 K), n is the number of electrons transferred in the reaction (here n=2), and F is the Faraday constant (9.649×104 C mol−1). Plots and curve fits were made with Origin (OriginLab Corp., Northampton, MA). The error bars in Figure 2 represent the range of measurements; that is, the highest and lowest values recorded in the series. The measurement error was estimated to be 1 mV by fitting extreme curves to the minimal and maximal fractions of reduced protein at each point.
Figure 2.

Determination of reduction potentials of cSelS and cSelS U188C in GSH/GSSG redox buffers. The ratio of reduced and oxidized protein was determined by electrospray ionization mass spectroscopy. (A) A representative electrospray ionization mass spectrum of cSelS following incubation in a redox buffer poised at −232 mV and subsequent alkylation. The panel shows the charge state distribution of multiply charged ions [M+18H]18+ to [M+22H]22+ from m/z 680 to 870. (B) Deconvoluted spectrum. Molecular weights: alkylated cSelS 188Δ 15149 Da, oxidized cSelS 15300 Da and alkylated cSelS 15414 Da. (C) A representative electrospray ionization mass spectrum of cSelS U188C following incubation in a redox buffer poised at −209 mV and subsequent alkylation. The panel shows the charge state distribution of multiply charged ions [M+18H]18+ to [M+22H]22+ from m/z 685 to 870. (D) Deconvoluted spectrum. Molecular weights: oxidized cSelS U188C 15253 Da and alkylated cSelS U188C 15365 Da. Panels E and F display the resulting titration curves for (E) cSelS, reduction potential −234±1 mV; and (F) cSelS U188C, reduction potential −211±1 mV. The fraction of reduced protein is plotted against the buffer redox potential poised by the ratio of GSH to GSSG. The error bars represent the range of measurements (that is, highest and lowest values) among three repetitions, using two independent protein preparations.
Measurements of Reduction Potential by Gel Shift Assay
The same sample preparation described above for ESI mass spectrometry-based assays was employed for gel shift assays and visualization by SDS-PAGE. Following an 18 h incubation in the respective redox buffer, the protein was precipitated with 100% TCA and dried by acetone. The pellet was dissolved in 50 μl 10 mM Methyl-PEG24-Maleimide (MM(PEG)24) solution in 2x non-reducing Tricine-SDSPAGE loading buffer. The samples were boiled for 5 min prior to loading on 16% Tricine-SDSPAGE gels. The gels were Coomassie-stained and the bands intensities were visualized and quantified using a FluorChem Q gel imager (ProteinSimple, Santa Clara, CA). The ratio of reduced protein to oxidized protein was used in conjunction with the Nernst equation to calculate the reduction potential.
Insulin Reduction Assay
The insulin reduction assay was performed as previously described by Holmgren.31 Briefly, the reaction mixture included 100 mM sodium phosphate buffer (pH 7.0), 2 mM EDTA, 0.13 mM bovine insulin and 0.33 mM dithiothreitol (DTT). The reaction was initiated by addition of hTrx, hPDI, cSelS, or cSelS mutants. The reaction’s progress was monitored by recording the increase in turbidity at 650 nm against time. The temperature was kept constant at 25 °C.
Peroxidase Assays
Peroxidase activity was measured via a coupled reaction with rTrxR and hTrx.32 The reaction mixture contained 100 mM potassium phosphate, pH 7.0, 2 mM EDTA, 150 μM NADPH, 2 nM rTrxR, 5 μM hTrx and either no enzyme or 5 μM of cSelS, cSelS U188C, cSelS U188S, or cSelS 188Δ. Enzymatic oxidation of NADPH was initiated by adding 200 μM H2O2 after incubation for 3 minutes to the sample cuvette and monitoring the consumption of NADPH spectroscopically at 340 nm. The rate of NADPH oxidation for each condition was calculated from 3 repeats.
cSelS peroxidase activity was also assayed with hGrx and glutathione reductase. The reaction mixture contained 100 mM potassium phosphate (pH 7.0), 2 mM EDTA, 150 μM NADPH, 0.5 mM GSH, 1 mU/μl yeast glutathione reductase, 5 μM hGrx, and 5 μM wild type or mutant cSelS. The reaction was initiated by adding 200 μM H2O2, and the change in absorbance at 340 nm was recorded. Glutathione peroxidase (GPx) from human erythrocytes served as a positive control.
Steady-state kinetic analysis was performed using the assay described above. To measure the kinetic parameters of cSelS peroxidase activity, H2O2 was added at concentrations of 12.5–1600 μM to a reaction mixture containing 8 nM rTrxR, 5 μM cSelS, 5 μM hTrx and 150 μM NADPH. For measurements with hTrx as a substrate, hTrx at concentrations of 0.625–20 μM was added to a reaction mixture containing 8 nM rTrxR, 5 μM cSelS, 150 μM NADPH and 200 μM H2O2. Each condition was repeated three times.
To test the substrate specificity of cSelS’ peroxidase activity, a variety of potential peroxide substrates were tested in a similar assay as described above. The soluble H2O2, cumene hydroperoxide (COOH) and tert-Butyl hydroperoxide (tBuOOH) were dissolved in 100 mM potassium phosphate (pH 7.0) and employed at a concentration of 200 μM. To prepare the hydrophobic 15-HETE and 15-HpETE, ethanol was evaporated by purging with a N2 stream, dissolved in 100 mM potassium phosphate (pH 7.0), and added to the reaction buffer at a concentration of 30 μM. The rate of the initial NADPH oxidation was calculated by monitoring the absorbance at 340 nm. Experiments were repeated three times for each substrate.
Oxidase and Isomerase Assays
The ability of cSelS to act as either an oxidase or isomerase was followed by detecting the recovery of either reduced and denatured or scrambled RNase activity.33 The reaction was monitored at 25 °C, by following the hydrolysis of cCMP by newly oxidized and properly folded RNase A at 296 nm.34 For the oxidase assays, reduced and denatured RNase A was prepared by adding 100 molar excess of DTT to RNase A in 50 mM Tris-HCl (pH 7.5), 6 M guanidine hydrochloride and 1 mM EDTA. After an overnight incubation at 4 °C, the excess DTT and guanidine were removed by Nap-5 column and the buffer exchanged into oxidase assay buffer: 50 mM HEPES-NaOH (pH 7.0), 150 mM sodium chloride, 75 mM imidazole, 2 mM EDTA and 0.5% Tween 20. The free thiol count of fully reduced RNase A, determined by thiol titer using Ellman’s reagent 5,5'–dithiolbis-2-nitrobenzoic acid (DTNB),35 was close to 8 thiols per protein, demonstrating that RNase was fully reduced. The assay buffer included 50 mM HEPES-NaOH (pH 7.0), 150 mM sodium chloride, 75 mM imidazole, 2 mM EDTA, 0.5% Tween 20, 4.5 mM cCMP, 4 μM reduced and denatured RNase A, 0.4 mM GSSG and 1.2 mM GSH as a redox buffer ([GSSG]: [GSH] = 1:3). The reaction was initiated by adding 1 μM cSelS. Human protein disulfide isomerase (hPDI) served as a positive control.
cSelS isomerase activity was measured using scrambled RNase, prepared as previously described by Thorpe.25 Briefly, native RNase A was added to 50 mM Tris-HCl (pH 7.5), 6 M guanidine hydrochloride with an equimolar amount of DTT for 30 h under anaerobic conditions. This solution was then exposed to air and allowed to slowly reoxidize in the dark, at room temperature. The reaction was monitored until less than 1% free thiol per RNase molecule remained. The buffer was then exchanged to 50 mM HEPES-NaOH (pH 7.0), 150 mM sodium chloride, 75 mM imidazole, 2 mM EDTA and 0.5% Tween 20. Scrambled RNase was stored in −20 °C until use. The assay mixture contained 50 mM HEPES-NaOH (pH 7.0), 150 mM sodium chloride, 75 mM imidazole, 2 mM EDTA, 0.5% Tween 20, 4.5 mM cCMP, 0.4 mM GSSG, and 1.2 mM GSH as a redox buffer and 8 μM scrambled RNase A. The reaction was initiated by adding 1 μM cSelS and monitored at 25 °C. hPDI served as a positive control.
RESULTS
Incorporation of Sec in proteins at a unique position remains an experimental challenge and a barrier for their characterization.36 To develop preparation strategies for the Sec-containing cSelS, we have first optimized strategies for obtaining high yield and purity of both the full length and the cytoplasmic portion of SelS U188C in E. coli.21 In the remainder of this paper, we focus on the characterization of the cytoplasmic portion of SelS (residues 52–189), abbreviated as cSelS. cSelS was fused to a binding partner, cytoplasmic maltose binding protein (cMBP), and expressed at 18 °C, to aid solubility and folding. Using this strategy, it is possible to prepare cSelS U188C with over 98% purity (Figure S1A of the Supporting Information) at a yield of 10 mg/L (0.6 μmol). The expected molecular weight was confirmed by electrospray mass spectrometry (Figure S1B of the Supporting Information). cSelS U188C dimerized in the presence and absence of reducing agents, even though the transmembrane region was deleted (Figure S2 of the Supporting Information). Hence, the coiled-coil region is sufficient for dimerization, as was previously shown.11 With the high-yield expression and purification strategy at hand, the production of the native Sec-containing cSelS was undertaken by utilizing E. coli’s innate Sec incorporation machinery.27 Incorporation of Sec into SelS in a genetically encoded fashion required numerous modifications to the expression vector and growth conditions. E. coli makes use of an mRNA structure termed the SEC Insertion Sequence (SECIS). This stem-loop architecture coordinates the recruitments to the ribosome proteins essential for Sec synthesis on its dedicated tRNA. In E. coli, those include: SelB, an elongation factor; SelC, the Sec tRNA; SelA, a selenocysteine synthase and SelD, a selenophosphate synthase. Arner and Bock demonstrated that it is possible to introduce Sec into recombinant proteins in E. coli by utilizing the SECIS element borrowed from its formate dehydrogenase H and coexpressing SelA, B and C.27 We have introduced the UGA codon at position 188, as well as E. coli formate dehydrogenase H SECIS element past the stop codon of the cMBP-cSelS coding DNA. The pSUABC vector expressing SelA, B and C under the control of their own natural promoter was a gift from Prof. Arner. The incorporation of Sec is further augmented by inducing expression at a late exponential phase, in which the level of release factor 2 is decreased.37 The incorporation ratio of Sec was about 3% of the total expressed protein. We have tested several methods to separate the full-length, selenium-containing protein, cSelS, from the truncated enzyme cSelS 188Δ (188Δ indicates that residues 188 and 189 are missing due to misreading of the Sec codon as a stop codon). The separation of cSelS and cSelS 188Δ is challenging since they are identical for all but two amino acids and form a dimer. To obtain a sample rich in cSelS, we have relied on the tendency of the cSelS 188Δ, which has only one cysteine, to aggregate in the absence of reducing agents. We have extracted and purified the protein expressed with a UGA codon using the procedures optimized for the cSelS U188C mutant but deliberately excluded reducing agents. The truncated protein, cSelS 188Δ, aggregated during purification, due to the formation of intermolecular disulfide bonds. The last step relied on size-exclusion chromatography to separate the tetramer and higher oligomeric forms of SelS from the dimeric Sec-containing form. Subsequently, only Sec-rich dimeric fractions were retained. Using this approach, samples were enriched from a starting ratio of 3% cSelS to about 50%. Figure 1A displays elution profile from size-exclusion column of a typical purification, in which the first peak eluted contains tetramer and higher order aggregates while the peak eluted last contains the dimeric form of cSelS and cSelS 188Δ (Figure 1B). On average, 50% of the protein in that fraction contained Sec, as detected by inductively coupled plasmon (ICP) spectroscopy. The yield for the selenium-rich fraction was about 0.6 mg per Liter media growth. The resulting protein is pure of contaminations, other than the truncated cSelS 188Δ. The CD spectra of cSelS and cSelS U188C are nearly identical (Figure S3 of the Supporting Information). However, as detailed in the following sections, only cSelS has enzymatic activity.
Figure 1.

Separation of dimeric forms rich in cSelS from higher order oligomers rich in cSelS 188Δ by size exclusion chromatography. (A) Elution profiles of cSelS (solid line) and cSelS U188C (dotted line) from a sephacryl S-100 under non-reducing condition. Peak 2, eluted last, contains the selenium-containing form of the enzyme, cSelS. (B) SDS-PAGE analysis of cSelS / cSelS 188Δ mixtures following separation by size exclusion chromatography. Lanes 1–3 were run under non-reducing conditions. Lane 1: cSelS U188C (control). Lane 2: cSelS first peak. Lane 3: cSelS second peak. M: Protein molecular weights standard (the molecular mass in kDa is noted on the right). Lanes 4–6 were run under reducing conditions. Lane 4: cSelS U188C (control). Lane 5: cSelS first peak. Lane 6: cSelS second peak.
cSelS reduction potential
The reduction potential of a given protein, a measurement of its tendency to gain electrons, is an important gauge of its ability to interact with other proteins. The differences in reduction potentials of selenoproteins and their Cys-containing homologues can be substantial; in the case of E. coli glutaredoxin 3, the reduction potential of the Cys-containing form was reported to be −194 mV while that of the forms containing Sec in the N-terminal and C-terminal positions of the active site’s redox motif are −260 mV and −275 mV.38 The reduction potential of cSelS U188C was previously reported to be −200 mV, as established by gel shift assays using maleimide alkylation.11 We were interested in measuring the reduction potential of the native selenium-containing cSelS. However, since our samples contained both cSelS and cSelS 188Δ, and the two dimerize, it was not possible to differentiate between different protein forms in the sample and quantify the percent of reduced and oxidized species by gel shift assays or by separating the reduced and oxidized forms chromatographically.30 Instead, we have opted to determine the redox properties by electrospray ionization mass spectrometry, where the different chemical species can be directly identified. The protein is equilibrated in different buffers whose reduction potential is posed by fixing the ratio between GSH and GSSG. Following an 18 h incubation period to ensure equilibrium was reached, the protein is denatured with acid and subsequently alkylated using iodoacetamide. Only exposed forms of Cys and Sec, but not those in selenenylsulfide bonds, can be alkylated by iodoacetamide. The fraction of reduced protein acquired a mass shift of 116 Da, due to alkylation of C174 and U188. The oxidized protein is unmodified. Subsequent acquisition of an electrospray ionization mass spectrum allows the identification of the ratio of oxidized to reduced and alkylated cSelS. Figure S4 demonstrates that similar ionization efficiencies for the two protein forms were achieved when testing a mixture of 1:1 oxidized cSelS and reduced and alkylated cSelS. Hence, the mass spectrum reflects their original concentrations in the sample. The identification of other forms, such as the alkylated cSelS 188Δ, simplified data interpretation.
To role out the formation of intermolecular selenenylsulfide bond that may be present due to the presence of cSelS 188Δ, we have isolated the dimeric form of cSelS using size exclusion chromatography and checked by thiol titer and by alkylated by iodoacetamide that there are no exposed Cys or Sec residues. This conclusion is further validated by the reproducibility of titrations regardless of a varying percent of cSelS 188Δ in samples.
Using this approach, the reduction potential of cSelS was determined to be −234±1 mV (Figure 2 and Figure S5 of the Supporting Information) and that of cSelS U188C to be −211±1 mV (Figure 2 and Figure S6 of the Supporting Information). The reduction potential of cSelS U188C was also measured by gel shift assays (Figure S7 of the Supporting Information) and found to be identical to that measured by the mass spectrometry based method. It is within a reasonable margin of error from the value reported by Christensen et al. 11 The truncated form cSelS 188Δ, which forms an intermolecular disulfide bond, is fully reduced and alkylated at −180 mV. It is easily distinguished by its mass and subsequently can be neglected from analysis. Titration curves shown in Figure 2 represent the average of three measurements using two independent sample preparations. The implications of the reduction potential of cSelS are further discussed in the conclusions.
Reductase activity
To test SelS’ potential reductase activity, we compared two activity assays that are based on reducing the disulfide bond between insulin’s chains A and B: by monitoring insulin reduction by NADPH consumption in glutathione reductase coupled assays39 and by following the process of aggregation of the free A and B chains by turbidity.31 The former assay relies on GSH as a redox couple, while the latter is compatible with other reducing agents. A reductase activity was detected only in insulin turbidity assays that were carried out with dithiothreitol (DTT) as a reducing agent. GSH is not an efficient electron donor. Interestingly, the reductase activity is present only when Sec is incorporated and is absent for the Cys mutant (Figure 3). When the amount of enzyme was doubled, the interval for the onset of turbidity halved (A 650 > 0.02) and the reaction rate in the linear region doubled. cSelS 188Δ causes rapid aggregation in the absence of reducing agents. This form can only be isolated as a tetramer or higher order oligomers prone to aggregation. The reductase activity did not depend on metals or cofactors. As the insulin turbidity assays cannot used to extract the catalytic rate, we used hTrx and hPDI to evaluate cSelS efficiency. For comparable concentrations of hTrx, cSelS and hPDI the turbidity onset was 12, 16 and 20 minutes, respectively. The reaction rates in the linear region were 0.027, 0.025 and 0.014 ΔA650 min−1 μM−1 respectively. Based on the turbidity assays, cSelS reductase activity is comparable to that of human thioredoxin 1 (hTrx)40 but slightly lower. Evidently, cSelS is an effective reductase in vitro.
Figure 3.

Reductase activity assays. The ability of cSelS to reduce insulin’s intermolecular disulfide bond is monitored by recording the increasing turbidity due to insulin’s chain B aggregation. The reaction mixture includes insulin and DTT. Insulin reduction was initiated by adding the protein of interest. The reaction was recorded with 2 μM hTrx (circle), 2 μM cSelS (square), 2 μM hPDI (star), 1 μM cSelS (triangle), 10 μM cSelS U188C (solid line), or in without additional enzymes (dotted line).
Peroxidase activity
Other plausible enzymatic functions for SelS are oxidase, isomerase and peroxidase. cSelS had neither an oxidase nor an isomerase activity for ribonuclease A (RNase A) (Figure S8 of the Supporting Information). This is not surprising, since the selenosulfide active site of SelS is located in the cytosol, while disulfide bond formation and oxidative folding processes take place in the ER. A peroxidase activity is more plausible since several selenoproteins possess a peroxidases activity.1 Specifically, there are at least two selenoprotein families that specialize in the removal of oxidative species: glutathione peroxidases and selenoproteins with minimal thioredoxin fold.41–44 cSelS itself does not efficiently accept electrons from GSH, like the former, nor does it have a known fold. As we show in this section, cSelS is also capable of reducing hydrogen peroxide efficiently - but not hydrophobic peroxides substrates.
We have measured the ability of cSelS to reduce hydrogen peroxide using the rat thioredoxin reductase (rTrxR) / hTrx or the yeast glutathione reductase (yGR) / human glutaredoxin 1 (hGrx) coupled enzymatic assays. cSelS efficiently utilized reducing equivalents from the rTrxR / hTrx system (Figure 4A); while the yGR / hGrx system is less efficient (Figure S9 of the Supporting Information). While cSelS can accept electrons directly from rTrxR, it is a low affinity substrate. The peroxidase activity was solely dependent on the presence of Sec (Figure 4B). The specificity of cSelS was tested for: hydrogen peroxide (H2O2), tert-butyl hydroperoxide (tBOOH), cumene hydroperoxide (COOH), 15S-hydroperoxy-6E, 8Z, 11Z, 14Z-eicosatetraenoic acid (HpETE) and 15S-hydroxy-6E, 8Z, 11Z, 14Z- eicosatetraenoic acid (HETE) (Figure 5). Only hydrogen peroxide and cumene hydroperoxide have turnover rates significantly higher than the control (p<0.001). Hence, cSelS is not a broad specificity peroxidase. Still, the specificity may depend on cSelS interactions with binding partners, which are likely to influence the structure of the active site.
Figure 4.

Peroxidase activity assays. (A) cSelS catalyze the reduction of H2O2 by coupling with the thioredoxin system. The reaction with 2 nM rTrxR, 5 μM cSelS and 5 μM hTrx is shown in circles, in the absence of cSelS in squares and in the absence of hTrx in triangles. (B) The activity of different mutants was compared: 5 μM cSelS (WT), 5 μM cSelS U188C (U188C), 5 μM cSelS U188S (U188S) and 5 μM cSelS 188Δ (188Δ). cSelS peroxidase activity depends on the presence of Sec (***, P<0.001).
Figure 5.

Substrate specificity of cSelS peroxidase activity. (A) Reaction scheme. (B) cSelS substrate specificity. Reaction with cSelS and hTrx (black bars), reaction excluding hTrx (striped bars) and reaction excluding cSelS (dotted bars). Only H2O2 and COOH have significant rates compared to the control (××, P<0.001, n=3). The affinity for H2O2 is the highest among the four substrates (****, P<0.001, n=3).
Next, we measured the steady-state kinetic parameters of cSelS’ peroxidase activity (Figure 6). cSelS exhibits a two substrate ping-pong mechanism with Michaelis-Menten-type saturable kinetics (Scheme 1).
Figure 6.

Kinetic parameters of cSelS peroxidase activity. (A) With H2O2 as substrate: Km = 52±5 μM, kcat = 0.110±0.003 s−1 and kcat/Km = (2.1±0.8)×103 M−1 s−1. (B) With hTrx as substrate: Km = 1.1±0.1 μM, kcat = 0.098±0.003 s−1 and kcat/Km = (9±1)×104 M−1 s−1. Means ± SD of three independent experiments are shown.
Scheme 1.
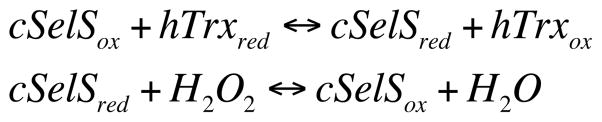
With H2O2 as substrate, cSelS exhibited a Michaelis constant (Km) of 58±5 μM, a catalytic constant (kcat) of 0.110±0.003 s−1, and a catalytic efficiency (kcat/Km) of (2.1±0.8)×103 M−1 s−1 (Figure 6A). We have also determined the kinetic parameters with hTrx as a substrate: Km = 1.1±0.1 μM, kcat = 0.098±0.003 s−1, and kcat/Km = (9±1)×104 M−1 s−1 (Figure 6B). The catalytic efficiency of cSelS with H2O2 as substrate is lower than that reported for GPx and peroxiredoxin. The kcat/Km is of the order of 107 M−1 s−1 for the selenium-containing GPx,45 while that of peroxiredoxins is of the order of 104–105 M−1 s−1.46 However, kcat/Km was determined for cSelS in isolation. Since it is an intrinsically disordered enzyme, this value may change in the presence of its binding partner.
DISCUSSION
The experiments establish that human SelS has a reductase as well as peroxidase activities in vitro. This is consistent with the observation that most selenoproteins are oxidoreductases. SelS reductase activity is comparable to that of hTrx. It does not, however, efficiently reduce peroxides compared to GPx or peroxiredoxins and is not able to reduce complex peroxides. Since it is neither an efficient nor a broad-spectrum peroxidase, then SelS peroxidase activity (at least in the absence of its protein partners) should be seen as an extension of SelS's activity as a reductase. This is analogous to the other selenium-containing reductase TrxR.47 TrxR reduces, in addition to protein disulfides, hydrogen peroxide, selenite, dehydroascorbate, α-lipoic acid, and lipid hydroperoxides.48 In this way, it provides a broad antioxidative capacity even though its peroxidase activity is lower than that of SelS. Its catalytic efficiency (kcat/Km) with H2O2 as substrate is 6.7×102 M−1 s−1 compared to cSelS’s 2.1×103 M−1 s−1. Furthermore, while SelS peroxidase efficiency is not on par with that of GPx or peroxiredoxins, it is possible that since it is a selenoprotein it is more resistant to damage by H2O2 and hence able to eliminate reactive oxygen species (ROS) even under oxidative stress. Overall, it is yet unclear what role the peroxidase activity of SelS’s plays in vivo.
Our assays show that the selenenylsulfide bond is preferentially reduced by hTrx and not by hGrx. Surprisingly, SelS reduction potential, −234 mV, is similar to that of its partner, hTrx, whose reduction potential is −230 mV.49 Typically, redox pairs have a difference in reduction potential driving the reduction of one by the other. This does not appear to be the case for SelS/hTrx. But the abundance of hTrx in the cytoplasm is significantly higher than that of SelS and its steady-state reduction potential is −280 mV.50 SelS is most likely to encounter the reduced hTrx and subsequently be reduced by it.
The rare amino acid Sec is essential for enzymatic activity; both the reductase and peroxidase activities require the presence of Sec and are not present when Sec is substituted with Cys. Why the selenium is critical for activity is not easy to explain. The enzymatic activity of only a subset of selenoproteins was characterized2 and the unique role of Sec in catalysis still awaits a unified description.51, 52 In the case of SelS, the differences do not stem from a large change in reduction potentials; the reduction potentials of cSelS and cSelS U188C are −234±1 mV vs. −211±1 mV. A difference in reduction potential of similar magnitude - between 20 to 25 mV - was recorded by our group for substitution of Sec to Cys at proteins’ C-terminal.53 Both forms are efficiently reduced by hTrx. Differences in pKa may be a potential explanation for the reliance on Sec, but pKa is easily fine tuned by the protein microenvironment and can be shifted as needed to introduce activity for the cSelS U188C. Certainly other members of the SelS/SelK eukaryotic family do not rely on Sec.3, 54 Perhaps a better explanation would invoke the formation of a particularly short-lived selenenic acid (Se-OH) or seleninic acid (SeO2−) as part of the mechanism. Indeed, the mechanism of the peroxidase activity appears to be related to atypical 2- cysteine peroxiredoxin where the resolving cysteine is in the same subunit and in which a sulfenic acid (S-OH) intermediate was identified.55 However, this is beyond the scope of the data presented here.
The finding that SelS is primarily a reductase suggests future directions in delineating its biological mechanism of action. SelS is a member of the ERAD machinery and presumably may take part in unfolding and transporting target proteins. However, SelS, whose active site is in the cytoplasm, is unlikely to remove disulfide bonds in ER-residing soluble misfolded proteins as several ER residing reductases are involved in that process prior to the transport in the channel.56, 57 Similarly, management of disulfide bonds in misfolded transmembrane proteins is not well understood, but SelS does not appear to be a critical component of the machinery.58 A unique feature of SelS is that it is an intrinsically disordered enzyme, a class that relies on order to disorder transition or conformational selection to restrict the conformational space upon binding the substrate.59 Most intrinsically disordered proteins take part in signaling or regulation. This suggests that SelS' physiological partner(s) is a signaling protein, possibly a member of the unfolded protein response (UPR),60, 61 a pathway responsible for combating oxidative stress. The ERAD path is linked to the UPR response via amplification loops in the cytoplasm.62 Many of the proteins in the path are also single-pass transmembrane proteins, raising the possibility that they interact with a membrane-bound reductase, such as SelS. Indeed, several selenoproteins act as stress sensors in regulatory pathways.63, 64 One notable binding partner of SelS that was also implicated in signalling is selenoprotein K (SelK), a single pass transmembrane enzyme with unknown function.3, 65 SelK does not contain a conventional seleno-redox motif where the Sec is coupled with a Cys, Ser or Thr in close proximity, and the reactive Sec may be able to interact with SelS’ active site. We are currently testing whether SelK binding influences SelS’ structure and enzymatic activity.
In summary, here we describe the enzymatic function and reduction potential of SelS. We demonstrate that it is enzymatically active in the absence of its binding partner and that the Sec is critical to cSelS function. However, why only the Sec-containing form is enzymatically active remains an open question.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the National Center for Research Resources (5P30RR031160-03) and the National Institute of General Medical Sciences (8 P30 GM103519-03). This material is based upon work supported by the National Science Foundation under Grant No. MCB-1054447 “CAREER: Reactivity of Selenoproteins” (S.R.).
We thank Dr. Colin Thorpe for helpful discussions. We acknowledge service from the University of Delaware’s protein characterization core facility.
ABBREVIATIONS
- SelS
selenoprotein S
- SelK
selenoprotein K
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- Grx
glutaredoxin
- ERAD
ER-assisted protein degradation
- TEV
tobacco etch virus
Footnotes
ASSOCIATED CONTENT
Supporting Information. Expression, purification and characterization of cSelS U188C, m/z spectra of reduction potential measurements, gel shifts assay, oxidase and isomerase activity and peroxidase assays with hGrx. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lu J, Holmgren A. Selenoproteins. J Biol Chem. 2009;284:723–727. doi: 10.1074/jbc.R800045200. [DOI] [PubMed] [Google Scholar]
- 2.Kasaikina MV, Hatfield DL, Gladyshev VN. Understanding selenoprotein function and regulation through the use of rodent models. Biochim Biophys Acta Mol Cell Res. 2012;1823:1633–1642. doi: 10.1016/j.bbamcr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shchedrina VA, Everley RA, Zhang Y, Gygi SP, Hatfield DL, Gladyshev VN. Selenoprotein K binds multiprotein complexes and is involved in the regulation of endoplasmic reticulum homeostasis. J Biol Chem. 2011;286:42937–42948. doi: 10.1074/jbc.M111.310920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curran JE, Jowett JBM, Elliott KS, Gao Y, Gluschenko K, Wang JM, Azim DMA, Cai GW, Mahaney MC, Comuzzie AG, Dyer TD, Walder KR, Zimmet P, MacCluer JW, Collier GR, Kissebah AH, Blangero J. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37:1234–1241. doi: 10.1038/ng1655. [DOI] [PubMed] [Google Scholar]
- 5.Du SQ, Liu HM, Huang KX. Influence of SelS gene silence on beta-Mercaptoethanol-mediated endoplasmic reticulum stress and cell apoptosis in HepG2 cells. Biochim Biophys Acta. 2010;1800:511–517. doi: 10.1016/j.bbagen.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Ye YH, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 7.Lilley BN, Ploegh HL. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci USA. 2005;102:14296–14301. doi: 10.1073/pnas.0505014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith MH, Ploegh HL, Weissman JS. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 10.Shchedrina VA, Zhang Y, Labunskyy VM, Hatfield DL, Gladyshev VN. Structure-function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxid Redox Signal. 2010;12:839–849. doi: 10.1089/ars.2009.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christensen LC, Jensen WJ, Vala A, Kamarauskaite J, Johansson L, Winther JR, Hofmann K, Teilum K, Ellgaard L. The human selenoprotein VCP-interacting membrane protein (VIMP) is non-globular and harbors a reductase function in an intrinsically disordered region. J Biol Chem. 2012;287:26388–26399. doi: 10.1074/jbc.M112.346775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haenzelmann P, Schindelin H. The structural and functional basis of the p97/Valosin-containing protein (VCP)-interacting motif (VIM) J Biol Chem. 2011;286:38679–38690. doi: 10.1074/jbc.M111.274506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 14.Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2012;14:93–U176. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JN, Kim H, Yao H, Chen Y, Weng K, Ye J. Identification of Ubxd8 protein as a sensor for unsaturated fatty acids and regulator of triglyceride synthesis. Proc Natl Acad Sci USA. 2010;107:21424–21429. doi: 10.1073/pnas.1011859107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, Hashimoto H, Taniguchi H, Fujimoto T. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell. 2012;23:800–810. doi: 10.1091/mbc.E11-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madsen L, Kriegenburg F, Vala A, Best D, Prag S, Hofmann K, Seeger M, Adams IR, Hartmann-Petersen R. The tissue-specific Rep8/UBXD6 tethers p97 to the endoplasmic reticulum membrane for degradation of misfolded proteins. Plos One. 2011;6 doi: 10.1371/journal.pone.0025061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chin KT, Xu HT, Ching YP, Jin DY. Differential subcellular localization and activity of kelch repeat proteins KLHDC1 and KLHDC2. Mol Cell Biochem. 2007;296:109–119. doi: 10.1007/s11010-006-9304-6. [DOI] [PubMed] [Google Scholar]
- 19.Uversky VN. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol. 2011;43:1090–1103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Yoshizawa S, Bock A. The many levels of control on bacterial selenoprotein synthesis. Biochim Biophys Acta. 2009;1790:1404–1414. doi: 10.1016/j.bbagen.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Srinivasan P, Pham DN, Rozovsky S. Expression and purification of the membrane enzyme selenoprotein K. Protein Expr Purif. 2012;86:27–34. doi: 10.1016/j.pep.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blommel PG, Fox BG. A combined approach to improving largescale production of tobacco etch virus protease. Protein Expr Purif. 2007;55:53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cormier CY, Mohr SE, Zuo DM, Hu YH, Rolfs A, Kramer J, Taycher E, Kelley F, Fiacco M, Turnbull G, LaBaer J. Protein Structure Initiative Material Repository: an open shared public resource of structural genomics plasmids for the biological community. Nucleic Acids Res. 2010;38:D743–D749. doi: 10.1093/nar/gkp999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell DA, Morton SU, Fernhoff NB, Marletta MA. Thioredoxin is required for S-nitrosation of procaspase-3 and the inhibition of apoptosis in Jurkat cells. Proc Natl Acad Sci USA. 2007;104:11609–11614. doi: 10.1073/pnas.0704898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rancy PC, Thorpe C. Oxidative protein folding in vitro: A study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochemistry. 2008;47:12047–12056. doi: 10.1021/bi801604x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jao SC, Ospina SME, Berdis AJ, Starke DW, Post CB, Mieyal JJ. Computational and mutational analysis of human glutaredoxin (thioltransferase): Probing the molecular basis of the low pK(a) of cysteine 22 and its role in catalysis. Biochemistry. 2006;45:4785–4796. doi: 10.1021/bi0516327. [DOI] [PubMed] [Google Scholar]
- 27.Arner ESJ, Sarioglu H, Lottspeich F, Holmgren A, Bock A. High-level expression in Escherichia coli of selenocysteine-containing rat thioredoxin reductase utilizing gene fusions with engineered bacterial-type SECIS elements and coexpression with the selA, selB and selC genes. J Mol Biol. 1999;292:1003–1016. doi: 10.1006/jmbi.1999.3085. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Q, Stone-Elander S, Arner ESJ. Tagging recombinant proteins with a Sel-tag for purification, labeling with electrophilic compounds or radiolabeling with C-11. Nature Protocols. 2006;1:604–613. doi: 10.1038/nprot.2006.87. [DOI] [PubMed] [Google Scholar]
- 29.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 30.Aslund F, Berndt KD, Holmgren A. Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J Biol Chem. 1997;272:30780–30786. doi: 10.1074/jbc.272.49.30780. [DOI] [PubMed] [Google Scholar]
- 31.Holmgren A. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J Biol Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 32.Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain - alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci USA. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyles MM, Gilbert HF. Catalysis of the oxidative folding of ribonuclease-A by protein disulfide isomerase - pre-steady state kinetics and the utilization of the oxidizig equivalents of the isomerase. Biochemistry. 1991;30:619–625. doi: 10.1021/bi00217a005. [DOI] [PubMed] [Google Scholar]
- 34.Crook EM, Mathias AP, Rabin BR. Spectrophotometric assay of bovine pancereatic ribonuclease-A by the use of cytisine 2'-3'-phosphate. Biochem J. 1960;74:234–238. doi: 10.1042/bj0740234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 36.Johansson L, Gafvelin G, Arner ESJ. Selenocysteine in proteins - properties and biotechnological use. Biochim Biophys Acta. 2005;1726:1–13. doi: 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 37.Rengby O, Johansson L, Carlson LA, Serini E, Vlamis-Gardikas A, Karsnas P, Arner ESJ. Assessment of production conditions for efficient use of Escherichia coli in high-yield heterologous recombinant selenoprotein synthesis. Appl Environ Microbiol. 2004;70:5159–5167. doi: 10.1128/AEM.70.9.5159-5167.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metanis N, Keinan E, Dawson PE. Synthetic seleno-glutaredoxin 3 analogues are highly reducing oxidoreductases with enhanced catalytic efficiency. J Am Chem Soc. 2006;128:16684–16691. doi: 10.1021/ja0661414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lambert N, Freedman RB. The latency of rat-liver microsomal protein disulfide isomerase. Biochem J. 1985;228:635–645. doi: 10.1042/bj2280635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren XL, Bjornstedt M, Shen B, Ericson ML, Holmgren A. Mutagenesis of structural half-cysteine residues in human thioredoxin and effects on the regulation of activity by selenodiglutathione. Biochemistry. 1993;32:9701–9708. doi: 10.1021/bi00088a023. [DOI] [PubMed] [Google Scholar]
- 41.Flohe L, Toppo S, Cozza G, Ursini F. A comparison of thiol peroxidase mechanisms. Antioxid Redox Signaling. 2011;15:763–780. doi: 10.1089/ars.2010.3397. [DOI] [PubMed] [Google Scholar]
- 42.Flohe L, Ursini F. Peroxidase: A term of many meanings. Antioxid Redox Signaling. 2008;10:1485–1490. doi: 10.1089/ars.2008.2059. [DOI] [PubMed] [Google Scholar]
- 43.Saito Y, Hayashi T, Tanaka A, Watanabe Y, Suzuki M, Saito E, Takahashi K. Selenoprotein P in human plasma as an extracellular phospholipid hydroperoxide glutathione peroxidase - Isolation and enzymatic characterization of human selenoprotein P. J Biol Chem. 1999;274:2866–2871. doi: 10.1074/jbc.274.5.2866. [DOI] [PubMed] [Google Scholar]
- 44.Novoselov SV, Kryukov GV, Xu XM, Carlson BA, Hatfield DL, Gladyshev VN. Selenoprotein H is a nucleolar thioredoxin-like protein with a unique expression pattern. J Biol Chem. 2007;282:11960–11968. doi: 10.1074/jbc.M701605200. [DOI] [PubMed] [Google Scholar]
- 45.Toppo S, Flohe L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim Biophys Acta. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 46.Trujillo M, Ferrer-Sueta G, Thomson L, Flohe L, Radi R. Kinetics of peroxiredoxins and their role in the decomposition of peroxynitrite. Subcell Biochem. 2007;44:83–113. doi: 10.1007/978-1-4020-6051-9_5. [DOI] [PubMed] [Google Scholar]
- 47.Arner ESJ. Focus on mammalian thioredoxin reductases - Important selenoproteins with versatile functions. Biochim Biophys Acta. 2009;1790:495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Nordberg J, Arner ESJ. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 49.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278:33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- 50.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1271–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arner ESJ. Selenoproteins-What unique properties can arise with selenocysteine in place of cysteine? Exp Cell Res. 2010;316:1296–1303. doi: 10.1016/j.yexcr.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 52.Ruggles EL, Snider GW, Hondal RJ. Chemical basis for the use of selenocysteine. In: Hatfield DL, Berry MJ, Gladyshev VN, editors. Selenium: Its Molecular Biology and Role in Human Health. Springer; New York: 2012. pp. 73–83. [Google Scholar]
- 53.Li F, Lutz PB, Pepelyayeva Y, Arnér ESJ, Bayse CA, Rozovsky S. Redox active motifs in selenoproteins. 2013 doi: 10.1073/pnas.1319022111. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roseler A, Prieto JH, Iozef R, Hecker B, Schirmer RH, Kulzer S, Przyborski J, Rahlfs S, Becker K. Insight into the selenoproteome of the malaria parasite plasmodium falciparum. Antioxid Redox Signaling. 2012;17:534–543. doi: 10.1089/ars.2011.4276. [DOI] [PubMed] [Google Scholar]
- 55.Rhee SG, Woo HA. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid Redox Signaling. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 56.Hagiwara M, Nagata K. Redox-dependent protein quality control in the endoplasmic reticulum: Folding to degradation. Antioxid Redox Signaling. 2012;16:1119–1128. doi: 10.1089/ars.2011.4495. [DOI] [PubMed] [Google Scholar]
- 57.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nature Reviews Molecular Cell Biology. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 58.Houck SA, Cyr DM. Mechanisms for quality control of misfolded transmembrane proteins. Biochim Biophys Acta, Biomembr. 2012;1818:1108–1114. doi: 10.1016/j.bbamem.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li E, Wimley WC, Hristova K. Transmembrane helix dimerization: Beyond the search for sequence motifs. Biochim Biophys Acta, Biomembr. 2012;1818:183–193. doi: 10.1016/j.bbamem.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Claessen JHL, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walter P, Ron D. The unfolded protein response: From stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 62.Higa A, Chevet E. Redox signaling loops in the unfolded protein response. Cell Signal. 2012;24:1548–1555. doi: 10.1016/j.cellsig.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 63.Hawkes WC, Alkan Z. Regulation of redox signaling by selenoproteins. Biol Trace Elem Res. 2010;134:235–251. doi: 10.1007/s12011-010-8656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brigelius-Flohe R, Flohe L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signaling. 2011;15:2335–2381. doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verma S, Hoffmann FW, Kumar M, Huang Z, Roe K, Nguyen-Wu E, Hashimoto AS, Hoffmann PR. Selenoprotein K knockout mice exhibit deficient calcium flux in immune cells and impaired immune responses. J Immunol. 2011;186:2127–2137. doi: 10.4049/jimmunol.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.