

Abstract
Background
The epidermal growth factor receptor (EGFR) and vascular endothelial growth factor receptor (VEGFR) have been implicated as therapeutic targets for head and neck squamous cell carcinoma (HNSCC). Vandetanib is a small-molecule tyrosine kinase inhibitor (TKI) with dual specificity for EGFR and VEGFR. Here we characterize the phenotypic and biochemical effects of vandetanib on various HNSCC cell lines.
Methods
In vitro models were used for studying tumor cell viability, invasion, and signaling as well as in vivo xenograft models.
Results
Treatment with vandetanib reduced viability, invasion, and tumor growth of HNSCC cell lines. Phosphorylation levels of mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription 3 (STAT3) were reduced in vandetanib-treated HNSCC cells. Additionally, vandetanib abrogates EGF-induced STAT3 activity and STAT3 target gene expression.
Conclusions
We demonstrated that vandetanib inhibits the growth of head and neck cancer cell lines. The antitumor effects of vandetanib appear to be exerted via the EGFR inhibitory effect of the compound.
Keywords: vandetanib, HNSCC, EGFR, angiogenesis, molecular targeted therapy
There are over 30,000 new cases of head and neck squamous cell carcinoma (HNSCC) each year. Despite recent advances, the overall survival of HNSCC patients has remained unchanged in the past 30 years. Patients with recurrent or metastatic cancer have a poorer prognosis and a decreased overall survival. Surgical resection of these cancers often leads to severe permanent cosmetic deformities and functional deficits. Therefore, there is a critical need for more effective agents to treat HNSCC.
The epidermal growth factor receptor (EGFR) has shown significant potential as a therapeutic target in solid malignancies, including those of the head and neck. Dys-regulation of EGFR contributes to HNSCC progression. EGFR is overexpressed in approximately 90% of HNSCC tumor specimens where increased expression is correlated with poor clinical outcome.1 Antitumor effects have been observed upon inhibition of the EGFR tyrosine kinase in both in vitro and in vivo models. Among the various approaches to EGFR inhibition, cetuximab (Erbitux, IMC-C225), a chimeric monoclonal antibody against EGFR, has been the most studied.2,3 The 2006 study reported by Bonner et al,3 which examined the efficacy of cetuximab when combined with radiotherapy in patients with locoregionally advanced head and neck cancer, led to the FDA approval of cetuximab for HNSCC.
There have been parallel developments with targeted therapies that inhibit angiogenesis. A key regulator of angiogenesis is vascular endothelial growth factor (VEGF), which is secreted by tumor cells in hypoxic conditions. After VEGF is secreted, it binds its receptors (VEGFR1–3) on endothelial cells, leading to endothelial cell proliferation and tumor angiogenesis.4 Beva-cizumab (Avastin, Genentech), a recombinant human monoclonal antibody to VEGF, has been studied in many cancers and has been FDA-approved in colorectal and breast carcinomas.5
Inhibition of EGFR and/or VEGFR has individually shown promise as an anticancer strategy; thus, it is not surprising that the concomitant inhibition of both targets has been proposed as an anticancer therapy.6–8 Tonra et al6 showed that combined treatment with cetuximab and DC101, a humanized monoclonal antibody to VEGFR-2, had more significant antitumor effects in xenografts of colon and pancreatic cancer than each agent alone. AEE788, a small-molecule tyrosine kinase inhibitor (TKI) with dual specificity against EGFR and VEGFR, proved to be an effective therapeutic strategy in a murine oral cancer model.7 Bevacizumab in combination with erlotinib (a small-molecule inhibitor of the EGFR tyrosine kinase) has been studied in phase I/II clinical trials involving patients with recurrent HNSCC. Vokes et al9,10 showed that this combination treatment resulted in a response rate of approximately 70% of patients enrolled. A novel small-molecule TKI, vandetanib, ZD6474 (Astra-Zeneca, Macclesfield, UK), is an oral dual inhibitor of both EGFR and VEGFR developed by AstraZeneca.11 In non-small cell lung cancer, vandetanib showed promising results when used in combination with docetaxel, a anti-mitotic chemotherapy drug.12 Additionally, vandetanib is in an ongoing open-label phase I study in combination with radiation therapy with or without cisplatin for untreated, unresected, locally advanced HNSCC.13
The antitumor effects of EGFR inhibition on HNSCC have been well documented. It has also been observed in vitro that inhibition of EGFR on tumor cells decreases production of VEGF by tumor cells.8 Consistent with this observation, EGFR inhibitors such as gefitinib and cetuximab have shown antiangiogenic properties when used in vivo.14 In several cancer types, signal transducer and activator of transcription 3 (STAT3) has been shown to mediate VEGF production.15,16 STAT3 mediates intracellular signaling pathways in tumor cells that are downstream of EGFR via activation of STAT3 target genes including VEGF.17 It has been demonstrated that EGFR inhibition decreases STAT3 activity and that in vitro suppression of STAT3, in breast and cervical cancers, decreases VEGF production.16,18 Therefore, it is our hypothesis that EGFR inhibition in HNSCC using vandetanib will result in antitumor effects in vitro and in vivo, and these effects are mediated by the activation of STAT3. We hypothesize that the anti-tumor effects of EGFR inhibition will be exerted, in part, through the downregulation of STAT3 and the STAT3 target gene, VEGF. We also anticipate that the decreased production of VEGF will potentiate the effects of direct VEGFR inhibition on the tumor endothelium. HNSCC tumor cells express significant levels of EGFR, but VEGFR expression is limited primarily to the endothelium. Although the specificity of vandetanib to VEGFR is approximately 10-fold greater than that of EGFR, we hypothesized that vandetanib will still have a significant antitumor effect on HNSCC due to the high EGFR expression.19 In this study, we demonstrate that vandetanib exerts significant in vitro and in vivo antitumor effects via inhibition of the EGFR pathway.
MATERIALS AND METHODS
Chemicals and reagents
Vandetanib was provided by AstraZeneca (Macclesfield, UK). The STAT3 monoclonal antibody used for Western blotting was obtained from Cell Signaling (Beverly, MA). The EGFR antibody for immunoprecipitation was obtained from Upstate Biotechnology (Lake Placid, NY). Antibodies against p44/42 mitogen-activated protein kinase (MAPK), phospho-p44/42 MAPK, phosphor-STAT3, and STAT3 were obtained from Cell Signaling (Danvers, MA). The PY99 antibody was obtained from Santz Cruz Biotechnologies (Santa Cruz, CA).
Cell culture
HNSCC cell lines (UM-22A, UM-22B, PCI-15B, PCI-37A, PCI-37B) used in this study are part of a large collection established in the Department of Otolaryngology at the University of Pittsburgh and Pittsburgh Cancer Institute. UM-22A and UM-22B are well-characterized HNSCC cell lines developed at the University of Michigan.18 Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. All cells have been genotyped.
Cell viability assay
To examine the ability of vandetanib to decrease the viability of HNSCC cell lines in vitro, we used a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)–based assay. Two thousand cells per well were grown in DMEM supplemented with 10% FBS in 96-well tissue-culture plates. After 24 hours, the cells were treated with various concentrations of vandetanib (up to 6 μM) in DMEM supplemented with 2% FBS. Because of the concern that the dimethyl sulfoxide (DMSO) in the vandetanib preparation could affect the experiments, the concentration of DMSO in all the wells was standardized and kept below 0.2% vol/vol. To measure the number of metabolically active cells after a 3-day incubation period, we used an MTT assay measured by a 96-well microtiter plate reader.
Immunoprecipitation
Cell lysate was prepared and combined with Protein-G beads and EGFR antibody (Upstate Biotech, Billerica, MA). The lysate was washed in lysis buffer and then 300 μg of protein was loaded onto an 8% SDS-PAGE gel and transferred to a nitrocellulose membrane using a semidry transfer machine (Bio-Rad Laboratories, Hercules, CA). After protein transfer, the membrane was blocked overnight with a blocking solution containing 5% nonfat dry milk in TBST (0.6% dry milk powder, 0.9% NaCl, 0.5% Tween 20, and 50 mmol/L Tris [pH 7.4]). The membrane was incubated with a phosphotyrosine (PY99) antibody (Santa Cruz Biotechnology) overnight and then washed with TBST solution 3 times for 10 minutes each. The membrane was then incubated with mouse immunoglobulin-G (IgG) with a horseradish peroxidase conjugate (Bio-Rad Laboratories) for 1 hour and then washed 3 times for 5 minutes each in TBST. The blot was developed with luminol reagents (Santa Cruz Biotechnology).
Western blot analysis
Approximately 40 μg of protein was resolved in an 8% SDS-PAGE gel and transferred onto a nitrocellulose membrane using a semidry transfer machine (Bio-Rad Laboratories). After protein transfer, the membrane was blocked overnight with a blocking solution containing 5% nonfat dry milk and 0.2% Tween 20 in 1× phosphate-buffered saline. The membrane was incubated with the primary antibodies (1:1000 phospho-p44/42 MAPK or p44/42 MAPK, phospho-Akt473, total Akt, and phosphor-STAT3, total STAT3, phospho-EGFR, and total EGFR) overnight and then washed with TBST solution (0.6% dry milk powder, 0.9% NaCl, 0.5% Tween 20, and 50 mmol/L Tris [pH 7.4]) 3 times for 5 minutes each. The membrane was then incubated with the secondary antibody (goat anti-rabbit/mouse IgG–horseradish peroxidase conjugate; Bio-Rad) for 1 hour and washed 3 times for 5 minutes each. The blot was developed with luminol reagent (Santa Cruz Biotechnology). The band intensity was quantitated with DigiDoc1000 software (Alpha Innatech Corporation, San Leandro, CA).
VEGF enzyme-linked immunosorbent assay
HNSCC cell lines were grown in serum-free medium and treated with vandetanib at 0 and 1 μg/mL concentrations. After 24 hours, the media was collected. The cells were also trypsinized, collected, and counted. The concentration of VEGF in the conditioned media was examined using a VEGF Quantikine enzyme-linked immunosorbent assay (ELISA) kit (R&D Systemsm Minneapolis, MN) in accord with the manufacturer’s instructions. Absorbance and concentration (pg/mL) were measured using a microplate reader at 450 nm, with 570 nm correction. Results were plotted as the ratio of the concentration to the total number of cells. These experiments were performed in triplicate.
Invasion assay
Cell invasion was evaluated in vitro using Matrigel-coated semipermeable modified Boyden inserts with a pore size of 8 μm (Becton Dickinson/Biocoat, Bedford, MA). Cells were plated in duplicate at a density of 5 × 103 cells per well in DMEM in the chamber or insert. Both the insert and the containing well were subjected to the same medium composition, except the insert contained serum-free medium and the well contained 10% FBS that served as a chemoattractant. The medium in the inserts was supplemented with DMSO, 0.5 μM vandetanib, or 1.0 μM vandetanib. After 24 hours of treatment at 37°C in a 5% CO2 incubator, the cells in the insert were removed by wiping gently with a cotton swab. Cells on the reverse side of the insert were fixed and stained with Hema 3 (Fisher Scientific, Hampton, NH) in accord with the manufacturer’s instructions. Invading cells in the entire chamber were counted using light microscopy at a magnification of ×20.
Luciferase assay
The UM-22B cell line stably transfected with a STAT3 luciferase reporter vector was kindly provided by Dr. Jennifer Grandis. In this vector, the luciferase gene is under the control of a high-affinity serum inducible element (hSIE) that binds STAT3. Therefore, the degree of luciferase activity correlates with the degree of intracellular STAT3 activity. For the assay, 2 × 105 cells were plated in standard 6-well plates for each type of treatment/cell type in triplicate in 3 mL 10% FBS DMEM then allowed to adhere and grow for 24 hours. Cells were then serum starved in DMEM for an additional 48 hours. The cells were treated with EGF, vandetanib, or both for 24 hours. The cells then were lysed and luciferase activity was measured as previously described.19
Xenografts in nude mice
HNSCC cell line UM-22B was cultured in DMEM containing 10% FBS. Cells were trypsinized and cell number and viability of the cells were determined using trypan blue dye exclusion using a hemocytometer. A suspension of 3 × 106 HNSCC cells in 50 μL serum-free medium was injected subcutaneously on the right flanks of nu/nu athymic nude mice (n = 40; Harlan Sprague–Dawley, Indianapolis, IN). Although an orthotopic model may have held more translational relevance, a limit of orthotopic models for head and neck cancer is that the maximal tumor size is limited due to anatomical proximity to vital organs. Therefore, we chose subcutaneous ectopic xenografts as the model, to establish larger tumors for a longer experimental time point. After palpable tumors were present in the majority of the mice, the mice with palpable tumors were randomized into treatment groups. Randomization was stratified by initial tumor volume. The vandetanib-treated mice received 30 mg/kg daily via oral gavage, the cisplatin-treated mice received 5 mg/kg weekly via intraperitoneal (IP) injection, and control mice were treated with the respective drug solvents. Tumor volumes were measured in 2 dimensions with calipers. Tumor volumes were calculated using the formula: (3.14/6) × larger diameter × (small diameter)2. Animal use and care were in strict compliance with institutional guidelines established by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Statistics
Tumor volumes were log transformed. The transformed data were fit to a mixed quadratic regression model, with individual animals described as random effects. Residuals were examined to assess the adequacy of the model. An expected growth curve was then estimated for each treatment group. Omnibus F tests were used to determine if the growth curves differed from zero and were not simultaneously parallel. Growth curves were tested for equality and test p values were adjusted with the step-down Bonferroni method.
RESULTS
Vandetanib decreases the viability of HNSCC cell lines in a dose-dependent fashion
It is known that EGFR inhibition decreases the viability of head and neck cancer cells.1 To confirm this phenotypic effect due to EGFR inhibition, we performed cell viability assays on head and neck cancer cell lines using vandetanib, a dual inhibitor of EGFR and VEGFR. The effect of vandetanib on the viability of HNSCC cell lines was examined using an MTT assay (see Figure 1). Vandetanib decreased the viability of HNSCC cell lines dose dependently. The IC50 of the cell lines used ranged from 0.13 to 2.0 μM. At a concentration range of 2.0 to 3.0 μM there was an inhibitory effect of >90%. These data show that our cell lines display greater sensitivity to other HNSCC cell lines previously reported to have IC50 in the range of 4.4–26.4 μM.22
FIGURE 1.

ZD6474 (vandetanib) inhibits the growth of HNSCC cell lines. HNSCC cell lines were plated in 96-well plates at density of 2 × 103 cells/well. The cells were then treated with various concentrations of ZD6474 for 72 hours. MTT assay was then performed in a standard fashion. These results are from the repetition of 3 independent experiments. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Vandetanib inhibits the activation of the EGFR tyrosine kinase in HNSCC cell lines leading to the downregulation of STAT and MAPK
The effects of EGFR inhibition are mediated through the activation of its downstream signaling molecules. A subset of these molecules includes STAT3 and MAPK, and the activation of these molecules in HNSCC is correlated with increased proliferation and invasion.23 Therefore, we examined the effects of vandetanib on the phosphorylation of EGFR as well as its downstream signaling molecules (Figures 2A–2C). As seen in Figure 2A, vandetanib efficiently inhibited the activation of the EGFR tyrosine kinase and also decreased the expression of phosphorylated forms of the downstream signaling elements, STAT3 and MAPK, in all 3 HNSCC cell lines examined. In PCI-15B cells, >40% inhibition was achieved at a concentration of 1 μM and 90% inhibition was achieved at 3 μM. At a 1–3 μM concentration, vandetanib also decreased the phosphorylation of EGFR, STAT3, and MAPK in these cells. We also found that vandetanib decreased EGFR and STAT3 phosphorylation more efficiently in UM-22A compared with UM-22B or PCI-15B cell lines. The decrease in activated STAT3 observed here alludes to the possibility of STAT3 mediating the phenotypic effect observed with vandetanib use in head and neck cancer cell lines.
FIGURE 2.
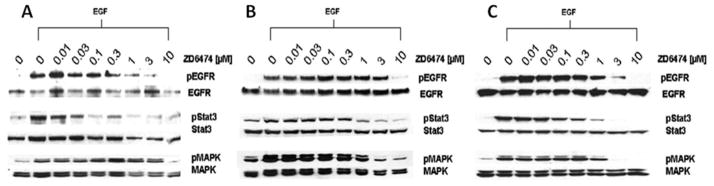
Vandetanib inhibits activation of EGFR signaling and downregulates STAT3 activity in HNSCC cell lines. HNSCC cell lines (A) UM-22A, (B) UM-22B, and (C) PCI-15B were treated with various concentration of vandetanib in media containing 2% FBS. The cells were then treated with EGF for 15 minutes prior to being harvested for Western blot analysis using appropriate antisera. These results are a representation of 3 independent experiments.
Vandetanib downregulates VEGF production in HNSCC
Knowing that VEGF is a STAT3 target gene, we wanted to further verify that vandetanib decreases the production of VEGF by HNSCC cells. HNSCC cell lines were treated with 0 or 1 μM of vandetanib for 24 hours. Media was collected after the 24-hour treatment period with vandetanib. The concentration of VEGF was then determined using an ELISA (see Figure 3). Compared with vehicle control, treatment with 1 μM of vandetanib decreased the production of VEGF in all examined cell lines, with the exception of PCI-37B, with the greatest decrease being observed with the PCI-15B cell line.
FIGURE 3.

Vandetanib downregulates the production of the Stat3 target gene VEGF. HNSCC cell lines were treated with 1 μM of vandetanib or an equal volume of DMSO for 24 hours and ELISA was performed on the conditioned media for VEGF levels. Compared with the untreated cells, there was a decrease in the levels of VEGF secretion after treatment with 1 μM of vandetanib a, except in PCI-37B. These results are the repetition of 3 independent experiments. *p ≥ .05.
Vandetanib decreases the invasion of HNSCC cell lines
It has been demonstrated previously that EGFR modulates the invasion of HNSCC cells.25 Therefore, we examined the effects of vandetanib on the invasion of HNSCC cell lines (see Table 1). HNSCC cells were plated on Matrigel-coated chambers and treated with solvent, 0.5 or 1.0 μM vandetanib, and incubated for 24 hours (see Figure 4). Treatment of vandetanib resulted in a dose-dependent decrease in the invasion of HNSCC cell lines with a >50% decrease at 1.0 μM in the UM-22A cell line. Each invasion assay was controlled for viability by performing a concurrent MTT assay, where no changes in viability were observed at the time points and concentrations used for the invasion studies.
TABLE 1.
Effects of half-maximal effective concentrations (EC50) of vandetanib on the invasion of HNSCC cell lines.
| HNSCC cell line | Vandetanib EC50, nM |
|---|---|
| PCI-15B | 558 |
| PCI-37A | 1695 |
| UM-22A | 0.3 |
| SCC-25 | 10 |
| UM-22B | 2424 |
| PCI-37B | 1726 |
Abbreviation: HNSCC, head and neck squamous cell carcinoma.
FIGURE 4.
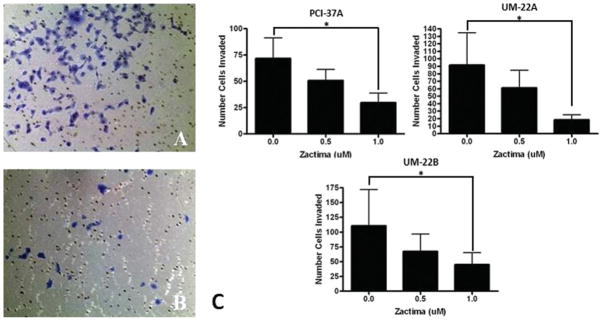
Invasion of HNSCC cell lines after treatment with vandetanib. HNSCC cell were treated with 0.5 μM, 1.0 μM and compared with DMSO control. A matrigel invasion assay was performed; 5 × 103 cells were plated in each insert. The cells were then fixed and stained and the number of invaded cells were counted and compared with the DMSO treatment group. (A) Representative micrograph of the control matrigel invasion insert of UM-22B (original magnification, ×10). (B) Representative micrograph of the matrigel invasion insert of UM-22B treated with 1 μM of vandetanib (original magnification, ×10). (C) Graph depicting the relative cell count of the treated inserts with respect to the untreated inserts. These data are the repetition of 3 independent experiments. The p values for all cell lines compared with the DMSO control were <0.05 for the 1 μM concentration.
Vandetanib decreases tumor volume of HNSCC xenografts in nude mice
To study the in vivo effects of vandetanib in HNSCC xenografts, the HNSCC cell line UM-22B was used to establish subcutaneous xenografts in nude mice because it was predetermined to be tumorigenic. Once tumors were established, the mice were treated with vandetanib, cis-platin, or both agents (see Figure 5). The cisplatin group was treated with cisplatin (IP) once a week and the vandetanib group was treated daily with vandetanib via oral gavage. The combination treatment group received both agents. When the tumor growth curves were compared with each other, vandetanib-treated animals had a significantly slower tumor growth than that of cisplatin-treated animals (p = .0020). Furthermore, the group treated with the combination of cisplatin and vandetanib showed a statistically significant decrease in tumor growth compared with either agent alone (p ≤ .00001 and p = .0298, respectively; effects additive). There is a limitation to the conclusions that can be made regarding dual therapy with vandetanib and cisplatin in that only a single cisplatin concentration was used, but the comparison of vandetanib alone to the combination indicates the potential effectiveness of vandetanib or other dual EGFR and VEGFR targeting approaches for head and neck cancer.
FIGURE 5.
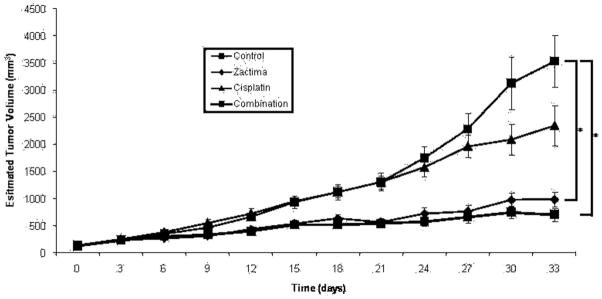
Vandetanib decreases tumor volume EGFR TKI sensitive and resistant cell lines in a murine model. HNSCC cell line UM-22B was used to establish subcutaneous xenografts in nude mice. The mice were then randomized into 4 groups after a 2-week period of tumor establishment. Fifteen mice were included in each group. The cisplatin group was treated with of cisplatin intraperitoneally (IP) once per week. The vandetanib group was treated with 30 mg/kg of vandetanib daily via oral gavage. The combination group received both 5 mg/kg of cisplatin and30 mg/kg of vandetanib. The control group received PBS (IP), once a week and 1% Tween 80, daily, via oral gavage. Values of p for vandetanib and the combination group were <0.005 compared with control.
DISCUSSION
Previously established methods in cancer therapeutics have used single target agents as treatment options. Cetuximab is used as an EGFR inhibitor and has proven to be an effective therapy for patients with HNSCC.3 However, there are still only a subset of patients that respond to EGFR inhibition and the response rates for patients with recurring or metastatic HNSCC ranges from only 10% to 20%.27 In addition to EGFR inhibition, VEGFR and VEGF inhibition have been used as anti-cancer strategies in various malignancies intending to reduce tumor-induced neoangiogenesis. Bevacizumab (Avastin), a monoclonal antibody to VEGF, is intended to reduce angiogenesis in patients with HNSCC but is only an effective therapy in a subgroup of cancer patients.
It has been suggested in several studies that EGFR and VEGFR pathways are not independent of each other but are interrelated in promoting tumor growth and progression. In a study by Viloria-Petit et al,28 the authors generated cetuximab-resistant squamous cell carcinoma xenografts in nude mice by progressively increasing dosing of cetuximab in mice with recurrent tumors. Cell lines derived from these resistant tumors showed high levels of VEGF when compared with the parental cell lines. Furthermore, HNSCC cell lines that overexpressed VEGF showed significant in vivo resistance to cetuximab therapy.23 In another study, cetuximab-resistant breast cancer and colorectal cancer cell lines were found to express high levels of VEGFR-1 when compared with their respective parental cell lines. Furthermore, downregulation of VEGFR-1 in these cell lines using short interfering (si)RNA resulted in restoration of cetuximab sensitivity.29
Vandetanib, an orally administered TKI, targets both EGFR and VEGFR transmembrane tyrosine kinases. In this study, we showed that vandetanib inhibits the viability of HNSCC cell lines. We also showed that vandetanib downregulates the activity of STAT3 and the production of VEGF. Additionally, this is the first report of the effect of vandetanib on the invasion of head and neck cell lines. To confirm in vitro results, we administered vandetanib to nude mice bearing HNSCC xenografts. The mice were treated with vandetanib, either alone or in combination with cisplatin, and showed a significant reduction in tumor growth compared with untreated control animals. Vandetanib was superior to cisplatin in decreasing tumor growth. This is translationally relevant, given that the efficacy of a new agent is often determined in combination with traditional chemotherapy. A number of mechanisms have been proposed for the antitumor effects seen with dual blockade of the EGFR and VEGFR pathway. As confirmed by data in this study, treatment with vandetanib results in diminished tumor cell production of the ligand VEGF. A study by Pore et al30 showed that the inhibition of EGFR leads to the downregulation of Sp1, a transcriptional activator with binding sites within the VEGF promoter region. Other studies have shown that STAT3, a protein involved in signal transduction downstream from EGFR, also binds to the VEGF promoter region and activates the transcription of VEGF. Inhibition of EGFR with AG1478 has been shown to result in the downregulation of STAT3 activity and the production of VEGF.31 Niu et al15 examined a number of human cancer cell lines and showed that VEGF expression correlated with STAT3 activity; furthermore, Niu and colleagues showed that downregulating STAT3 activity with dominant-negative STAT3 protein led to the downregulation of VEGF expression. Our study also showed that vandetanib induced a dose-dependent decrease in the phosphorylation of STAT3 along with a decrease in VEGF production, suggesting that STAT3 is important in mediating the effects of EGFR inhibition.
Another mechanism for the antitumor effects of vandetanib may be due to the direct inhibition of EGFR on tumor endothelium. Although the exact role of endothelial cell-expressed EGFR in tumor angiogenesis remains to be elucidated, it has been shown that endothelial cells express EGFR and undergo angiogenic responses to EGF. Conversely, the inhibition of EGFR using small-molecule TKI such as Iressa has resulted in antiangiogenic effects.32 Finally, it has been shown that VEGF receptors are expressed by HNSCC cell lines and tumors.28–30 The functional significance of tumor expression of VEGFR in HNSCC is not known. However, tumor-expressed VEGFR has been shown to be involved in mediating the migration and invasion of tumor cells in several types of cancer, including breast and colorectal cancer.33–35 In the case of HNSCC, a study by Bock et al34 suggests not only that VEGF-C may contribute to the invasion and migration of HNSCC cells, but also that downregulation of VEGF-C using siRNA strategy in HNSCC cell lines decreased their motility and invasion. Therefore, it is possible that the inhibition of tumor-expressed VEGFR by vandetanib may contribute to the antitumor effects of this inhibitor. This constitutes future work that is currently being explored. Additionally, vandetanib represents a possible approach to inhibiting EGFR and VEGFR concomitantly, and it represents a laboratory tool and a research model to study this dual inhibition. The success of vandetanib in the clinic is still under investigation, and the studies here further prove the potential efficacy of EGFR TKIs and dual EGFR and VEGFR as an approach to inhibit the growth of head and neck tumors.
CONCLUSIONS
The antitumor effects and EGFR targeting capacity of vandetanib provide evidence for the use of vandetanib as an exciting new treatment alternative in the management of HNSCC. We have demonstrated in this study that vandetanib exerts a antitumor effect in HNSCC both in vitro and in vivo. Although this study was designed to demonstrate the EGFR inhibitory effect of vandetanib, the drug has additional specificity for VEGFR. As this dual inhibition strategy is transitioned from research setting into clinical use, it is imperative that we further understand the mechanisms through which the inhibition of both EGFR and VEGFR could potentiate an antitumor effect. Although vandetanib has been well tolerated in clinical trials, there are associated side effects including diarrhea, hypertension, and skin rash.36 Identification of appropriate biomarkers will allow for proper selection of patients in the use of vandetanib.
Acknowledgments
Contract grant sponsor: National Institutes of Health; contract grant number: K08 DE019201.
References
- 1.Grandis JR, Melhelm MF, Gooding WE, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90:824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 2.Kies M, Arquette M, Nabell L, et al. Final report of the efficacy and safety of the anti-epidermal growth factor antibody Erbitux (IMC-225), in combination with cisplatin in patients with recurrent squamous cell carcinoma of the head and neck (SCCHN) refractory to cisplatin containing chemotherapy. Proc ASCO. 2002;21:226a. (abstract) [Google Scholar]
- 3.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 4.Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 5.Sandler A, Herbst R. Combining targeted agents: blocking the epidermal growth factor and vascular endothelial growth factor pathways. Clin Cancer Res. 2006;12:4421s–4425s. doi: 10.1158/1078-0432.CCR-06-0796. [DOI] [PubMed] [Google Scholar]
- 6.Tonra JR, Deevi DS, Corcoran E, et al. Synergistic antitumor effects of combined epidermal growth factor receptor and vascular endothelial growth factor receptor-2 targeted therapy. Clin Cancer Res. 2006;12:2197–2207. doi: 10.1158/1078-0432.CCR-05-1682. [DOI] [PubMed] [Google Scholar]
- 7.Yigitbasi OG, Younes MN, Doan D, et al. Tumor cell and endothelial cell therapy of oral cancer by dual tyrosine kinase receptor blockade. Cancer Res. 2004;64:7977–7984. doi: 10.1158/0008-5472.CAN-04-1477. [DOI] [PubMed] [Google Scholar]
- 8.Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol Cancer Res. 2007;5:203–220. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 9.Mauer AM, Cohen E, Wong SJ, et al. Phase I study of epidermal growth factor receptor (EGFR) inhibitor, erlonitib, and vacular endothelial growth factor monoclonal antibody, bevacizumab, in recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) Proc Am Soc Clin Oncol. 2004:A5539. (abstract) [Google Scholar]
- 10.Vokes EE, Cohen E, Mauer AM, et al. A phase I study of erlotinib and bevacizumab for recurrent or metastatic squamous cell carcinoma of the head and neck (HNC) Proc Am Soc Clin Oncol. 2005:A5504. (abstract) [Google Scholar]
- 11.Ciardiello F, Caputo R, Damiano V, et al. Antitumor effects of ZD6474, a small molecule vascular endothelial growth factor receptor tyrosine kinase inhibitor, with additional activity against epidermal growth factor receptor tyrosine kinase. Clin Cancer Res. 2003;9:1546–1556. [PubMed] [Google Scholar]
- 12.Heymach JV, Johnson BE, Prager D, et al. Randomized, placebo-controlled phase II study of vandetanib plus docetaxel in previously treated non small-cell lung cancer. J Clin Oncol. 2007;25:4270–4277. doi: 10.1200/JCO.2006.10.5122. [DOI] [PubMed] [Google Scholar]
- 13.Papadimitrakopoulou V, Frank SJ, Blumenschein GR, et al. Phase I evaluation of vandetanib with radiation therapy (RT) plus or minus cisplatin in previously untreated advanced head and neck squamous cell carcinoma (HNSCC) J Clin Oncol. 2009;27(Suppl 15s):Abstract 6016. [Google Scholar]
- 14.Ciardiello F, Bianco R, Damiano V, et al. Antiangiogenic and antitumor activity of anti-epidermal growth factor receptor C225 monoclonal antibody in combination with vascular endothelial growth factor antisense oligonucleotide in human GEO colon cancer cells. Clin Cancer Res. 2000;6:3739–3747. [PubMed] [Google Scholar]
- 15.Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 16.Xu Q, Briggs J, Park S, et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005;24:5552–5560. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 17.Leeman RJ, Lui VW, Grandis JR. STAT3 as a therapeutic target in head and neck cancer. Expert Opin Biol Ther. 2006;6:231–241. doi: 10.1517/14712598.6.3.231. [DOI] [PubMed] [Google Scholar]
- 18.He Y, Zeng Q, Drenning SD, et al. Inhibition of human squamous cell carcinoma growth in vivo by epidermal growth factor receptor antisense RNA transcribed from the U6 promoter. J Natl Cancer Inst. 1998;90:1080–1087. doi: 10.1093/jnci/90.14.1080. [DOI] [PubMed] [Google Scholar]
- 19.Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–4655. [PubMed] [Google Scholar]
- 20.Grénman R, Carey TE, McClatchey KD, et al. In vitro radiation resistance among cell lines established from patients with squamous cell carcinoma of the head and neck. Cancer. 1991;67:2741–2747. doi: 10.1002/1097-0142(19910601)67:11<2741::aid-cncr2820671105>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 21.Leong PL, Andrews GA, Johnson DE, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci U S A. 2003;100:4138–4143. doi: 10.1073/pnas.0534764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sano D, Kawakami M, Fujita K, et al. Antitumor effects of ZD6474 on head and neck squamous cell carcinoma. Oncology Reports. 2007;17:289–295. [PubMed] [Google Scholar]
- 23.Chai RL, Grandis JR. Advances in molecular diagnostics and therapeutics in head and neck cancer. Curr Treat Options Oncol. 2006;7:3–11. doi: 10.1007/s11864-006-0027-4. [DOI] [PubMed] [Google Scholar]
- 24.Lui VW, Boehm AL, Koppikar P, et al. Antiproliferative mechanisms of a transcription factor decoy targeting signal transducer and activator of transcription (STAT) 3: the role of STAT1. Mol Pharmacol. 2007;71:1435–1443. doi: 10.1124/mol.106.032284. [DOI] [PubMed] [Google Scholar]
- 25.Koppikar P, Choi SH, Egloff AM, et al. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin Cancer Res. 2008;14:4284–4291. doi: 10.1158/1078-0432.CCR-07-5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller S, Su L, Tighiouart M, et al. Distinctive E-cadherin and epidermal growth factor receptor expression in metastatic and nonmetastatic head and neck squamous cell carcinoma: predictive and prognostic correlation. Cancer. 2008;113:97–107. doi: 10.1002/cncr.23557. [DOI] [PubMed] [Google Scholar]
- 27.Cohen EE. Role of epidermal growth factor receptor pathway-targeted therapy in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2006;24:2659–2665. doi: 10.1200/JCO.2005.05.4577. [DOI] [PubMed] [Google Scholar]
- 28.Viloria-Petit A, Crombet T, Jothy S, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090–5101. [PubMed] [Google Scholar]
- 29.Bianco R, Rosa R, Damiano V, et al. Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin Cancer Res. 2008;14:5069–5080. doi: 10.1158/1078-0432.CCR-07-4905. [DOI] [PubMed] [Google Scholar]
- 30.Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 2006;66:3197–3204. doi: 10.1158/0008-5472.CAN-05-3090. [DOI] [PubMed] [Google Scholar]
- 31.Johns TG, Luwor RB, Murone C, et al. Antitumor efficacy of cytotoxic drugs and the monoclonal antibody 806 is enhanced by the EGF receptor inhibitor AG1478. Proc Natl Acad Sci U S A. 2003;100:15871–15876. doi: 10.1073/pnas.2036503100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirata A, Uehara H, Izumi K, Naito S, Kuwano M, Ono M. Direct inhibition of EGF receptor activation in vascular endothelial cells by gefitinib (“Iressa,” ZD1839) Cancer Sci. 2004;95:614–618. doi: 10.1111/j.1349-7006.2004.tb02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svensson S, Jirstrom K, Rydén L, et al. ERK phosphorylation is linked to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene. 2005;24:4370–4379. doi: 10.1038/sj.onc.1208626. [DOI] [PubMed] [Google Scholar]
- 34.Mulkeen AL, Silva T, Yoo PS, et al. Short interfering RNA-mediated gene silencing of vascular endothelial growth factor: effects on cellular proliferation in colon cancer cells. Arch Surg. 2006;141:367–374. doi: 10.1001/archsurg.141.4.367. [DOI] [PubMed] [Google Scholar]
- 35.Fan F, Wey JS, McCarty MF, et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene. 2005;24:2647–2653. doi: 10.1038/sj.onc.1208246. [DOI] [PubMed] [Google Scholar]
- 36.Holden SN, Eckhardt SG, Basser R, et al. Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and EGF receptor signaling, in patients with solid, malignant tumors. Ann Oncol. 2005;16:1391–1397. doi: 10.1093/annonc/mdi247. [DOI] [PubMed] [Google Scholar]