

Abstract
Dominant congenital spinal muscular atrophy (DCSMA) is a disorder of developing anterior horn cells and shows lower-limb predominance and clinical overlap with hereditary spastic paraplegia (HSP), a lower-limb-predominant disorder of corticospinal motor neurons. We have identified four mutations in bicaudal D homolog 2 (Drosophila) (BICD2) in six kindreds affected by DCSMA, DCSMA with upper motor neuron features, or HSP. BICD2 encodes BICD2, a key adaptor protein that interacts with the dynein-dynactin motor complex, which facilitates trafficking of cellular cargos that are critical to motor neuron development and maintenance. We demonstrate that mutations resulting in amino acid substitutions in two binding regions of BICD2 increase its binding affinity for the cytoplasmic dynein-dynactin complex, which might result in the perturbation of BICD2-dynein-dynactin-mediated trafficking, and impair neurite outgrowth. These findings provide insight into the mechanism underlying both the static and the slowly progressive clinical features and the motor neuron pathology that characterize BICD2-associated diseases, and underscore the importance of the dynein-dynactin transport pathway in the development and survival of both lower and upper motor neurons.
Main Text
Spinal muscular atrophies (SMAs) are a clinically and genetically heterogeneous group of disorders affecting motor neurons of the anterior horn of the spinal cord. The most common form of SMA is caused by autosomal-recessive mutations in survival of motor neuron 1 (SMN1 [MIM 600354]). Severe cases present in infancy with progressive weakness of proximal and axial muscles and early death. In contrast, autosomal-dominant congenital SMA (DCSMA) is characterized by nonprogressive congenital or early-onset lower-limb-predominant weakness1–3 and often results in significant mobility impairment. Hereditary spastic paraplegia (HSP) is also a heterogeneous predominantly lower-limb disorder that affects the upper (corticospinal) motor neurons (UMNs). Onset is usually in adulthood, and clinical features are progressive.4 Many forms of DCSMA and HSP remain genetically uncharacterized, and the pathological mechanisms that underlie these disorders have not been fully elucidated. Treatment is essentially supportive, given that effective medical therapies have not yet been developed.
We undertook a genome-wide Illumina SNP-based linkage study by using DNA from four affected and five unaffected family members from a large genetically uncharacterized DCSMA-affected kindred (AUS1, shown in Figure 1) and identified six candidate linkage regions.5 Exome sequencing of genomic DNA from two affected family members (III.2 and IV.5) was performed with the Agilent SureSelect v.2 kit for capture and the Illumina HiSeq 2000 sequencer. Reads were processed by Picard and aligned to the human reference genome (UCSC Genome Browser hg19)6 with the Burrows-Wheeler Aligner.7 Calling of single-nucleotide variants (SNVs) and small indels was performed with the Genome Analysis Tool Kit.8 Variants were annotated with a modified version of the Ensembl Variant Effect Predictor9 and filtered for inheritance patterns and predicted functional severity with the xBrowse web server. xBrowse was used for identifying variants that (1) were predicted to have a functional impact on protein-coding regions (missense, nonsense, splice-disrupting, or frameshift mutations) relative to the gene models specified by Gencode v.12, (2) fell within regions shown by previous linkage analysis to be identical by descent in all affected individuals within the family, and (3) had a frequency of less than 0.1% in data obtained from the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server, the 1000 Genomes Project, and over 1,000 internal-control exome samples. xBrowse identified 162 rare (frequency < 1%) missense variants, in-frame insertions, deletions, and predicted truncating variants shared by both affected family members, but only the c.320C>T (p.Ser107Leu) variant (RefSeq accession number NM_001003800.1) in bicaudal D homolog 2 (Drosophila) (BICD2 [MIM 609797]), encoding dynein-binding protein BICD2, was present in the previously identified linkage regions (max LOD = 1.81).
Figure 1.
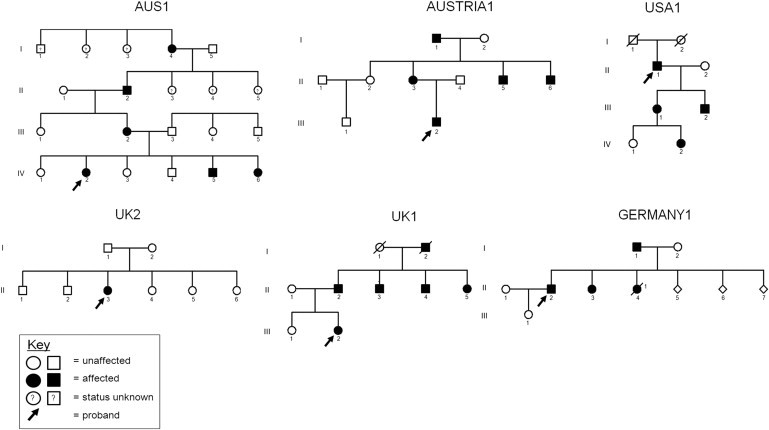
Pedigrees of Kindreds Affected by DCSMA, DCSMA+UMN, or HSP
A pattern consistent with autosomal-dominant inheritance is shown in five of the six kindreds. The sixth kindred, UK2, consists of a simplex case, confirmed to be de novo by segregation analysis. Although there is no documented male-to-male transmission in AUS1, linkage data had previously excluded X-linked loci in this kindred. The three pedigrees on the top row of the figure (AUS1, AUSTRIA1, and USA1) share the same N-terminal p.Ser107Leu substitution; haplotype analysis suggests that AUS1 and AUSTRIA1 share a common ancestry. USA1 also has European ancestry. The other three kindreds have kindred-specific mutations. The three c.320C>T (p.Ser107Leu) kindreds are affected by a classical DCSMA phenotype, and limited UMN features are present only in USA1. Affected individuals in UK1 and UK2 have more marked UMN features. Germany 1 segregates with an HSP phenotype.
Querying the Genome Variant Database for Human Diseases (GVDHD) for additional novel (defined as absent from exome variant databases) BICD2 variants resulted in the discovery of several UK- and Austria-based kindreds and cases in which BICD2 had been recently identified as a candidate gene for DCSMA. Whole-exome sequencing of the index case of a second DCSMA-affected kindred, showing additional UMN features (DCSMA+UMN) (UK1, Figure 1), was performed, and the variants were filtered as previously described.10,11 Of 96 filtered variants, the c.1502G>C (p.Arg501Pro) variant in BICD2 was identified as the likely causative mutation because the clinical phenotype caused by its mutation is similar to that of individuals with mutations in DYNC1H1 (MIM 600112), encoding a known BICD2 binding partner, cytoplasmic dynein 1 heavy chain 1.12
A genome-wide SNP (Affymetrix GeneChip Human Mapping 10K array)-based multipoint linkage analysis using genomic DNA from three affected and three unaffected family members had been performed previously in another DCSMA-affected kindred, AUSTRIA1 (Figure 1), revealing ten linked loci, including the BICD2 locus on chromosome 9. Subsequent whole-exome sequencing by GVDHD resulted in the identification of a BICD2 variant (c.320C>T [p.Ser107Leu]) as the likely causative mutation in this kindred. Whole-exome sequencing of a simplex DCSMA+UMN case (UK2, Figure 1) had been undertaken within the UK10K Consortium. The identified variant (c.565A>T [p.Ile189Phe]) was not present in either parent (de novo dominant). Additional novel heterozygous BICD2 variants with high in silico pathogenicity scores were identified by GVDHD exome sequencing of DNA from a further DCSMA+UMN-affected kindred (USA1 [c.320C>T (p.Ser107Leu)]) and a kindred affected by an HSP phenotype (GERMANY1 [c.1523A>C (p.Lys508Thr)]) (Figure 1).
The six BICD2 mutations (summarized in Table 1) were all confirmed by Sanger sequencing, and segregation with the disease phenotype was confirmed in all family members for whom DNA was available. The c.320C>T (p.Ser107Leu) mutation is in a CpG dinucleotide and thus most likely represents a methylcytosine-deamination mutation. No additional novel heterozygous variants were identified in 55 unrelated simplex and dominantly inherited UK and Australian DCSMA cases; however, additional novel variants have subsequently been identified by the GVDHD team in other DCSMA, HSP, and DCSMA+UMN mixed-phenotype cases, and Sanger sequencing and segregation confirmation are currently underway (S.Z., unpublished data).
Table 1.
Characteristics of BICD2 Mutations Identified in Six Kindreds Affected by DCSMA, HSP, or DCSMA+UMN
| Family | Phenotypea | Age of Onset | Number Affected | Nucleic Acid Mutation | Amino Acid Alteration | PolyPhe-2 Score | PolyPhen-2 Conservationb |
|---|---|---|---|---|---|---|---|
| AUS1 | DCSMA | birth | 6 | c.320C>T | p.Ser107Leu | 0.999 | 39/40 |
| AUSTRIA1 | DCSMA | birth | 5 | c.320C>T | p.Ser107Leu | 0.999 | 39/40 |
| USA1 | DCSMA+UMN | birth | 4 | c.320C>T | p.Ser107Leu | 0.999 | 39/40 |
| UK2 | DCSMA+UMN | birth | 1 | c.565A>T | p.Ile189Phe | 1.00 | 44/44 |
| UK1 | DCSMA+UMN | birth | 6 | c.1502G>C | p.Arg501Pro | 0.98 | 34/44 |
| GERMANY1 | HSP | adulthood | 4 | c.1523A>C | p.Lys508Thr | 0.912 | 32/44 |
In silico prediction of the functional consequences of the six mutations was performed with PolyPhen-2, and all six mutations were compared against the NHLBI Exome Sequencing Project Exome Variant Server for confirmation of novelty.
DCSMA+UMN indicates that UMN features were present in at least one family member.
The numerator indicates the number of species conserved for the unaltered amino acid at this location, and the denominator indicates the number of species for which the amino acid sequence is known at this location (according to PolyPhen-2 data).
Because the same mutation (c.320C>T [p.Ser107Leu]) is present in AUS1 and AUSTRIA1, we analyzed the local haplotype structure in a 200 kb window surrounding BICD2 in AUS1 and AUSTRIA1 by using genotype data from a genome-wide Illumina CytoSNP array. We determined that the mutation falls on a shared eight-SNP haplotype with a background frequency in the European population of approximately 2%, suggesting that the two families share a relatively recent founder mutation. USA1, which also segregates for c.320C>T (p.Ser107Leu), has European ancestry and might also be related to AUS1 and AUSTRIA1.
The DCSMA-affected individuals presented with congenital or early-onset hip dislocation, lower-limb contractures, foot deformities, and predominantly distal leg wasting (clinical images in Figure 2 and summary of clinical features in Table 2). Weakness was often more marked proximally in the lower limbs; upper-limb involvement was mild. UMN signs, including brisk reflexes and extensor plantar responses, were present in childhood in severely affected individuals. Motor and sensory nerve conduction studies were normal apart from reduced or absent peroneal compound muscle action potentials representing secondary axonal loss in some individuals. Electromyography (EMG) confirmed chronic denervation and reinnervation. Lower-limb MRI demonstrated a consistent pattern of muscle involvement (Figure 2) similar to that seen in individuals with DYNC1H1 mutations. The clinical features of AUS1 have previously been reported.5 Postmortem findings in an affected infant showed reduced numbers of lumbar and, to a lesser extent, cervical anterior horn motor neurons, but there was no evidence of ongoing cell-body or axonal degeneration.5 Members of GERMANY1, affected by an HSP phenotype, did not develop features until adulthood (20–70 years) (Table 1). As is typical for this disorder, affected kindred members showed lower-limb spasticity and hyperreflexia. Contractures, weakness, and wasting were slowly progressive and less severe than in DCSMA-affected kindreds. Because there is only one family affected by pure UMN features, it is premature to comment on genotype-phenotype correlations. This of course will be of considerable interest as more families are identified.
Figure 2.
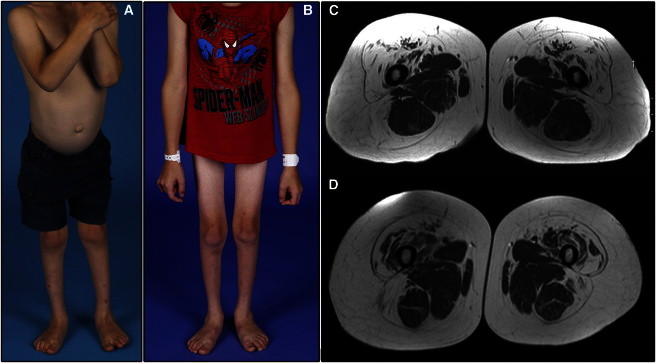
Clinical Features in Kindreds Affected by DCSMA Due to BICD2 Mutations
(A) A severely affected 4-year-old member of AUS1 (IV.5) shows marked wasting of the buttocks and entire lower limb, pes planus, and calcaneovalgus foot deformities but preserved truncal and upper-limb muscle bulk. This child has a wide-based waddling gait and hyperlordotic posture and uses a wheelchair to travel distances of more than a few hundred meters.
(B) AUS1 IV.5, now aged 7 years, shows stable wasting of the lower limbs and stable preservation of upper-limb muscle bulk. His sister (IV.6) shares similar features. Both siblings have required noninvasive nocturnal ventilation since 4 years of age for obstructive sleep-disordered breathing.
(C) MRI of the 32-year-old mother of siblings IV.5 and IV.6 shows a specific pattern of muscle involvement and sparing with or without relative hypertrophy in the upper thigh and hallmark preservation of medial adductors and semitendinosus.
(D) The MRI features of the mother in (C) are identical to those of a 24-year-old male AUSTRIA1 member, who shares the same c.320C>T (p.Ser107Leu) mutation. Members of UK1 and UK2 have more severe muscle involvement, but sparing of the same subset of muscles (e.g., medial adductors and semitendinosus in the upper thigh) is still evident. The lower-limb MRI features are almost identical to those seen in individuals with DYNC1H1-associated DCSMA.
Table 2.
Clinical Features in Kindreds Affected by BICD2 Mutations
|
Kindred |
||||||
|---|---|---|---|---|---|---|
| AUS1 | AUSTRIA1 | USA1 | UK2 | UK1 | GERMANY1 | |
| Mutation | c.320C>T (p.Ser107Leu) | c.320C>T (p.Ser107Leu) | c.320C>T (p.Ser107Leu) | c.565A>T (p.Ile189Phe) | c.1502G>C (p.Arg501Pro) | c.1523A>C (p.Lys508Thr) |
| Clinical phenotype | DCSMA | DCSMA | DCSMA+UMNa | DCSMA+UMNa | DCSMA+UMNa | HSP |
| Number of generations in pedigree | 4 | 3 | 4 | 2 | 3 | 3 |
| Number of affected and unaffected individuals (clinically confirmed) | 6 (4) | 5 (3) | 4 (4) | 1 de novo (1) | 5 (5) | 4 (1) |
| Congenital and Early-Onset Contracture Dislocations | ||||||
| Congenital hip dysplasia and/or joint dislocation | 1 | 0 | 2 | 1 | 1 | 0 |
| Congenital or early-onset hip contracture | 1 | 0 | 0 | 1 | 0 | 0 |
| Congenital or early-onset knee contracture | 2 | 0 | 0 | 1 | 0 | 0 |
| Congenital or early-onset Achilles tendon contractures | 2 | 2 | 0 | 1 | 5 | 0 |
| Congenital talipes or early-onset deformities | CV = 4, EV = 0 | CV = 2, EV = 0 | CV = 0, EV = 0 | CV = 1, EV = 0 | CV = , EV = 2 | CV = 0, EV = 0 |
| Foot Features and Abnormalities | ||||||
| High arch (H), pes cavus (C), or pes planus (P) | H = 0, C = 0, p = 4 | H = 0, C = 0, p = 0 | H = 0, C = 3, p = 0 | H = 0, C = 0, p = 0 | H = 1, C = 0, 1 p = 0 | H = 1, C = 1, p = 0 |
| Lower-Limb Features: Weakness and Wasting | ||||||
| Proximal = distal weakness | 3 | 3 | 2 | 1 | 2 | 1 |
| Proximal > distal weakness | 0 | 0 | 2 | 0 | 0 | 0 |
| Distal > proximal weakness | 1 | 0 | 0 | 0 | 2 | 0 |
| Proximal = distal wasting | 3 | 3 | 2 | 0 | 3 | 0 |
| Proximal > distal wasting | 0 | 0 | 1 | 0 | 0 | 0 |
| Distal > proximal wasting | 1 | 0 | 1 | 1 | 1 | 1 |
| No weakness or wasting but other features (e.g., contractures) | 0 | 0 | 0 | 0 | 1 | 0 |
| Adult-onset lower-limb contractures, weakness, and wasting | 0 | 0 | 0 | NA (child) | 0 | hip, Achilles tendon |
| Lower-Limb Deep-Tendon Reflexes | ||||||
| Reduced or absent in lower limbs | 4 | 2 (not tested in the third) | 1 | 1 | 1 | 0 |
| Increased in lower limbs | 0 | 0 | 0 | 0 | 2 | 1 |
| Mixed (reduced and increased) in lower limbs | 0 | 0 | 2 | 0 | 2 | 0 |
| Plantar Responses | ||||||
| Normal | 4 | 2 (not tested in the third) | 4 | 0 | 3 | 0 |
| Upgoing or equivocal | 0 | 0 | 1 | 1 | 2 | 1 |
| Upper-Limb Features: Weakness and Wasting | ||||||
| Mild weakness in one or more regions | 0 | 2 (shoulder, hand) | 3 (shoulder, fingers) | 1(elbow) | 2 (scapular winging) | 0 |
| Mild wasting in one or more regions | 0 | 2 (shoulder, hand) | 0 | 0 | 0 | 0 |
| No weakness or wasting | 4 | 1 | 1 | 0 | 0 | 1 |
| Adult-onset upper-limb contractures, weakness, and wasting | 0 | 0 | 0 | NA (child) | 0 | 0 |
| Upper-Limb Deep-Tendon Reflexes | ||||||
| Reduced or absent in upper limbs | 1 | 2 (not tested in the third) | 0 | 0 | 0 | 0 |
| Increased in upper limbs | 0 | 0 | 0 | 1 | 5 | 1 |
| Mixed in upper limbs | 0 | 0 | 0 | 0 | 0 | 0 |
| Motor Development and Ambulation | ||||||
| Delayed early motor milestones | 4 | 1 | 4 | 1 | 2 | 0 |
| Wide-based gait and/or hyperlordotic gait | 2 | 2 | 1 | 1 | 1 | 0 |
| High-stepping gait | 0 | 3 | 0 | 0 | 0 | 0 |
| Fatigues quickly when walking and falls often | 3 | 1 | 2 | 1 | 4 | 1 |
| Uses orthoses and/or walking aide, including frame | 2 | NA | 3 | 0 | 0 | 1 |
| Uses wheelchair | 2 | 1 | 0 | 1 | 1 | 2 |
| Respiratory Abnormalities | ||||||
| Obstructive sleep apnea symptoms and/or signs and/or need for NiPV | 3 | − | 1 | 0 | 0 | − |
| Restrictive lung disease and/or reduced FVC | 0 | − | 1 | 0 | 0 | − |
| Other Clinical Features | ||||||
| Vibration abnormality (V) and/or foot paraesthesias (P) | 0 | 0 | 3 (V: 1 with diabetes) | 0 | 0 | 1 (V and P) |
| Mild to moderate intellectual disability | 1 | 0 | 0 | 0 | 1 | 0 |
| Scoliosis (S), kyphosis (K), pectus excavatum (P), bulbar weakness (B) | S = 1, K = 0, p = 1, B = 1 | S = 1, K = 0, p = 0, B = 0 | S = 0, K = 1, p = ?, B = 0 | S = 0, K = 0, p = 0, B = 0 | S = 0, K = 1, p = 0, B = 0 | S = 0, K = 0, p = 0, B = 0 |
| Surgery Required | ||||||
| Surgical relocation of one or both hip and/or femoral osteotomies | 1 | − | 0 | 1 | 1 | 0 |
| Tendon-release surgery on foot and/or other lower limb | 1; 2 planned | − | 0 | 1 | 3 | 0 |
Shown are the clinical features in six kindreds affected by DCSMA, HSP, or DCSMA+UMN due to BICD2 mutations. Abbreviations are as follows: CV, calcaneovalgus; EV, equinovarus; NA, not available; NiPV, noninvasive pulmonary ventilation; and FVC, forced vital capacity.
UMN signs present in lower limbs, upper limbs, or both in at least one affected kindred member.
Bicaudal D (BICD), meaning “two tails,” was named for the strikingly abnormal anterior-to-posterior body patterning observed in BicD mutant Drosophila.13 The BICD proteins (BicD in Drosophila and BICD1 and BICD2 in mammals) are adaptor proteins, which interact with the dynein-dynactin motor complex and with the small GTPase RAB614,15 to facilitate trafficking of key cellular cargos, including mRNA, Golgi, and secretory vesicles,14,16 all of which are critical to motor neuron development and/or maintenance. During interphase, BICD2 has properties of a peripheral coat protein,15 which suggests a preference for membrane-bound cargos.
Human BICD2 contains five predicted coiled-coil binding domains grouped into three binding regions,15,17 as shown in Figure 3A. The first N-terminal binding region (CC1) interacts directly with cytoplasmic dynein,15 the second (CC2) interacts with the kinesin motor complex (kinesin-1: isoforms KIF5A and KIF5B),18 and the third (CC3) interacts with RAB6.14 All four disease-causing protein alterations identified by this study lie within the first two binding regions (Figure 3A).
Figure 3.
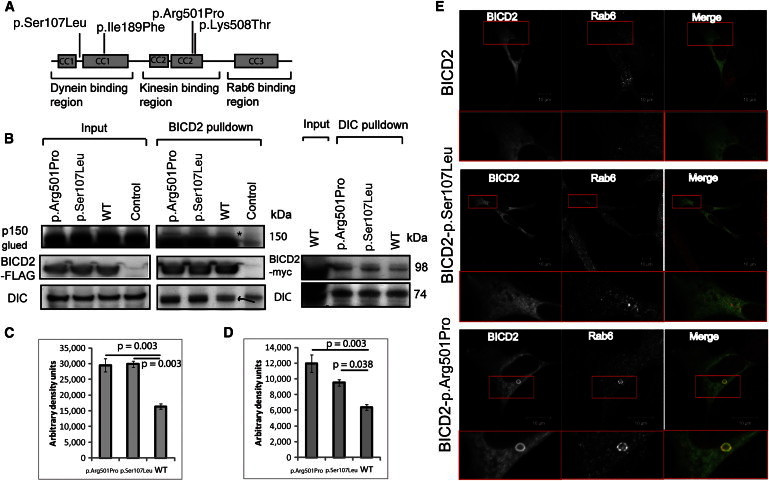
Functional Characterization of BICD2 Mutations
(A) A schematic diagram demonstrating the three BICD2 regions composed of five coiled-coil domains, the four identified alterations, and known interacting proteins.
(B) Coimmunoprecipitation studies of HEK293 cells transfected with BICD2 C terminally tagged with FLAG-Myc show a higher interaction between the p.Arg501Pro and p.Ser107Leu altered proteins and DIC and the p150Glued subunit of dynactin (band shown with ∗). The control represents a nontransfected cell lysate. For the FLAG-BICD2 pulldown, the pulled down DIC is highlighted by an arrow (the upper band is present in the nontransfected control). Reverse immunoprecipitation with DIC antibody confirmed the increased interaction between the p.Arg501Pro and p.Ser107Leu variants and DIC.
(C and D) Optical densitometry for DIC and dynactin p150Glued pulldown for a single immunoprecipitation performed in triplicate. ANOVA p = 0.003 in (C), and ANOVA p = 0.004 in (D). Error bars represent the SEM.
(E) Immunocytochemistry of SH-SY5Y cells transfected with constructs encoding p.Arg501Pro and p.Ser107Leu and stained for Myc and RAB6 show the presence of a BICD2 p.Arg501Pro-RAB6 ring structure.
In view of the known interaction between BICD2 and the dynein-dynactin motor complex and the similarities in clinical features between BICD2-associated DCSMA and DYNC1H1-associated DCSMA,12 we sought to determine what effect two BICD2 variants (BICD2p.Ser107Leu in CC1 and BICD2p.Arg501Pro in CC2) had on BICD2-dynein-complex binding affinity. Full-length C-terminal Myc-DDK-tagged human BICD2 cDNA in a p.CMV6 entry plasmid (RC209960) was obtained from Origene Technologies. BICD2 mutations were introduced into the transcript by site-directed mutagenesis with a QuikChange II XL kit (Stratagene) (primers are available upon request) and were transfected into human embryonic kidney (HEK) 293 cells with lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For immunoprecipitation, precleared cell lysate was incubated with either anti-FLAG M2 affinity gel (Sigma) or dynein intermediate chain (DIC) antibody (a kind gift from K.K. Pfister, University of Virginia) bound by Sepharose 4B protein A beads (Invitrogen) on a shaker overnight at 4°C. Proteins were eluted into SDS-PAGE sample loading buffer and loaded onto a NuPAGE 12% Bis-Tris gel (Invitrogen) for immunoblot analysis.
HEK293 cells transfected with either wild-type or altered Myc-DDK-tagged BICD2 demonstrated higher binding affinity between BICD2 and DIC for both mutations (Figures 3B and 3C). Both altered BICD2 proteins also had higher binding affinity for the p150Glued subunit of dynactin (Figures 3B and 3D). Because the p.Ser107Leu substitution lies within the dynein (CC1) binding region, altered binding is likely to be a direct result of altered BICD2-dynein interaction. Increased binding between the dynein-dynactin complex and BICD2p.Arg501Pro might be an indirect result of altered-BICD2-kinesin-1 binding, which secondarily favors dynein binding.
In order to determine whether the increased affinity for BICD2p.Ser107Leu and BICD2p.Arg501Pro with the dynein-dynactin complex has any functional consequences in vitro, we transfected SH-SY5Y cells with plasmids containing the same two altered BICD2 constructs. In SH-SY5Y cells, overexpressed wild-type BICD2 was diffusely present throughout the cytoplasm (Figure 3E). In contrast, in cells transfected with BICD2p.Arg501Pro, the altered protein accumulated in the perinuclear region, where it formed ring-like structures that colocalized with RAB6 (Figure 3E). These ring-like structures were not observed in cells transfected with BICD2p.Ser107Leu (Figure 3E). For BICD2p.Arg501Pro, it is possible that its higher affinity for the dynein-dynactin complex results in increased sequestration of RAB6-positive vesicles to the pericentrosomal region and decreased anterograde trafficking of secretory vesicles to the plasma membrane.
The bicaudal proteins play an important role in neuronal development. In Drosophila, loss of BicD leads to a congenital locomotor defect in larvae.19 During neuronal differentiation, rapid outgrowth of neurite extensions is dependent upon anterograde secretory trafficking.20 BICD2 and a closely related member of the BICD family, BICD-related protein 1 (BICDR-1), have opposing dynein-dynactin-complex-dependent trafficking roles that compete with one another to allow precise temporal regulation of neurite outgrowth.17 During very early neuronal development, BICDR-1 sequesters RAB6-positive secretory vesicles to a pericentrosomal location, which inhibits neurite outgrowth17 (Figure S1, available online). As neurons mature, BICDR-1 expression falls, releasing RAB6-positive secretory vesicles to the BICD2-adapted axonal-transport pathways for anterograde trafficking to the plasma membrane to facilitate neurite outgrowth.18,21 We propose that BICD2p.Ser107Leu and BICD2p.Arg501Pro perturb this process by favoring retrograde secretory-vesicle trafficking away from the cell periphery and back toward the pericentrosomal region (Figure S1). This would impair embryonic development of motor neurons, a pathological hallmark of DCSMA.13 Although DCSMA is primarily a nonprogressive anterior horn cell disorder, features suggestive of UMN loss become more prominent with age in DCSMA and are a characteristic feature of HSP. This suggests that mutations in BICD2 also have a detrimental effect on the maintenance of a subpopulation of UMNs.
In conclusion, we have identified causative mutations in BICD2, which encodes a dynein-dynactin trafficking adaptor protein, in six kindreds segregating for DCSMA, DCSMA+UMN, or HSP. Mutations resulting in amino acid substitutions in two binding regions increase binding affinity between BICD2 and the dynein-dynactin complex, which is likely to result in altered specificity and/or efficiency of key BICD2-mediated cellular trafficking processes. We postulate that perturbation of BICD2-mediated trafficking of RAB6-positive secretory vesicles leads to impaired development and maintenance of a key subset of anterior horn cells and UMNs and thus results in both the static and the slowly progressive features that characterize BICD2-associated disorders.
Ethical Approval
The procedures used in this study were in accordance with the ethical standards of the responsible human experimentation committees (institutional and national). This study was approved by The Children’s Hospital at Westmead (Sydney, Australia) Human Ethics Committee (#10/CHW/45), the institutional ethical review board of the UCL Institute of Child Health and Great Ormond Street Hospital in the UK (in accordance with the UK10K project ethical framework), the National Hospital for Neurology and Neurosurgery Research Ethics Committee/Central London (REC 3 09/H0716/61), the ethics committee of the Medical University of Graz (EK-Nr. 23-165 ex 10/11), and the University of Rochester Research Subjects Review Board (Inherited Neuropathies Consortium Rare Disease Clinical Research Network protocol). Informed consent was obtained from all individuals included in this research.
Acknowledgments
We would like to thank Leigh Waddell for coordination of AUS1 exome samples, Katy Eichinger for research evaluation of USA1, and Garth Nicholson, Marina Kennerson, Giovanni Stevanin, and Matthew Harms for further investigating additional BICD2 variants that ultimately did not segregate with the disease phenotype in their kindreds. AUS1 exome sequencing was supported by a grant from the National Human Genome Research Institute, National Institutes of Health (Medical Sequencing Program grant U54 HG003067 to E.S. Lander, the principal investigator at the Broad Institute of Harvard and MIT). Genetic testing of AUSTRAI1, UK1, USA1, and GERMANY1 was supported by the Inherited Neuropathies Consortium (Rare Disease Clinical Research Network, National Institute of Neurological Disorders and Stroke 1U54NS0657). The study makes use of data (from sample set NM5061932 [UK2]) generated by the UK10K Consortium with funding from the Wellcome Trust (award WT091310). Investigators who contributed to the generation of these data are listed at www.UK10K.org. Funding sources for individual authors are summarized in the Supplemental Acknowledgments.
Supplemental Data
Supplemental Data include Supplemental Acknowledgments and one figure and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Genomes Management Application (GEM.app), https://genomics.med.miami.edu/
Human Genome Variation Society, http://www.hgvs.org/mutnomen/
NCBI CCDS Database, http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi?REQUEST=CCDS&DATA=CCDS35064
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
UK10K, http://www.uk10k.org/
xbrowse server, http://atgu.mgh.harvard.edu/xbrowse
References
- 1.Fleury P., Hageman G. A dominantly inherited lower motor neuron disorder presenting at birth with associated arthrogryposis. J. Neurol. Neurosurg. Psychiatry. 1985;48:1037–1048. doi: 10.1136/jnnp.48.10.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mercuri E., Messina S., Kinali M., Cini C., Longman C., Battini R., Cioni G., Muntoni F. Congenital form of spinal muscular atrophy predominantly affecting the lower limbs: a clinical and muscle MRI study. Neuromuscul. Disord. 2004;14:125–129. doi: 10.1016/j.nmd.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Reddel S., Ouvrier R.A., Nicholson G., Dierick I., Irobi J., Timmerman V., Ryan M.M. Autosomal dominant congenital spinal muscular atrophy—a possible developmental deficiency of motor neurones? Neuromuscul. Disord. 2008;18:530–535. doi: 10.1016/j.nmd.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 4.Salinas S., Proukakis C., Crosby A., Warner T.T. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- 5.Oates E.C., Reddel S., Rodriguez M.L., Gandolfo L.C., Bahlo M., Hawke S.H., Lamandé S.R., Clarke N.F., North K.N. Autosomal dominant congenital spinal muscular atrophy: a true form of spinal muscular atrophy caused by early loss of anterior horn cells. Brain. 2012;135:1714–1723. doi: 10.1093/brain/aws108. [DOI] [PubMed] [Google Scholar]
- 6.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.J., Makhijani V., Roth G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 7.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Züchner S., Dallman J., Wen R., Beecham G., Naj A., Farooq A., Kohli M.A., Whitehead P.L., Hulme W., Konidari I. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 2011;88:201–206. doi: 10.1016/j.ajhg.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez M.A., Acosta Lebrigio R.F., Van Booven D., Ulloa R.H., Powell E., Speziani F., Tekin M., Schüle R., Züchner S. GEnomes Management Application (GEM.app): A New Software Tool for Large-Scale Collaborative Genome Analysis. Hum. Mutat. 2013 doi: 10.1002/humu.22305. Published online March 5, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harms M.B., Ori-McKenney K.M., Scoto M., Tuck E.P., Bell S., Ma D., Masi S., Allred P., Al-Lozi M., Reilly M.M. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78:1714–1720. doi: 10.1212/WNL.0b013e3182556c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bullock S.L., Ish-Horowicz D. Conserved signals and machinery for RNA transport in Drosophila oogenesis and embryogenesis. Nature. 2001;414:611–616. doi: 10.1038/414611a. [DOI] [PubMed] [Google Scholar]
- 14.Matanis T., Akhmanova A., Wulf P., Del Nery E., Weide T., Stepanova T., Galjart N., Grosveld F., Goud B., De Zeeuw C.I. Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat. Cell Biol. 2002;4:986–992. doi: 10.1038/ncb891. [DOI] [PubMed] [Google Scholar]
- 15.Hoogenraad C.C., Akhmanova A., Howell S.A., Dortland B.R., De Zeeuw C.I., Willemsen R., Visser P., Grosveld F., Galjart N. Mammalian Golgi-associated Bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J. 2001;20:4041–4054. doi: 10.1093/emboj/20.15.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swan A., Suter B. Role of Bicaudal-D in patterning the Drosophila egg chamber in mid-oogenesis. Development. 1996;122:3577–3586. doi: 10.1242/dev.122.11.3577. [DOI] [PubMed] [Google Scholar]
- 17.Schlager M.A., Kapitein L.C., Grigoriev I., Burzynski G.M., Wulf P.S., Keijzer N., de Graaff E., Fukuda M., Shepherd I.T., Akhmanova A., Hoogenraad C.C. Pericentrosomal targeting of Rab6 secretory vesicles by Bicaudal-D-related protein 1 (BICDR-1) regulates neuritogenesis. EMBO J. 2010;29:1637–1651. doi: 10.1038/emboj.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grigoriev I., Splinter D., Keijzer N., Wulf P.S., Demmers J., Ohtsuka T., Modesti M., Maly I.V., Grosveld F., Hoogenraad C.C., Akhmanova A. Rab6 regulates transport and targeting of exocytotic carriers. Dev. Cell. 2007;13:305–314. doi: 10.1016/j.devcel.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Li X., Kuromi H., Briggs L., Green D.B., Rocha J.J., Sweeney S.T., Bullock S.L. Bicaudal-D binds clathrin heavy chain to promote its transport and augments synaptic vesicle recycling. EMBO J. 2010;29:992–1006. doi: 10.1038/emboj.2009.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfenninger K.H. Plasma membrane expansion: a neuron’s Herculean task. Nat. Rev. Neurosci. 2009;10:251–261. doi: 10.1038/nrn2593. [DOI] [PubMed] [Google Scholar]
- 21.Swift S., Xu J., Trivedi V., Austin K.M., Tressel S.L., Zhang L., Covic L., Kuliopulos A. A novel protease-activated receptor-1 interactor, Bicaudal D1, regulates G protein signaling and internalization. J. Biol. Chem. 2010;285:11402–11410. doi: 10.1074/jbc.M110.105403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.