

Abstract
Proteoglycans (PGs) are a major component of the extracellular matrix in many tissues and function as structural and regulatory molecules. PGs are composed of core proteins and glycosaminoglycan (GAG) side chains. The biosynthesis of GAGs starts with the linker region that consists of four sugar residues and is followed by repeating disaccharide units. By exome sequencing, we found that B3GALT6 encoding an enzyme involved in the biosynthesis of the GAG linker region is responsible for a severe skeletal dysplasia, spondyloepimetaphyseal dysplasia with joint laxity type 1 (SEMD-JL1). B3GALT6 loss-of-function mutations were found in individuals with SEMD-JL1 from seven families. In a subsequent candidate gene study based on the phenotypic similarity, we found that B3GALT6 is also responsible for a connective tissue disease, Ehlers-Danlos syndrome (progeroid form). Recessive loss-of-function mutations in B3GALT6 result in a spectrum of disorders affecting a broad range of skeletal and connective tissues characterized by lax skin, muscle hypotonia, joint dislocation, and spinal deformity. The pleiotropic phenotypes of the disorders indicate that B3GALT6 plays a critical role in a wide range of biological processes in various tissues, including skin, bone, cartilage, tendon, and ligament.
Main Text
Skeletal dysplasias represent a vast collection of genetic disorders of the skeleton, currently divided into 40 groups.1 Spondyloepimetaphyseal dysplasia (SEMD) is one group (group 13) of skeletal dysplasia that contains more than a dozen distinctive diseases. SEMD with joint laxity (SEMD-JL) is a subgroup of SEMD that consists of type 1 (SEMD-JL1 [MIM 271640]) and type 2 (SEMD-JL2 [MIM 603546]). SEMD-JL1 or SEMD-JL Beighton type is an autosomal-recessive disorder that shows mild craniofacial dysmorphism (prominent eye, blue sclera, long upper lip, small mandible with cleft palate) and spatulate finger with short nail.2 The large joints of individuals with SEMD-JL1 are variably affected with hip dislocation, elbow contracture secondary to radial head dislocation, and clubfeet. Joint laxity is particularly prominent in the hands. Skeletal changes of SEMD-JL1 are characterized by moderate platyspondyly with anterior projection of the vertebral bodies, hypoplastic ilia, and mild metaphyseal flaring.3 Kyphoscoliosis progresses with age, leading to a short trunk, whereas platyspondyly become less conspicuous and the vertebral bodies appear squared in shape with age. Recently, dominant kinesin family member 22 (KIF22 [MIM 603213]) mutations have been found in SEMD-JL2;4,5 however, the genetic basis of SEMD-JL1 remains unknown.
To identify the SEMD-JL1-causing mutation, we performed whole-exome sequencing experiments. We recruited seven individuals with SEMD-JL1 from five unrelated Japanese families (F1–F5) and a Singapore/Japanese family (F6) (Table 1). One family (F1) had a pair of affected sibs (P1 and P2) from nonconsanguineous parents. Genomic DNA was extracted by standard procedures from peripheral blood, saliva, or Epstein-Barr virus-immortalized lymphocyte of the individuals with SEMD-JL1 and/or their parents after informed consent. The study was approved by the ethical committee of RIKEN and participating institutions. We captured the exomes of the seven subjects as previously described.6,7 In brief, we sheared genomic DNA (3 μg) by Covaris S2 system (Covaris) and processed with a SureSelect All Exon V4 kit (Agilent Technologies). We sequenced DNAs captured by the kit with HiSeq 2000 (Illumina) with 101 base pair-end reads. We performed the image analysis and base calling by HiSeq Control Software/Real Time Analysis and CASAVA1.8.2 (Illumina) and mapped the sequences to human genome hg19 by Novoalign. We processed the aligned reads by Picard to remove PCR duplicate. The mean depth of coverage for reads was 132.8×, and, on average, 91.0% of targeted bases had sufficient coverage (20× coverage) and quality for variant calling (Table S1 available online). The variants were called by Genome Analysis Toolkit 1.5-21 (GATK) with the best practice variant detection with the GAKT v.3 and annotated by ANNOVAR (2012 February 23).
Table 1.
Clinical and Radiographic Findings of the Individuals with B3GALT6 Mutations
| Subject ID | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | P12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID | F1 | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F9 | F10 |
| Clinical diagnosis | SEMD-JL1 | SEMD-JL1 | SEMD-JL1 | SEMD-JL1 | SEMD-JL1 | SEMD-JL1 | SEMD-JL1 | SEMD-JL1 | EDS-PF | EDS-PF | EDS-PF | EDS-PF |
| General Information | ||||||||||||
| Ethnicity | Japanese | Japanese | Japanese | Japanese | Japanese | Japanese | Japanese/Singaporean | Vietnamese | Italian | Italian/Canadian | Italian/Canadian | Brazilian |
| Gender | M | M | F | M | F | F | M | M | M | F | F | F |
| Age | 34 years | 31 years | 12 years, 7 months | 6 years | 5 years, 1 month | 12 years | 2 years, 9 months | 34 years | 8 months | 7 years | 1 month | 5 years, 1 month |
| Gestational age | 39 weeks, 2 days | full term | 37 weeks | 40 weeks, 1 day | 39 weeks, 5 days | full term | 39 weeks | full term | ND | 36 weeks | 37 weeks | 39 weeks |
| Birth length (cm) | ND | ND | 36 | ND | 43.1 | 42 | 43 | (average) | ND | 44 | 44 | 44 |
| Birth weight (g) | ND | 2,200 | 2,124 | 2,832 | 2,535 | 2,222 | 2,485 | 3,500 | ND | 2,097 | 2,790 | 3,300 |
| Clinical Features | ||||||||||||
| Height (cm) (SD)a | 127.7 (−7.4) | 130 (−7.0) | 88.8 (−10.7) | 94 (−4.0) | 90 (−4.0) | 118.4 (−5.1) | 78.2 (−4.0) | 118 (−9.1) | 66 (−1.6) | 90 (−6.8) | 45 (−3.7) | 81 (−5.9) |
| Weight (kg) (SD)a | 40.3 (−2.2) | 36.9 (−2.5) | 13.2 (−3.7) | 15.4 (−1.5) | 14.4 (−1.3) | 23.2 (−2.0) | 10.6 (−1.9) | 28 (−3.3) | 5.65 (−3.0) | 13.9 (−2.2) | 2.65 (−2.8) | 8.5 (−8.4) |
| Craniofacial | ||||||||||||
| Flat face with prominent forehead | ND | ND | + | + | + | + | + | – | + | + | + | + |
| Prominent eyes, proptosis | ND | ND | + | – | – | + | + | – | + | + | + | + |
| Blue sclerae | ND | ND | + | + | + | – | + | – | + | + | + | – |
| Long upper lip | ND | ND | – | + | + | – | + | + | + | + | + | – |
| Micrognathia | ND | ND | + | + | + | + | – | + | – | – | – | – |
| Cleft palate | ND | ND | – | – | – | – | – | – | – | – | – | + |
| Musculoskeletal | ||||||||||||
| Kyphoscoliosisb | + (7 months) | + (1.2 years) | + (8 months) | + (infancy) | + (2 years) | + (3 months) | + (8 months) | + (1 year) | + (6 months) | ++ (prenatal) | ++ (prenatal) | ++ (2 years) |
| Spatulate finger | – | ND | + | + | + | + | – | – | + | + | + | – |
| Finger laxity | ND | ND | + | + | – | – | + | – | ++ | + | + | + |
| Large joint laxity | ND | ND | + | + | – | – | + | – | ++ | ++ | ++ | + |
| Restricted elbow movement | + | ND | + | + | + | – | – | + | + | + | + | + |
| Hand contracture | – | – | – | – | – | + | – | – | – | + | + | – |
| Hip dislocation | – | – | – | + | – | + | – | – | – | + | + | + |
| Clubfeet | – | – | + | – | – | – | + | – | – | + | + | – |
| Muscular hypotonia | – | – | + | – | – | – | – | – | ++ | ++ | ++ | ++ |
| Skin and Hair | ||||||||||||
| Doughy skin | ND | ND | + | – | – | – | + | – | ++ | + | + | + |
| Hyperextensibility | ND | ND | + | – | – | – | + | – | ++ | + | + | – |
| Cutis laxa | ND | ND | – | – | – | – | – | – | + | + | – | + |
| Sparse hair | ND | ND | – | – | – | – | – | – | + | + | + | – |
| Others | MR, DD | camptodactyly | DD | pectus excavatum | ||||||||
| Radiological Features | ||||||||||||
| Platyspondyly | +c | +c | +c | + | + | + | + | + | + | + | + | + |
| Anterior beak of vertebral bodyb | + | + | – (4 years) | – (5 years) | + | + | + | – | + | + | + | + |
| Short ilia | + | + | + | + | + | + | + | + | + | + | + | + |
| Prominent lesser trochanter | + | + | + | – | + | + | + | + | + | + | + | + |
| Metaphyseal flaring | + | + | + | + | + | + | + | + | + | – | + | + |
| Epiphyseal dysplasia of femoral head | – | – | – | + | – | + | – | – | – | – | + | + |
| Elbow malalignment | ND | ND | + | + | + | + | + | + | + | + | + | + |
| Advanced carpal ossificationb | – (9 years) | ND | – (12 years) | + | + | + | + | ND | + | – (7 years) | – | – (5 years) |
| Carpal fusion | ND | ND | + | – | – | – | – | – | – | – | – | – |
| Metacarpal shortening | ND | ND | + | + | + | + | + | + | – | – | + | – |
| Overtubulation | – | – | – | – | – | – | – | – | + | + | + | + |
Abbreviations are as follows: SEMD-JL1, spondyloepimetaphyseal dysplasia with joint laxity type 1; EDS-PF, Ehlers-Danlos syndrome, progeroid form; ND, no data; MR, mitral regurgitation; DD, developmental delay.
At last presentation.
Age at medical attention provided in parentheses.
Absent at age 20 years in P1 and P2 and at age 12 years in P3.
Based on the hypothesis that SEMD-JL1 is inherited in an autosomal-recessive fashion, we filtered variants with the script created by BITS (Tokyo, Japan) according to following conditions: (1) variants registered in ESP5400, (2) variants found in our in-house controls (n = 274), (3) synonymous changes, (4) rare variants registered in dbSNP build 135 (MAF < 0.01), and (5) variants associated with segmental duplication. After combining variants selected by the homozygous mutation model and the compound heterozygous mutation model, we selected genes shared by individuals from three or more families. The analysis of the next-generation sequencing identified possible compound heterozygous variants in B3GALT6 in individuals from three families (Table S2). In addition, two other subjects had possible causal heterozygous variants of B3GALT6.
B3GALT6 (RefSeq accession number NM_080605.3) is a single-exon gene on chromosome 1p36.33. It encodes UDP-Gal:βGal β1,3-galactosyltransferase polypeptide 6 (or galactosyltransferase-II: GalT-II), an enzyme involved in the biosynthesis of the glycosaminoglycan (GAG) linker region.8 The biosyntheses of dermatan sulfate (DS), chondroitin sulfate (CS), and heparin/heparan sulfate (HS) GAGs start with the formation of a tetrasaccharide linker sequence, glucuronic acid-β1-3-galactose-β1-3-galactose-β1-4-xylose-β1 (GlcUA-Gal-Gal-Xyl), which is covalently attached to the core protein. The linker region synthesis involves a single linear pathway composed of four successive steps catalyzed by distinctive enzymes (Figure 1). The first step is the addition of xylose to the hydroxy group of specific serine residues on the core protein by xylosyltransferases from UDP-Xyl, followed by two distinct galactosyltransferases (GalT-I and II) and a glucuronosyltransferase from UDP-Gal and UDP-GlcUA, respectively. The next hexosamine addition is critical because it determines which GAG (i.e., CS, DS, or HS) is assembled on the linker region. GalT-II encoded by B3GALT6 functions in the third step of the linker formation (Figure 1).
Figure 1.
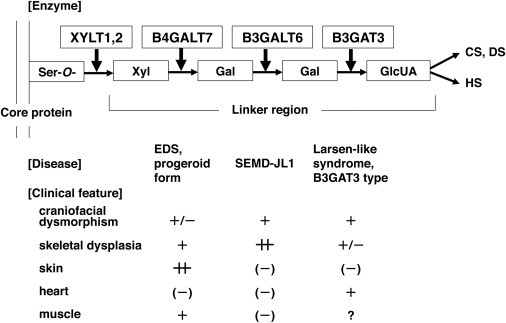
Enzymes Involved in Biosynthesis of the Glycosaminoglycan Linker Region and Summary Features of Diseases Caused by Their Defects Based on a Conventional Concept for the Diseases
The biosyntheses of GAGs start with the formation of a common tetrasaccharide linker sequence covalently attached to the core protein. The linker region synthesis involves a single linear pathway composed of four successive steps catalyzed by distinctive enzymes. Abbreviations are as follows: XYLT, β-xylosyltransferase; B4GALT7, xylosylprotein β1,4-galactosyltransferase, polypeptide 7 (β1,4-galactosyltransferase-I); B3GALT6, UDP-Gal, βGal β1,3-galactosyltransferase polypeptide 6 (β1,3-galactosyltransferase-II); B3GAT3, β-1,3-glucuronyltransferase 3 (glucuronosyltransferase I); Ser-O, the serine residue of the GAG attachment site on the proteoglycan core protein; Xyl, xylose; Gal, galactose; GlcUA, D-glucuronic acid; CS, chondroitin sulfate; DS, dermatan sulfate; HS, heparan sulfate; EDS, Ehlers-Danlos syndrome; SEMD-JL1, spondyloepimetaphyseal dysplasia with joint laxity type 1.
To confirm the results obtained by the next-generation sequencing, we examined the seven subjects used for the next-generation sequencing and an additional subject from a Vietnamese family (F7) by direct sequence of the PCR products from genomic DNAs using 3730xl DNA Analyzer (Applied Biosystems). The Sanger sequencing confirmed all B3GALT6 mutations found by the next-generation sequencing and identified additional B3GALT6 mutations. The results indicated that B3GALT6 mutations were found in all subjects (Tables 2 and S1). All but P4 from F3 were compound heterozygotes of missense mutations. In P4, only a heterozygous c.1A>G (p.Met1?) mutation was found, although we searched for a B3GALT6 mutation in the entire coding region, 5′ and 3′ UTRs, and flanking regions of B3GALT6. Most of the mutations are predicted to be disease causing by in silico analysis. The c.1A>G (p.Met1?) mutation was found in individuals from five of the seven families.
Table 2.
B3GALT6 Mutations in Spondyloepimetaphyseal Dysplasia with Joint Laxity Type 1 and Ehlers-Danlos Syndrome, Progeroid Form
| Family | Clinical Diagnosis | Nucleotide Change | Amino Acid Change |
|---|---|---|---|
| F1 | SEMD-JL1 | c.1A>G | p.Met1? |
| c.694C>T | p.Arg232Cys | ||
| F2 | SEMD-JL1 | c.1A>G | p.Met1? |
| c.466G>A | p.Asp156Asn | ||
| F3a | SEMD-JL1 | c.1A>G | p.Met1? |
| F4 | SEMD-JL1 | c.1A>G | p.Met1? |
| c.694C>T | p.Arg232Cys | ||
| F5 | SEMD-JL1 | c.694C>T | p.Arg232Cys |
| c.899G>C | p.Cys300Ser | ||
| F6 | SEMD-JL1 | c.1A>G | p.Met1? |
| c.193A>G | p.Ser65Gly | ||
| F7 | SEMD-JL1 | c.200C>T | p.Pro67Leu |
| c.694C>T | p.Arg232Cys | ||
| F8 | EDS-PF | c.353delA | p.Asp118Alafs∗160 |
| c.925T>A | p.Ser309Thr | ||
| F9 | EDS-PF | c.588delG | p.Arg197Alafs∗81 |
| c.925T>A | p.Ser309Thr | ||
| F10 | EDS-PF | c.16C>T | p.Arg6Trp |
| c.415_423del | p.Met139Ala141del |
The nucleotide changes are shown with respect to B3GALT6 mRNA sequence. The corresponding predicted amino acid changes are numbered from the initiating methionine residue.
Only a heterozygous mutation was found.
Although mutations affecting initiation codons have been reported to be pathogenic in several diseases,9 the effects of initiation codon mutations on the encoded protein are variable among the genes. We therefore investigated the effect of the c.1A>G (p.Met1?) mutation on the protein by using C-terminally FLAG-tagged B3GALT6 with and without the mutation expressed in HeLa cells (RIKEN Cell Bank). We detected the mutant B3GALT6 protein with a molecular weight ∼4 kD lower compared with the wild-type (WT) protein (Figure 2A). These results suggest that translation initiation at the second ATG of the coding sequence, at position c.124, would become the initiation codon because of the mutation, probably resulting in an N-terminal deletion of 41 amino acids (p.Met1_Ala41del), in the same open reading frame that contains the transmembrane domain. We then examined the subcellular localization of the mutant B3GALT6 protein by immunocytochemistry. The immunofluorescence for WT-B3GALT6 was observed in a perinuclear region overlapping with that for α-mannosidase II, a marker of the Golgi as previously reported.8 In contrast, the immunofluorescence for the mutant B3GALT6 protein was observed in the nucleus and cytoplasm (Figure 2B). Therefore, the mutant protein can be considered to be functionally null because of the mislocalization.
Figure 2.
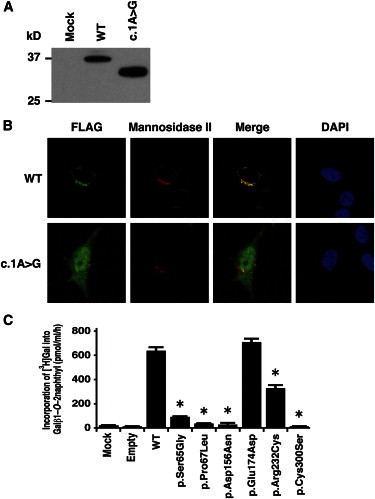
Analyses of B3GALT6 Missense Mutant Proteins Identified in Individuals with SEMD-JL1 In Vitro
(A) Immunoblot analysis of lysates from HeLa cells expressing transfected wild-type (WT) and mutant (c.1A>G) B3GALT6. The mutant B3GALT6 yields a shortened protein. The difference of the molecular sizes between WT and mutant proteins is approximately ∼4 kD.
(B) Subcellular localization of B3GALT6. HeLa cells were transfected with WT and mutant (c.1A>G) B3GALT6. Cells were stained with anti-FLAG (green), anti-α-mannosidase II (red), and 4′,6-diamidino-2-phenylindole (DAPI; blue). WT was expressed in the Golgi, but the mutant was found in cytoplasm and nucleus.
(C) Decreased enzyme activities of the missense mutant proteins (p.Ser65Gly, p.Pro67Lys, p.Asp156Asn, p.Arg232Cys, and p.Cys300Ser). p.Glu174Asp is a common polymorphism in the public database. The GalT-II activity is measured by incorporation of [3H]Gal into Galβ1-O-2naphthyl (pmol/ml/hr) and represents the averages of three independent experiments performed in triplicate. Empty and mock indicate the GalT-II activity obtained with the conditioned medium transfected with or without an empty vector. ∗p < 0.0001 versus WT (one-way analysis of variance with Dunnett’s adjustment).
To investigate the causality of other B3GALT6 missense mutations, we also examined the subcellular localization of the mutant B3GALT6 proteins by immunocytochemistry. c.193A>G (p.Ser65Gly), c.200C>T (p.Pro67Leu), and c.694C>T (p.Arg232Cys) mutants showed mislocalization, whereas c.466G>A (p.Asp156Asn) and c.899G>C (p.Cys300Ser) mutants showed normal localization (Figure S1). To investigate whether the B3GALT6 missense mutations affect the enzyme function, the GalT-II activities of soluble FLAG-tagged proteins for WT and mutant B3GALT6 proteins were assayed. The GalT-II activities of p.Ser65Gly-, p.Pro67Leu-, p.Asp156Asn-, p.Arg232Cys-, and p.Cys300Ser-B3GALT6 were significantly decreased compared with WT-B3GALT6 (Figure 2C), indicating that these mutations resulted in a loss of enzyme function. On the other hand, there were no significant differences in the GalT-II activities between WT-B3GALT6 and p.Glu174Asp-B3GALT6, a common polymorphism (rs12085009) in the public database (Figure 2C).
All SEMD-JL1 individuals with the B3GALT6 mutation had the characteristic skeletal abnormalities, including platyspondyly, short ilia, and elbow malalignment (Table 1 and Figure S2); however, some had a range of extraskeletal and connective tissue abnormalities that overlapped with those seen in Ehlers-Danlos syndrome, progeroid form (EDS-PF [MIM 130070]). EDS-PF is an autosomal-recessive connective tissue disorder characterized by sparse hair, wrinkled skin, and defective wound healing with atrophic scars.10 In addition, skeletal abnormalities so far reported in EDS-PF are limited to generalized osteopenia and radial head dislocation, which are in contrast with the severe generalized dysplasias of the axial and appendicular skeleton observed in SEMD-JL1. Thus, both disorders at first glance appear as separate clinical entities, although they share the clinical features of short stature, joint laxity and dislocation, and facial dysmorphism. In two families with individuals with EDS-PF, recessive mutations of B4GALT7 (MIM 604327) have been found.11,12 B4GALT7 (RefSeq NM_007255.2) encodes an enzyme, xylosylated protein β-1,4-galactosyltransferase, that catalyzes the second step of the GAG linker region biosynthesis (Figure 1). Therefore, we speculated that B3GALT6 and B4GALT7 deficiencies might show similar phenotypes. We then examined B3GALT6 in four additional individuals (P9–P12) who had phenotypes compatible with EDS-PF (Table 1 and Figure S3) but in whom no B4GALT7 mutations had been found. Sanger sequencing of the EDS-PF-like subjects revealed that all were compound heterozygotes for B3GALT6 mutations (Table 2). There were two frameshift mutations and one missense mutation (c.925T>A [p.Ser309Thr]) common in two families (F8 and F9). We investigated the enzyme function of the missense mutation by using the same assay for SEMD-JL1 missense mutations. The GalT-II activities of p.Ser309Thr-B3GALT6 were significantly decreased (Figure S4).
Collectively, 11 different mutations in individuals from 10 families were identified in B3GALT6 by a combination of exome and targeted sequencing (Table 2 and Figure S5). None of these mutations were detected in more than 200 ethnicity-matched controls or in public databases, including the 1000 Genomes database, indicating that they are unlikely to be polymorphisms. SEMD-JL1 and EDS-PF-like individuals had no common mutations (Table 2). The individuals with B3GALT6 mutations were short at birth and their short stature worsened with age. Their common clinical features were a flat face with prominent forehead and kyphoscoliosis (Table 1). Kyphoscoliosis was noticed in infancy in most cases and even in utero in severe cases. Although skeletal changes were essentially the same, craniofacial and skin abnormalities, joint laxities, and muscular hypotonia were variable among the individuals with B3GALT6 mutations. Common radiographic features were platyspondyly that becomes less conspicuous with age, short ilia, and elbow malalignment (Table 1). Prominent lesser trochanters and metaphyseal flaring were seen in most cases. No individuals showed generalized osteoporosis. The disease phenotype was very variable between families (mutations), but in two familial cases, phenotypes were similar between the pair of the sibs. As a corollary, our results indicate that EDS-PF is genetically heterogeneous, with a proportion of cases being caused by mutations in B4GALT7 and another in B3GALT6.
Diseases caused by defects in enzymes involved in the biosynthesis of the GAG linker region are categorized as the GAG linkeropathy. The first member of GAG linkeropathy has been identified to arise from an EDS-PF/B4GALT7 deficiency. B4GALT7 mutations have been identified in homozygous c.808C>T (p.Arg270Cys)12 and compound heterozygous (c.557C>A [p.Ala186Asp] and c.617T>C [p.Leu206Pro])11 states. Another member of GAG linkeropathy manifests itself as Larsen-like syndrome, B3GAT3 type (MIM 245600). A family with individuals harboring a homozygous B3GAT3 (MIM 606374; RefSeq NM_012200.3) mutation (c.830G>A [p.Arg227Gln]) has been identified. The clinical features of five affected individuals of the family are characterized by dislocation and laxity of joints and congenital heart defects.11 The former considerably overlaps with the phenotypes of SEMD-JL1 and EDS-PF, two other GAG linkeropathies; however, the association of heart defects has critically differentiated this disease from the others (Figure 1).
Given that the linker region biosynthesis is nonparallel and that the defects in the three enzymes simply affect the amounts of the linker region available to form GAGs (CS, HS, DS), phenotypic similarities of the three diseases are quite understandable. The quantitative difference of the phenotypes (severity of the diseases) most probably results from the difference in the degree of enzyme defects resulting from mutations. On the other hand, qualitative differences of the three diseases (e.g., scoliosis caused by the B3GALT6 mutation, heart disease caused by the B3GAT3 mutation, etc.) suggest other explanations. Tissue expression patterns of the three genes do not entirely explain the differences. We examined their mRNA expression in various human tissues, including cartilage, bone, and connective tissues by quantitative real-time PCR (Figure S6). We detected strong expression of B3GALT6 in cartilage and bone but only weak expression in skin, ligament, and tendon. B4GALT7 expression was stronger in cartilage than B3GALT6 and also weak in skin and ligament. B3GAT3 expression was not specific to heart. The qualitative difference may result from the difference in the effects of the three genes on GAG formation.
To examine how B3GALT6 mutations affects the products of GAGs in vivo, we measured the amounts of CS and HS chains at the surface of lymphoblastoid cells from the subjects by flow cytometry by using CS-stub and HS-stub antibodies as previously described.13–15 In brief, purified GAG fractions were treated individually with a mixture of chondroitinases ABC and AC-II, a mixture of chondroitinases AC-I (EC 4.2.2.5) (Seikagaku Corp.) and AC-II (EC 4.2.2.5) (Seikagaku Corp.), chondroitinase B (EC 4.2.2.19) (IBEX Technologies), or a mixture of heparinases-I and -III (IBEX Technologies) for analyzing the disaccharide composition of CS/DS, CS, DS, and HS, respectively. The digests were labeled with a fluorophore 2-aminobenzamide (2AB) and aliquots of the 2AB derivatives of CS/DS/HS disaccharides were analyzed by anion-exchange HPLC on a PA-03 column (YMC Co.). The HS-stub antibody (3G10) showed a markedly reduced binding to the epitopes on the subjects’ cells (Figure S7). The relative numbers of the HS chains presented as the mean fluorescence intensity (MFI) of the cell population stained with the antibody for P1, P2, and P3 were 26%, 56%, and 35% of the control, respectively. On the other hand, the CS-stub antibody (2B6) showed a similar binding to the epitopes on the subjects’ cells relative to those of the control (Figure S7). The MFI for P1, P2, and P3 were 114%, 104%, and 106% of the control, respectively. Furthermore, we measured disaccharide of GAG chains from lymphoblastoid cells by using anion-exchange HPLC after digestion with chondroitinase and heparinase. The amounts of the disaccharide from HS chains were significantly decreased, whereas CS and DS chains were ∼5 times higher than those in the control (Table 3).
Table 3.
The Amount of GAGs in the Lymphoblastoid Cells from Individuals with Spondyloepimetaphyseal Dysplasia with Joint Laxity Type 1
| Subject |
GAG (Disaccharides/mg Acetone Powder)a[pmol] |
|||
|---|---|---|---|---|
| CS/DS | CS | DS | HS | |
| Control | 62 | 48 | 29 | 128 |
| SEMD-JL1 | ||||
| P1 | 313 | 295 | 118 | 15 |
| P2 | 345 | 175 | 60 | 21 |
| P3 | 270 | 162 | 28 | 20 |
Calculated based on the peak area in chromatograms of digests with a mixture of chondroitinases ABC and AC-II (CS/DS), chondroitinases AC-I and AC-II (CS), chondroitinase B (DS), and heparinases I and III (HS).
Previous biochemical studies on EDS-PF with B4GALT7 mutations show a reduction in the synthesis of DS chains.16,17 The c.830G>A (p.Arg227Gln) mutation in B3GAT3 causes a drastic reduction in GlcAT-I activity in fibroblasts of the individual with SEMD-JL1 and numbers of CS and HS chains on the core proteins at the surface of the fibroblasts are decreased to about half of the controls.11 Cultured lymphoblastoid cells from individuals with a c.419C>T (p.Pro140Leu) mutation in B3GAT3 show that defective synthesis is more pronounced for CS than for HS.11 Taken together with our results, these findings suggest that the effects of the deficiencies of the three enzymes on GAG synthesis are not identical. A possible explanation for the qualitative phenotypic differences may be that the biosynthesis of the GAG linker region is not a simple step-by-step addition but involves parallel processing and/or alternative pathways. Other glycosyltransferases may have similar biochemical functions to these three enzymes and thus complement their deficient activities to variable degrees in cell- and/or tissue-specific manners, leading to differences in the amount of GAGs in the tissues. It is known that B3GALT6 and B4GALT7 have several homologs.18 It must be noted that all biochemical studies so far have been performed in vitro or in cultured cells, and therefore there is a severe limitation to our understanding of the pathogenesis at tissue and organ levels.
By exome sequencing, we identified loss-of-function mutations in B3GALT6 in 12 individuals from 10 families. The mutations produced a spectrum of connective tissue disorders characterized by lax skin, muscle hypotonia, joint dislocation, and skeletal dysplasia and deformity, which include phenotypes previously known as SEMD-JL1 and EDS-PF (Figures S1 and S2). The pleiotropic phenotypes of B3GALT6 mutations indicate that B3GALT6 plays critical roles in development and homeostasis of various tissues, including skin, bone, cartilage, tendons, and ligaments. Biochemical studies that used lymphoblastoid cells of the individuals with B3GALT6 mutations showed a decrease of HS and a paradoxical increase of CS and DS of the cell surface. Further clinical, genetic, and biological studies are necessary to understand the pathological mechanism of the diseases caused by enzyme defects involved in the biosynthesis of the GAG linker region and roles of the region in GAG metabolism and function.
Acknowledgments
We thank the individuals with the disease and their family for their help to the study. We also thank the Japanese Skeletal Dysplasia Consortium. This study is supported by research grants from the Ministry of Health, Labor, and Welfare (23300101 to S.I. and N. Matsumoto; 23300201 to S.I.), by Grants-in-Aid for Young Scientists (23689052 to N. Miyake and 23790066 to S.M.) from the Japan Society for the Promotion of Science; by the Matching Program for Innovations in Future Drug Discovery and Medical Care (K.S.); by The Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT); by a Grant-in-aid for Encouragement from the Akiyama Life Science Foundation (S.M.); by Swiss National Science Foundation Grants (31003A_141241 and 310030_132940); by The CoSMO-B project (Brazil and Switzerland); by the Leenaards Foundation (Switzerland); and by Research on intractable diseases, Health and Labour Sciences Research Grants, H23-Nanchi-Ippan-123 (S.I.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Novoalign, http://www.novocraft.com/main/page.php?s=novoalign
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beighton P., Gericke G., Kozlowski K., Grobler L. The manifestations and natural history of spondylo-epi-metaphyseal dysplasia with joint laxity. Clin. Genet. 1984;26:308–317. doi: 10.1111/j.1399-0004.1984.tb01065.x. [DOI] [PubMed] [Google Scholar]
- 3.Nishimura G., Satoh M., Aihara T., Aida N., Yamamoto T., Ozono K. A distinct subtype of “metatropic dysplasia variant” characterised by advanced carpal skeletal age and subluxation of the radial heads. Pediatr. Radiol. 1998;28:120–125. doi: 10.1007/s002470050310. [DOI] [PubMed] [Google Scholar]
- 4.Boyden E.D., Campos-Xavier A.B., Kalamajski S., Cameron T.L., Suarez P., Tanackovic G., Andria G., Ballhausen D., Briggs M.D., Hartley C. Recurrent dominant mutations affecting two adjacent residues in the motor domain of the monomeric kinesin KIF22 result in skeletal dysplasia and joint laxity. Am. J. Hum. Genet. 2011;89:767–772. doi: 10.1016/j.ajhg.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Min B.J., Kim N., Chung T., Kim O.H., Nishimura G., Chung C.Y., Song H.R., Kim H.W., Lee H.R., Kim J. Whole-exome sequencing identifies mutations of KIF22 in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am. J. Hum. Genet. 2011;89:760–766. doi: 10.1016/j.ajhg.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyake N., Elcioglu N.H., Iida A., Isguven P., Dai J., Murakami N., Takamura K., Cho T.J., Kim O.H., Hasegawa T. PAPSS2 mutations cause autosomal recessive brachyolmia. J. Med. Genet. 2012;49:533–538. doi: 10.1136/jmedgenet-2012-101039. [DOI] [PubMed] [Google Scholar]
- 7.Tsurusaki Y., Okamoto N., Ohashi H., Kosho T., Imai Y., Hibi-Ko Y., Kaname T., Naritomi K., Kawame H., Wakui K. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 2012;44:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- 8.Bai X., Zhou D., Brown J.R., Crawford B.E., Hennet T., Esko J.D. Biosynthesis of the linkage region of glycosaminoglycans: cloning and activity of galactosyltransferase II, the sixth member of the beta 1,3-galactosyltransferase family (beta 3GalT6) J. Biol. Chem. 2001;276:48189–48195. doi: 10.1074/jbc.M107339200. [DOI] [PubMed] [Google Scholar]
- 9.Saunders C.J., Minassian B.E., Chow E.W., Zhao W., Vincent J.B. Novel exon 1 mutations in MECP2 implicate isoform MeCP2_e1 in classical Rett syndrome. Am. J. Med. Genet. A. 2009;149A:1019–1023. doi: 10.1002/ajmg.a.32776. [DOI] [PubMed] [Google Scholar]
- 10.Kresse H., Rosthøj S., Quentin E., Hollmann J., Glössl J., Okada S., Tønnesen T. Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid. Am. J. Hum. Genet. 1987;41:436–453. [PMC free article] [PubMed] [Google Scholar]
- 11.Baasanjav S., Al-Gazali L., Hashiguchi T., Mizumoto S., Fischer B., Horn D., Seelow D., Ali B.R., Aziz S.A., Langer R. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 2011;89:15–27. doi: 10.1016/j.ajhg.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faiyaz-Ul-Haque M., Zaidi S.H., Al-Ali M., Al-Mureikhi M.S., Kennedy S., Al-Thani G., Tsui L.C., Teebi A.S. A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type. Am. J. Med. Genet. A. 2004;128A:39–45. doi: 10.1002/ajmg.a.30005. [DOI] [PubMed] [Google Scholar]
- 13.Kinoshita A., Sugahara K. Microanalysis of glycosaminoglycan-derived oligosaccharides labeled with a fluorophore 2-aminobenzamide by high-performance liquid chromatography: application to disaccharide composition analysis and exosequencing of oligosaccharides. Anal. Biochem. 1999;269:367–378. doi: 10.1006/abio.1999.4027. [DOI] [PubMed] [Google Scholar]
- 14.Miyake N., Kosho T., Mizumoto S., Furuichi T., Hatamochi A., Nagashima Y., Arai E., Takahashi K., Kawamura R., Wakui K. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010;31:966–974. doi: 10.1002/humu.21300. [DOI] [PubMed] [Google Scholar]
- 15.Mizumoto S., Sugahara K. Glycosaminoglycan chain analysis and characterization (Glycosylation/Epimerization). In Methods in Molecular Biology. In: Rédini F., editor. Proteoglycans: Methods and Protocols. Humana Press, Springer; New York, USA: 2012. pp. 99–115. [DOI] [PubMed] [Google Scholar]
- 16.Okajima T., Fukumoto S., Furukawa K., Urano T. Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 1999;274:28841–28844. doi: 10.1074/jbc.274.41.28841. [DOI] [PubMed] [Google Scholar]
- 17.Quentin E., Gladen A., Rodén L., Kresse H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. USA. 1990;87:1342–1346. doi: 10.1073/pnas.87.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Togayachi A., Sato T., Narimatsu H. Comprehensive enzymatic characterization of glycosyltransferases with a beta3GT or beta4GT motif. Methods Enzymol. 2006;416:91–102. doi: 10.1016/S0076-6879(06)16006-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.