

Abstract
Dowling-Degos disease (DDD), or reticular pigmented anomaly of the flexures, is a type of rare autosomal-dominant genodermatosis characterized by reticular hyperpigmentation and hypopigmentation of the flexures, such as the neck, axilla, and areas below the breasts and groin, and shows considerable heterogeneity. Loss-of-function mutations of keratin 5 (KRT5) have been identified in DDD individuals. In this study, we collected DNA samples from a large Chinese family affected by generalized DDD and found no mutation of KRT5. We performed a genome-wide linkage analysis of this family and mapped generalized DDD to a region between rs1293713 and rs244123 on chromosome 20. By exome sequencing, we identified nonsense mutation c.430G>T (p.Glu144∗) in POFUT1, which encodes protein O-fucosyltransferase 1, in the family. Study of an additional generalized DDD individual revealed the heterozygous deletion mutation c.482delA (p.Lys161Serfs∗42) in POFUT1. Knockdown of POFUT1 reduces the expression of NOTCH1, NOTCH2, HES1, and KRT5 in HaCaT cells. Using zebrafish, we showed that pofut1 is expressed in the skin and other organs. Morpholino knockdown of pofut1 in zebrafish produced a phenotype characteristic of hypopigmentation at 48 hr postfertilization (hpf) and abnormal melanin distribution at 72 hpf, replicating the clinical phenotype observed in our DDD individuals. At 48 and 72 hpf, tyrosinase activities decreased by 33% and 45%, respectively, and melanin protein contents decreased by 20% and 25%, respectively. Our findings demonstrate that POFUT1 mutations cause generalized DDD. These results strongly suggest that the protein product of POFUT1 plays a significant and conserved role in melanin synthesis and transport.
Introduction
Dowling-Degos disease (DDD [MIM 179850]) is an autosomal-dominant genodermatosis characterized by reticular pigmented anomaly. It was first described by Dowling and Freudenthal in 1938.1 Degos and Ossipowski confirmed this disease to be a distinct clinical entity in 1954.2 In classic DDD, individuals usually present with pigmentation with a flexural distribution.3,4 However, generalized DDD could also manifest numerous hypopigmented or erythematous macules and papules on the neck, chest, and abdomen.5–9 Generalized DDD resembles dyschromatosis universalis hereditaria (DUH [MIM 127500]).10 In contrast to those of DUH and dyschromatosis symmetrica hereditaria (MIM 127400), the histopathology of DDD reveals typical thin branch-like patterns of epidermal downgrowth.9–11
In 2006, Li et al. mapped the DDD-associated gene to chromosomal region 17p13.3.3 Betz et al. mapped a novel locus for DDD to chromosome 12q in two German families and identified loss-of-function mutations in keratin 5 (KRT5 [MIM 148040]).4 However, no KRT5 mutations have been identified in several familial and sporadic cases.4 Therefore, additional genetic mutations responsible for this disease should be investigated.
Here, we ruled out KRT5 mutations in two Chinese families affected by DDD and identified two mutations in protein O-fucosyltransferase 1 (POFUT1 [MIM 607491]) as the cause of the disease. Functional analysis revealed the role of POFUT1 in melanin synthesis and transport.
Material and Methods
Subjects and Clinical Presentation
This study investigated two Chinese generalized-DDD-affected families, one from Jiangsu Province and one from Chongqing. Family 1 includes 25 individuals (13 affected and 12 unaffected), and family 2 includes seven individuals (two affected and five unaffected) (Figures 1A and 1B). DDD was diagnosed by experienced dermatologists on the basis of typical manifestations and histopathological findings. After informed consent was obtained, genomic DNA was extracted from the peripheral-blood lymphocytes of DDD individuals. Skin biopsies were performed on the groin of the index individual (III:11) in family 1 and on the neck of the proband (II:3) in family 2. This study was approved by the ethics committees of the Shanghai Jiaotong University School of Medicine and was conducted in accordance with the principles of the Declaration of Helsinki.
Figure 1.
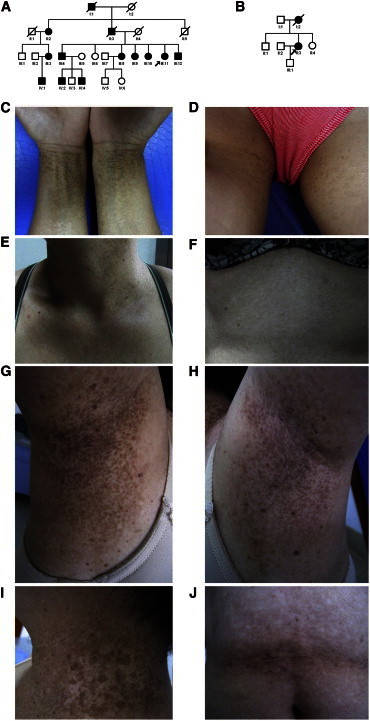
Family Pedigrees and Clinical Photographs of Individuals with Generalized DDD
(A and B) Pedigrees of families 1 (A) and 2 (B). The squares and circles denote males and females, respectively. Affected individuals are shown in black. The probands are denoted by arrows.
(C–F) Clinical photographs of individual III:11 in family 1. Reticular hyperpigmentation on the wrists (C) and inguinal regions (D), reticular hyperpigmentation and hyperkeratotic dark-brown papules on the neck (E), and reticular hypopigmentation on the abdomen (F) are shown.
(G–J) Clinical photographs of individual II:3 in family 2. Reticular hyperpigmentation and hypopigmentation on the bilateral axillae with hyperkeratotic dark-brown papules (G and H), reticular hyperpigmentation and hypopigmentation on the neck (I), and reticular hypopigmentation on the abdomen (J) are shown.
The proband of family 1 is a 49-year-old woman with a 20-year history of reticular hyperpigmentation on the skin of the neck, wrist, chest, inguinal regions, and inner sides of the thighs (Figures 1C–1E). Spots of pigmentation were initially found on the neck at the age of 23 years. These spots gradually developed in number and size and showed deeper coloration. Hypopigmented macules appeared on the abdomen and back 20 years ago without obvious subjective symptoms (Figure 1F). Skin lesions obviously worsened in summer and lightened in winter. All DDD individuals presented with pigmentation on the skin of the neck, wrist, chest, inguinal regions, and inner sides of the thighs (Figure S1, available online). Hypopigmented macules and hyperkeratotic darkbrown papules were found on the neck, chest, and back of some DDD individuals in family 1 (Figure S1). Hematoxylin and eosin staining showed hyperkeratosis with multiple horny follicular plugs and papillary epidermal downgrowth with abnormal basal pigment-granule distribution. Sporadic melanin granules and melanophages were seen in the superficial layer of dermis (Figure S2A). Masson-Fontana stain showed uneven distribution of melanin in the epidermis. A region of stratum basal showed a large number of melanin particles that were evenly distributed all over the layer of the epidermis (Figure S2C). Another region of the epidermis, however, rarely showed positive staining (Figure S2D).
Transmission electron microscopy (TEM) showed that melanocytes in the controls contained sufficient melanosomes at stages I–IV in the cytoplasm. By contrast, all of melanocytes of DDD individual III:11 in family 1 were small in size and lacked melanosomes (Figures 2A–2C). In addition, premelanosomes were notably rarely seen in melanocytes scattered with melanosomes (Figure 2A). Nevertheless, keratinocytes showed normal keratin filaments and interactions with hemidesmosomes and desmosomes (Figure 2D).
Figure 2.
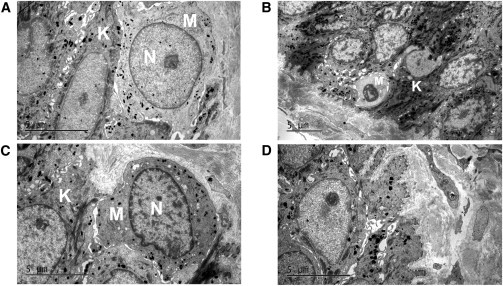
Electron Microscopy of Skin from DDD Individual III:11 in Family 1 and a Control
(A) Ultrathin section from the individual with DDD shows a few melanosomes in the melanocytes. Of note, premelanosomes are rarely seen. Abbreviations are as follows: M, melanocyte; K, keratinocyte; and N, nucleus.
(B and C) In contrast with control skin (C), the melanocyte of the individual with DDD (B) is small in size, has lighter cytoplasm, and lacks melanosomes.
(D) Normal keratin filaments and their interaction with hemidesmosomes and desmosomes of the individual with DDD.
In family 2, the proband is a 53-year-old woman who has presented with brown macules over flexural areas for over 32 years. Skin lesions initially developed on the neck and gradually increased in size. Pigmented and hypopigmented macules then spread to the face, bilateral axillae, popliteal fossa, and groin without subjective symptoms (Figures 1G–1J). Some of the macules merged and organized in a reticular pattern. Hyperkeratotic dark-brown papules were found on the bilateral axillae (Figures 1G and 1H). A skin biopsy from the neck showed epidermal papillomatous hyperkeratosis with keratotic plugs, trochanterellus elongation with abnormal basal pigment-granule distribution, and perivascular lymphohistiocytic infiltration of the superficial dermis (Figure S2B).
Sequence Analysis
Five micrograms of genomic DNA from an affected individual (III:8 in family 1) were sent to Genergy Biotech (Shanghai, China) for whole-exome capture and sequencing. A Sure Select Human All Exon Kit (Agilent) was used for exon capture. Exomes were then sequenced on an Illumina Hiseq2000 (Illumina) according to the manufacturer’s instructions. Variants were filtered against dbSNP135, the 1000 Genomes Project, and HapMap 8.
We designed primers flanking all seven coding exons and intron-exon boundaries of POFUT1 (RefSeq accession number NM_015352.1) (Table S1). PCR products were purified with a QIAquick PCR Purification Kit (QIAGEN). POFUT1 was sequenced with an ABI PRISM 3730 automated sequencer (Applied Biosystems). Sequence comparisons and analysis were performed with Phred-Phrap-Consed program version 12.0. Samples from 600 unrelated population-matched controls were sequenced for exclusion of the possibility that the mutation identified was a polymorphism in POFUT1.
Zebrafish Studies
The wild-type AB strain of zebrafish was used in this study. Zebrafish embryos were generated by natural pairwise mating in our aquaculture facility. All zebrafish studies were conducted under the guidance and approval of the institutional animal care and use committees at Shanghai Jiaotong University School of Medicine and Wenzhou Medical College.
The sequence of the exon 2-intron 2 splice pofut1 morpholino (pofut1-MO) was 5′-GACGTGTTGTTGTGTTTACATTAGC-3′, and the sequence for the standard control morpholino (scMO) was 5′-CCTCTTACCTCAGTTACAATTTATA-3′ (Gene Tools). Approximately 4 nl of 0.25 mM pofut1 or the scMO solution was microinjected into zebrafish embryos at the 1- to 2-cell stage with a microinjector (NARISHIGE IM-300) under a stereo microscope (Olympus SZX7). Zebrafish embryos of controls and pofut1 morphants were allowed to develop until 72 hr postfertilization (hpf) for the evaluation of melanin formation and distribution.
Primers spanning pofut1 exon 1 (forward primer: 5′-CAGACGTGACGTGGGATG-3′) and exon 6 (reverse primer: 5′-CTGTGGAGACGCCATAAAG-3′) were used for RT-PCR analysis for confirmation of the effectiveness of gene knockdown. The predicted size of the amplified cDNA fragment containing exon 2 was 722 bp, and a product generated from cDNA without part of exon 2 was estimated to be about 617 bp in size. The primer ef1α sequences used as the internal control were 5′-TCACCCTGGGAGTGAAACAGC-3′ (forward) and 5′-ACTTGCAGGCGATGTGAGCAG-3′ (reverse). All of the primers used for RT-PCR analysis were purchased from Invitrogen.
Zebrafish were photographed at 48 and 72 hpf under a stereo microscope (Olympus SZX7) coupled with a charge-coupled device (JVC) for obtaining quantitative information on melanin contents; changes in body length among different groups were analyzed with Image J software (National Institutes of Health). Approximately 30–40 zebrafish per group were used for body-length analysis.
Tyrosinase activity and melanin content were determined by a microplate reader (Mithras LB940, Berthold Technologies) as described previously with minor modifications.12 All experiments were repeated at least three times, and four to five samples were used per group.
Fifty embryos per sample were collected at 6, 12, 24, 28, 32, 48, 60, and 72 hpf for RT-PCR assessment of the time course of pofut1 expression. Primers used included forward primer 5′-CAGACGTGACGTGGGATG-3′ and reverse primer 5′-CTGTGGAGACGCCATAAAG-3′.
Cell-Culture Studies
HaCaT human keratinocyte cells were cultured at 37°C in a humidified 5% CO2 atmosphere in RPMI-1640 medium with 10% fetal calf serum (GIBCO, Invitrogen), 100 IU/ml penicillin G, and 100 mg/ml streptomycin sulfate (Sigma-Aldrich).
POFUT1 lentivirus expression vector pLV.Ex3d.P/puro-Pofut1 was constructed by replacement of the GFP fragment of the pLV.Ex3d.P/puro vector (Cyagen) with the POFUT1 (RefSeq NM_015352.1) coding sequence amplified from the plasmid. Oligonucleotides were synthesized for the generation of an annealing shRNA targeting the sequence of POFUT1 from positions 249–269 (5′-CCATGTGTCCTACCAGAAGTA-3′) and from positions 462–482 (5′-GTTTCATGTGAGTTTCAACAA-3′). The fragments were cloned separately into pLentiX1/puro (Cyagen).
Virus packaging was performed in human embryonic kidney (HEK) 293T cells after the cotransfection of pLV.Ex3d.P/puro-Pofut1 or pLentiX1/puro-shRNA-Pofut1 with the packaging plasmid (pLV/helper-SL3, pLV/helper-SL4, and pLV/helper-SL5) with the use of Lipofectamine 2000 (Invitrogen). Viruses were harvested 48 hr after transfection. HaCaT cells were infected with the filtered lentivirus in the presence of 6 μg/ml polybrene (Sigma-Aldrich). The knockdown efficiency was detected at the mRNA level by quantitative real-time PCR.
Quantitative Real-Time PCR Analysis
We used quantitative real-time PCR to determine the expression levels of pofut1 transcripts in adult zebrafish tissues or organs, including the eye, brain, skin, liver, heart, spleen, fin, trunk muscle, testis, and ovary. We also applied quantitative real-time PCR to quantify the relative mRNA expressions of pofut1, notch1, notch2, hey1, krt5, mitf, and tyr in 48 and 72 hpf embryos. The PCR primers are given in Table S2. We used TRIzol reagent (Invitrogen) to isolate total RNA from the samples. We prepared cDNA from 1 μg of total RNA per group by using RT-PCR kits (Toyobo) at a final volume of 20 μl. We carried out each PCR determination in triplicate at a final volume of 25 μl by using SYBR Premix Ex Taq Green PCR Mix (Takara) in a Light Cycler 480 real-time PCR machine (Roche Diagnostics).
We used the same techniques to determine the expression levels of POFUT1 in keratinocytes, HaCaT cells, melanocytes, and normal human skin.
To analyze mRNA changes in response to treatments, we used TRIzol reagent to extract total cellular RNA. The expression levels of POFUT1, NOTCH1 (MIM 190198), NOTCH2 (MIM 600275), HES1 (MIM 139605), and KRT5 (MIM 148040) transcripts were examined in HaCaT cells. The PCR primers are given in Table S3.
Statistical Analysis
We performed a one-way ANOVA to determine statistical significance and a Dunnett’s post hoc test to independently compare the treatment group with the control groups (SPSS). p < 0.05 was considered statistically significant. All of the data are presented as the mean ± SD unless otherwise stated.
Results
Linkage of DDD to Chromosome 20
In order to identify the candidate gene for generalized DDD, we genotyped 14 individuals from family 1 (II:2, III:2, III:3, III:4, III:6, III:7, III:8, III:10, III:11, III:12, IV:1, IV:2, IV:5, and IV:6) by using an Illumina Infinium HumanLinkage-24 panel after excluding pathogenic KRT5 mutations in the family 1 proband by conventional Sanger sequencing. Genome-wide linkage analysis was performed with a total of 5,913 SNP markers within average genetic and physical distances of 441 kb and 0.58 cM, respectively. SNPs with a call rate less than 90%, monomorphic SNPs, and non-Mendelian transmitted markers were removed. A total of 4,456 informative autosomal SNPs were retained in the linkage analysis. Multipoint parametric linkage analyses were performed with the MERLIN program version 1.1.2. A fully penetrant autosomal-dominant model was used with a rare disease frequency of 0.0001. Critical recombination events of the pedigree members were also determined through haplotype construction in MERLIN.
Genome-wide linkage analysis in family 1 revealed multipoint LOD scores > 3 for chromosome 20, indicating linkages to this chromosome (Figures 3A and 3B and Table S4). No other locus of suggestive linkage was detected. Recombination events were observed in individuals III:3, IV:1, and IV:5 (Figure 3A). These recombination events defined the susceptibility region in 20p11.21–20q13.12 to a 14.79 cM interval (54.04–68.83 cM between rs1293713 and rs244123). All affected individuals shared a common haplotype across this disease interval. The suggested haplotypes of this pedigree are shown in Figure 3A.
Figure 3.
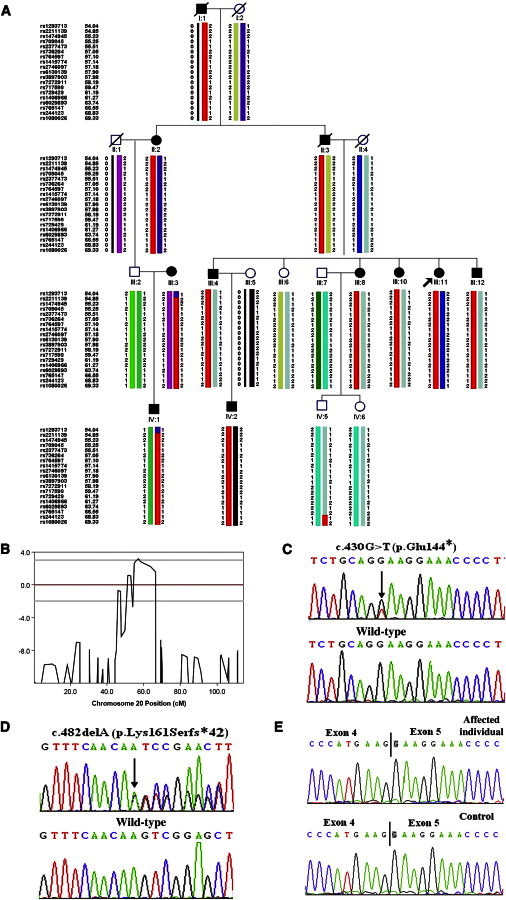
Identification of the Disease-Causing POFUT1 Mutations in Two Families Affected by Generalized DDD
(A) Haplotype analysis of family 1. Haplotype indicates heterozygosity between rs1293713 and rs244123 in affected family members. The recombination events defined the susceptibility region in 20p11.21–20q13.12 to a 14.79 cM interval. Red bars indicate the chromosomal region shared by affected members of the pedigree.
(B) Output of the multipoint-LOD-score analysis for chromosome 20.
(C and D) POFUT1 mutations in the two families: c.430G>T (p.Glu144∗) in family 1 (C) and c.482delA (p.Lys161Serfs∗42) in family 2 (D). The black arrows indicate mutations.
(E) The normal POFUT1 transcripts from the DDD individual and control skin.
POFUT1 Mutations
The map region contained 253 known genes. We then sequenced the entire exome in one affected member (III:8) of family 1. Exome sequencing generated 81,327,908 unique reads. The mean read depth across the exome was 71 reads. A total of 143,919 variants were identified in this DDD individual. After the identified heterozygous variants were filtered through the 1000 Genomes Project, HapMap 8, and dbSNP135, two variants remained in the 14.79 cM critical region. These remaining variants included missense mutation c.329G>A (p. Cys110Tyr) in exon 3 of CST7 (MIM 603253; RefSeq NM_003650.3) and nonsense mutation c.430G>T (p.Glu144∗) in exon 4 of POFUT1. The nonsense mutation was confirmed by conventional sequencing and cosegregated with generalized DDD in family 1 (Figure 3C) and was absent in 600 healthy unrelated Chinese individuals. The nonsense mutation was present in all affected members and absent in all unaffected family members within family 1. The missense mutation in CST7 did not cosegregate within the family. The affected members (III:3, IV:1, and IV:2) were not found to harbor this mutation.
To confirm whether POFUT1 is also mutated in an additional individual affected by generalized DDD, we screened POFUT1 coding sequence by Sanger sequencing in a 53-year-old woman from family 2 (from Chongqing, China). Deletion of an adenine (c.482delA [p.Lys161Serfs∗42]) was identified in exon 4 of POFUT1 in this DDD individual; this deletion was also absent in unaffected family members (II:1 and II:4) in family 2 and 600 healthy unrelated Chinese individuals (Figure 3D).
To determine whether the mutant allele is present in the RNA of affected individuals, we isolated RNA from immortalized lymphocytes of two affected individuals (III:8 and IV:2) and a normal member (IV:3) from family 1. After cDNA synthesis and PCR with gene-specific primers, we sequenced the obtained products. In the two affected individuals, normal transcripts were detected but the truncated POFUT1 transcripts were not detected, presumably because of nonsense-mediated decay (Figure 3E). We could not obtain a POFUT1 transcript from the DDD individual in family 2.
POFUT1 in Skin Biopsies of Affected Individuals
We also examined the localization patterns of POFUT1 in individual III:11 in family 1 by using rabbit polyclonal antibodies against POFUT1 (ab140205, Abcam). Immunohistochemical staining showed nonuniform localization of POFUT1 in the epidermis. The majority of the cells showed weak cytoplasmic staining, but local keratinocytes showed relatively stronger staining. In general, POFUT1 localization obviously decreased in the DDD individual compared with the control (Figures S2E–S2G).
Analysis of Pofut1 Knockdown in Zebrafish
Pofut1 was successfully knocked down in zebrafish embryos injected with pofut1-MO (termed the pofut1-MO group) as compared with zebrafish embryos injected with scMO (termed the scMO group) and untreated control groups, although no changes in ef1α expression were observed among the groups. Either no mRNA transcripts or two mRNA transcripts emerged in the pofut1-MO group from 12 to 72 hpf, whereas no differences were observed between the scMO and untreated control groups (Figure 4A). We screened pofut1 coding sequence by Sanger sequencing and identified the 105 bp deletion in exon 2 in the pofut1-MO group (Figure 4B).
Figure 4.

Identification of the Effectiveness of pofut1 Knockdown and the Effect of Tyrosinase Activity and Melanin Protein Contents of Zebrafish by Knockdown of Pofut1 Expression
(A and B) Effectiveness of pofut1 knockdown was confirmed by RT-PCR and Sanger sequencing. The expression of pofut1 decreased obviously in 72 hpf (A), and compared to the wild-type cDNA sequence, the cDNA sequence of the pofut1-MO group showed a lack of 105 bp from exon 2 (B). MO∗ indicates the pofut1-MO group.
(C and D) Zebrafish phenotype observation at 48 and 72 hpf. The melanin contents in the zebrafish tail at 48 hpf (C) and in both the tail and body axis at 72 hpf (D) decreased significantly in the pofut1-MO group compared with controls, and the melanin distribution in the zebrafish tail was presented as dispersion in the pofut1-MO group (D). The black arrow indicates abnormal melanin distribution in the zebrafish fin.
(E) Detection of tyrosinase activity and melanin protein contents at 48 and 72 hpf. Both tyrosinase activity and melanin protein contents were lower in the pofut1-MO group at 48 and 72 hpf, respectively, than in controls. The results are shown as the mean ± SD (n = 5), and similar results were obtained when the experiments and measurements were repeated three times. Error bars indicate the mean ± SD. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05.
(F) RT-PCR analyses of pofut1 expression in zebrafish from 6 to 72 hpf. Pofut1 was knocked down in all pofut1-MO groups from 12 to 72 hpf. The size of the amplified cDNA fragment containing exon 2 was 722 bp, and a product generated from cDNA without part of exon 2 of the pofut1-MO group was 617 bp in size.
The melanin contents in the zebrafish tail at 48 hpf (Figure 4C) and both the tail and body axis at 72 hpf (Figure 4D) decreased significantly in the pofut1-MO group compared with the scMO and untreated control groups. In addition, the melanin distribution in the zebrafish tail presented as dispersion in the pofut1-MO group at 72 hpf (Figure 4D). Zebrafish body lengths shortened significantly in the pofut1-MO group compared with the scMO and untreated control groups (data not shown).
At 48 and 72 hpf in the pofut1-MO group, tyrosinase activities decreased by 33% and 45%, respectively, whereas melanin protein contents decreased by 20% and 25%, respectively (Figure 4E).
The time course of pofut1 expression revealed that pofut1 transcripts were detectable at all designated time points from 6 to 72 hpf and that there was dramatically increased expression beginning at 32 hpf. Pofut1 was successfully knocked down in the entire pofut1-MO group from 12 to 72 hpf (Figure 4F).
We also applied quantitative real-time PCR to quantify the relative mRNA expressions of pofut1, notch1, notch2, hey1, krt5, mitf, and tyr at 48 and 72 hpf embryos. Morpholino knockdown of pofut1 in zebrafish resulted in a corresponding decrease in the expression levels of pofut1, notch2, hey1, krt5, mitf, and tyr. The expression of notch1 decreased at 48 hpf but did not show significant change between the pofut1-MO group and the controls at 72 hpf (Figures 5A and 5B).
Figure 5.
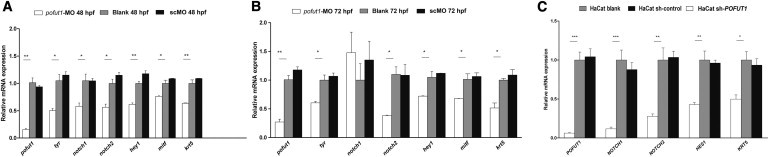
Analysis of Pofut1 Knockdown in Zebrafish and Reduced POFUT1 in HaCaT Cells
(A and B) RNA expression levels of pofut1, notch1, notch2, hey1, krt5, tyr, and mitf were determined by quantitative real-time PCR at 48 and 72 hpf in zebrafish. Morpholino knockdown of pofut1 in zebrafish resulted in a corresponding decrease in the expression of pofut1, notch2, hey1, krt5, tyr, and mitf at 48 (A) and 72 (B) hpf. The expression of notch1 decreased at 48 hpf, but not at 72 hpf. The results are shown as the mean ± SD (n = 3); one representative experiment of two is shown. Error bars indicate the mean ± SD. ∗∗p < 0.01, ∗p < 0.05.
(C) RNA expression levels of POFUT1, NOTCH1, NOTCH2, HES1, and KRT5 were determined by quantitative real-time PCR in HaCaT cells. The shRNA knockdown of POFUT1 in HaCaT cells resulted in a corresponding decrease in the expression of POFUT1, NOTCH1, NOTCH2, HES1, and KRT5. The results are shown as the mean ± SD (n = 3); one representative experiment of two is shown. Error bars indicate the mean ± SD. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05.
Pofut1 expression levels of transcripts were higher in the testis than in other tissues or organs in adult zebrafish (Figure S3).
Consequences of Reduced POFUT1 in Human Cells
To investigate whether POFUT1 is expressed in the skin, we examined POFUT1 expression in different human cell lines by using quantitative real-time PCR. POFUT1 expression was much higher in melanocytes than in other cell lines and normal skin (Figure S4).
The shRNA POFUT1 knockdown efficiency was detected by quantitative real-time PCR in HaCaT cells. The results indicated that the knockdown efficiency of the two fragments described above was 34.8% (5′-CCATGTGTCCTACCAGAAGTA-3′) and 95.4% (5′-GTTTCATGTGAGTTTCAACAA-3′). The higher efficiency of shRNA knockdown of POFUT1 in HaCaT cells resulted in a corresponding decrease in the expression of POFUT1, NOTCH1, NOTCH2, HES1, and KRT5 (Figure 5C).
Discussion
The Notch signaling pathway is a conserved signaling pathway that plays an important role in cell fate and tissue formation during embryogenesis.13 There are four Notch receptors (NOTCH1–NOTCH4) and five Notch ligands (JAG1, JAG2, DLL1, DLL3, and DLL4) in this signaling pathway in mammals.14 Notch receptors are transmembrane glycoproteins. About 29–36 epidermal growth factor (EGF)-like repeats contain a consensus sequence for O-fucosylation in their extracellular domain.15 POFUT1 adds O-linked fucose to EGF-like repeats of Notch receptors, after which Notch ligands bind to Notch receptors and induce the proteolytic release of Notch intracellular domain (NICD).14,15 NICD, in a complex with the transcriptional repressor RBP-J, activates target genes, such as HES1. Therefore, POFUT1 is an essential component of the canonical Notch signaling pathway.15 In order to validate the impact of POFUT1 loss of function on the Notch signaling pathway, we detected the expression of receptor-encoding genes NOTCH1 (MIM 190198) and NOTCH2 (MIM 600275) and the target gene, HES1, in the Notch pathway in shRNA POFUT1 in HaCaT cells. The expression levels of NOTCH1, NOTCH2, and HES1 were all downregulated. The same result was again demonstrated in the pofut1-MO group, indicating that loss of function of POFUT1 has a strong impact on the Notch pathway.
Aberrant gain or loss of function of Notch signaling components has been connected to several inherited human diseases since 1991.16–26 Mutations in NOTCH1 cause several types of cardiac disease and cutaneous and lung squamous cell carcinoma, whereas mutations in NOTCH2 and NOTCH3 (MIM 600276) cause Hajdu-Cheney syndrome (MIM 102500) and adult-onset cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (MIM 125310), respectively.16–20 Mutations in the ligand-encoding delta-like-3 (DLL3 [602768]) cause autosomal-recessive spondylocostal dysostosis.25 Alagille syndrome type 1 (MIM 118450) is caused by mutations in JAG1 (MIM 601920) and NOTCH2.27,28 In 2012, Hassed et al. reported that mutations in RBPJ (MIM 147183) cause Adams-Oliver syndrome (MIM 100300).29 Abrogation of Pofut1 in mice causes embryonic lethality with Notch-like phenotypes, including vasculogenic, cardiogenic, and neurogenic phenotypes.30 In this study, nonsense mutation c.430G>T (p.Glu144∗) was detected in exon 4 of family 1. Mutant POFUT1 transcripts were not detected. This mutation presumably leads to nonsense-mediated decay of POFUT1 transcripts. Our studies show that the mutation results in haploinsufficiency of POFUT1 with abnormal skin pigments; no obvious anomaly was found in other organs and systems. On the basis of this finding, we predict that POFUT1 can maintain normal functions in most tissues and organs under POFUT1 haploinsufficiency.
Melanocytes are well known to be responsible for the pigmentation of animals. A series of studies have indicated that the Notch signaling pathway plays an important role in the regulation of melanocyte lineage development.31–33 Kumano et al. found that mice harboring a double heterozygous deletion of Notch1 and Notch2 show gradual hair graying after birth.34 Schouwey et al. also found a progressive hair-graying phenotype induced by Notch1 and Notch2 mutations but without changes in the pigmentation of the dermis and choroid.32 The researchers thus speculated that both Notch1 and Notch2 contribute to the maintenance of melanoblasts and melanocyte stem cells and are essential for proper hair pigmentation.32 The DDD individuals in this study presented with pigmented anomalies on the skin of the neck, wrist, chest, back, inguinal regions, and inner sides of the thighs but had normal hair pigmentation. These results indicate that haploinsufficiency of POFUT1 influences the Notch signaling pathway more severely in skin than in hair in humans. We also observed the abnormal distribution of melanin in the 72 hpf zebrafish tail in the pofut1-MO group. This finding is consistent with that of Aubin-Houzelstein et al., who reported that melanoblasts and melanocytes might be observed in ectopic locations in the skin of cRBP-J-knockout mice.35 These results support the hypothesis that disruption of the Notch signaling pathway influences the migration of melanoblasts and melanocytes.
A previous study showed that KRT5 would be another pathogenic gene for DDD. Therefore, we evaluated the expression of KRT5 in the pofut1-MO group and in shRNA POFUT1 HaCaT cells. We found that KRT5 expression was lower in the shRNA POFUT1 cell model than in the controls, and MO knockdown of pofut1 in zebrafish also resulted in a corresponding decrease in the expressions of krt5. Betz et al. speculated that keratins are crucial in the organization of cell adhesion, melanosome uptake into keratinocytes, organelle transport, and nuclear anchorage.4 These results might explain why DDD individuals in this study presented with brown macules in the skin.
In this study, we found that melanin synthesis was strikingly affected after pofut1 knockdown in 48 hpf zebrafish. The melanin contents in the zebrafish tail at 48 hpf and in both the tail and body axis at 72 hpf significantly decreased in the pofut1-MO group compared with the scMO and untreated control groups. It was speculated that loss of function of pofut1 might influence the process of melanin synthesis, so we evaluated the expression of tyr and mitf. Both of them showed a remarkable reduction in expression at the mRNA level, indicating that hypopigmented macules in DDD individuals most likely result from an impairment of the ability to synthesize melanin in the melanocytes with loss-of-function mutations in pofut1. At 48 and 72 hpf in pofut1-MO zebrafish, tyrosinase activities decreased by 33% and 45%, respectively, whereas melanin protein contents decreased by 20% and 25%, respectively. We also found that premelanosomes were rarely observed by TEM in the proband of family 1. Microphthalmia-associated transcription factor (MITF) is regulated by multiple signals.36 The decrease of mitf expression in zebrafish embryos indicates the relationship between mitf and the Notch pathway. MITF is recognized as a master lineage regulator in melanocyte development, involving various cellular processes such as cell-cycle control, survival, motility, differentiation, and pigmentation.37 Therefore, the mitf expression reduction resulting from pofut1 knockdown in zebrafish, of course, influences the development of melanocytes. This might explain why the melanocyte from individual III:8 in family 1 in TEM was small in size and lacked melanosomes. These findings support the hypothesis that haploinsufficiency of POFUT1 influences melanin synthesis in melanocytes.
Acknowledgments
We thank all subjects for their ongoing participation in this study. We also thank Mianzhi Yao for critical reading and comments concerning this manuscript. We thank Tao Yang, Jiangbo Liu, Ronghu Ke, Guolong Zhang, Bo Ling, and Qianqian Wang for their support. This study was funded by a grant from the Shanghai Municipal Natural Science Foundation (#12ZR1420000) and a grant from the National Nature Science Foundation of China (81171544).
Contributor Information
Hong Yu, Email: smallgrass6@163.com.
Zhirong Yao, Email: dermatology.yao@sohu.com.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
NCBI RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Dowling G.B., Freudenthal W. Acanthosis Nigricans. Proc. R. Soc. Med. 1938;31:1147–1150. [PMC free article] [PubMed] [Google Scholar]
- 2.Degos R., Ossipowski B. [Reticulated pigmentary dermatosis of the folds: relation to acanthosis nigricans] Ann. Dermatol. Syphiligr. (Paris) 1954;81:147–151. [PubMed] [Google Scholar]
- 3.Li C.R., Xing Q.H., Li M., Qin W., Yue X.Z., Zhang X.J., Ma H.J., Wang D.G., Feng G.Y., Zhu W.Y., He L. A gene locus responsible for reticulate pigmented anomaly of the flexures maps to chromosome 17p13.3. J. Invest. Dermatol. 2006;126:1297–1301. doi: 10.1038/sj.jid.5700271. [DOI] [PubMed] [Google Scholar]
- 4.Betz R.C., Planko L., Eigelshoven S., Hanneken S., Pasternack S.M., Bussow H., Van Den Bogaert K., Wenzel J., Braun-Falco M., Rutten A. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am. J. Hum. Genet. 2006;78:510–519. doi: 10.1086/500850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pickup T.L., Mutasim D.F. Dowling-Degos disease presenting as hypopigmented macules. J. Am. Acad. Dermatol. 2011;64:1224–1225. doi: 10.1016/j.jaad.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 6.Sandhu K., Saraswat A., Kanwar A.J. Dowling-Degos disease with dyschromatosis universalis hereditaria-like pigmentation in a family. J. Eur. Acad. Dermatol. Venereol. 2004;18:702–704. doi: 10.1111/j.1468-3083.2004.01028.x. [DOI] [PubMed] [Google Scholar]
- 7.Lestringant G.G., Masouyé I., Frossard P.M., Adeghate E., Galadari I.H. Co-existence of leukoderma with features of Dowling-Degos disease: reticulate acropigmentation of Kitamura spectrum in five unrelated patients. Dermatology (Basel) 1997;195:337–343. doi: 10.1159/000245984. [DOI] [PubMed] [Google Scholar]
- 8.Lee S.J., Lee H.J., Kim D.W., Jun J.B., Chung S.L., Bae H.I. A case of Dowling-Degos disease suggesting an evolutional sequence. J. Dermatol. 2000;27:591–597. doi: 10.1111/j.1346-8138.2000.tb02234.x. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y.H., Lin Y.C. Generalized Dowling-Degos disease. J. Am. Acad. Dermatol. 2007;57:327–334. doi: 10.1016/j.jaad.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 10.Bukhari I.A., El-Harith E.A., Stuhrmann M. Dyschromatosis universalis hereditaria as an autosomal recessive disease in five members of one family. J. Eur. Acad. Dermatol. Venereol. 2006;20:628–629. doi: 10.1111/j.1468-3083.2006.01522.x. [DOI] [PubMed] [Google Scholar]
- 11.Li M., Yang L.J., Li C.R., Jin C., Lai M., Zhang G., Hu Y., Ji J., Yao Z. Mutational spectrum of the ADAR1 gene in dyschromatosis symmetrica hereditaria. Arch. Dermatol. Res. 2010;302:469–476. doi: 10.1007/s00403-010-1039-2. [DOI] [PubMed] [Google Scholar]
- 12.Choi T.Y., Kim J.H., Ko D.H., Kim C.H., Hwang J.S., Ahn S., Kim S.Y., Kim C.D., Lee J.H., Yoon T.J. Zebrafish as a new model for phenotype-based screening of melanogenic regulatory compounds. Pigment Cell Res. 2007;20:120–127. doi: 10.1111/j.1600-0749.2007.00365.x. [DOI] [PubMed] [Google Scholar]
- 13.Guruharsha K.G., Kankel M.W., Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012;13:654–666. doi: 10.1038/nrg3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stahl M., Uemura K., Ge C., Shi S., Tashima Y., Stanley P. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J. Biol. Chem. 2008;283:13638–13651. doi: 10.1074/jbc.M802027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao D., Huang Y., Huang X., Wang W., Yan Q., Wei L., Xin W., Gerson S., Stanley P., Lowe J.B., Zhou L. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood. 2011;117:5652–5662. doi: 10.1182/blood-2010-12-326074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang N.J., Sanborn Z., Arnett K.L., Bayston L.J., Liao W., Proby C.M., Leigh I.M., Collisson E.A., Gordon P.B., Jakkula L. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA. 2011;108:17761–17766. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garg V., Muth A.N., Ransom J.F., Schluterman M.K., Barnes R., King I.N., Grossfeld P.D., Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 18.Isidor B., Lindenbaum P., Pichon O., Bézieau S., Dina C., Jacquemont S., Martin-Coignard D., Thauvin-Robinet C., Le Merrer M., Mandel J.L. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat. Genet. 2011;43:306–308. doi: 10.1038/ng.778. [DOI] [PubMed] [Google Scholar]
- 19.Simpson M.A., Irving M.D., Asilmaz E., Gray M.J., Dafou D., Elmslie F.V., Mansour S., Holder S.E., Brain C.E., Burton B.K. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 20.Joutel A., Corpechot C., Ducros A., Vahedi K., Chabriat H., Mouton P., Alamowitch S., Domenga V., Cécillion M., Marechal E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 21.Li L., Krantz I.D., Deng Y., Genin A., Banta A.B., Collins C.C., Qi M., Trask B.J., Kuo W.L., Cochran J. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 22.Bauer R.C., Laney A.O., Smith R., Gerfen J., Morrissette J.J., Woyciechowski S., Garbarini J., Loomes K.M., Krantz I.D., Urban Z. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum. Mutat. 2010;31:594–601. doi: 10.1002/humu.21231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McElhinney D.B., Krantz I.D., Bason L., Piccoli D.A., Emerick K.M., Spinner N.B., Goldmuntz E. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106:2567–2574. doi: 10.1161/01.cir.0000037221.45902.69. [DOI] [PubMed] [Google Scholar]
- 24.Ellisen L.W., Bird J., West D.C., Soreng A.L., Reynolds T.C., Smith S.D., Sklar J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 25.Bulman M.P., Kusumi K., Frayling T.M., McKeown C., Garrett C., Lander E.S., Krumlauf R., Hattersley A.T., Ellard S., Turnpenny P.D. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 2000;24:438–441. doi: 10.1038/74307. [DOI] [PubMed] [Google Scholar]
- 26.McKellar S.H., Tester D.J., Yagubyan M., Majumdar R., Ackerman M.J., Sundt T.M., 3rd Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2007;134:290–296. doi: 10.1016/j.jtcvs.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 27.Lu F., Morrissette J.J., Spinner N.B. Conditional JAG1 mutation shows the developing heart is more sensitive than developing liver to JAG1 dosage. Am. J. Hum. Genet. 2003;72:1065–1070. doi: 10.1086/374386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamath B.M., Bauer R.C., Loomes K.M., Chao G., Gerfen J., Hutchinson A., Hardikar W., Hirschfield G., Jara P., Krantz I.D. NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 2012;49:138–144. doi: 10.1136/jmedgenet-2011-100544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassed S.J., Wiley G.B., Wang S., Lee J.Y., Li S., Xu W., Zhao Z.J., Mulvihill J.J., Robertson J., Warner J., Gaffney P.M. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am. J. Hum. Genet. 2012;91:391–395. doi: 10.1016/j.ajhg.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi S., Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc. Natl. Acad. Sci. USA. 2003;100:5234–5239. doi: 10.1073/pnas.0831126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriyama M., Osawa M., Mak S.S., Ohtsuka T., Yamamoto N., Han H., Delmas V., Kageyama R., Beermann F., Larue L., Nishikawa S. Notch signaling via Hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J. Cell Biol. 2006;173:333–339. doi: 10.1083/jcb.200509084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schouwey K., Delmas V., Larue L., Zimber-Strobl U., Strobl L.J., Radtke F., Beermann F. Notch1 and Notch2 receptors influence progressive hair graying in a dose-dependent manner. Dev. Dyn. 2007;236:282–289. doi: 10.1002/dvdy.21000. [DOI] [PubMed] [Google Scholar]
- 33.Lin H.Y., Kao C.H., Lin K.M., Kaartinen V., Yang L.T. Notch signaling regulates late-stage epidermal differentiation and maintains postnatal hair cycle homeostasis. PLoS ONE. 2011;6:e15842. doi: 10.1371/journal.pone.0015842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumano K., Masuda S., Sata M., Saito T., Lee S.Y., Sakata-Yanagimoto M., Tomita T., Iwatsubo T., Natsugari H., Kurokawa M. Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res. 2008;21:70–78. doi: 10.1111/j.1755-148X.2007.00423.x. [DOI] [PubMed] [Google Scholar]
- 35.Aubin-Houzelstein G., Djian-Zaouche J., Bernex F., Gadin S., Delmas V., Larue L., Panthier J.J. Melanoblasts’ proper location and timed differentiation depend on Notch/RBP-J signaling in postnatal hair follicles. J. Invest. Dermatol. 2008;128:2686–2695. doi: 10.1038/jid.2008.120. [DOI] [PubMed] [Google Scholar]
- 36.Shibahara S., Takeda K., Yasumoto K., Udono T., Watanabe K., Saito H., Takahashi K. Microphthalmia-associated transcription factor (MITF): multiplicity in structure, function, and regulation. J. Investig. Dermatol. Symp. Proc. 2001;6:99–104. doi: 10.1046/j.0022-202x.2001.00010.x. [DOI] [PubMed] [Google Scholar]
- 37.Levy C., Khaled M., Fisher D.E. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006;12:406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.