

Abstract
Spinal muscular atrophy (SMA) is a heterogeneous group of neuromuscular disorders caused by degeneration of lower motor neurons. Although functional loss of SMN1 is associated with autosomal-recessive childhood SMA, the genetic cause for most families affected by dominantly inherited SMA is unknown. Here, we identified pathogenic variants in bicaudal D homolog 2 (Drosophila) (BICD2) in three families afflicted with autosomal-dominant SMA. Affected individuals displayed congenital slowly progressive muscle weakness mainly of the lower limbs and congenital contractures. In a large Dutch family, linkage analysis identified a 9q22.3 locus in which exome sequencing uncovered c.320C>T (p.Ser107Leu) in BICD2. Sequencing of 23 additional families affected by dominant SMA led to the identification of pathogenic variants in one family from Canada (c.2108C>T [p.Thr703Met]) and one from the Netherlands (c.563A>C [p.Asn188Thr]). BICD2 is a golgin and motor-adaptor protein involved in Golgi dynamics and vesicular and mRNA transport. Transient transfection of HeLa cells with all three mutant BICD2 cDNAs caused massive Golgi fragmentation. This observation was even more prominent in primary fibroblasts from an individual harboring c.2108C>T (p.Thr703Met) (affecting the C-terminal coiled-coil domain) and slightly less evident in individuals with c.563A>C (p.Asn188Thr) (affecting the N-terminal coiled-coil domain). Furthermore, BICD2 levels were reduced in affected individuals and trapped within the fragmented Golgi. Previous studies have shown that Drosophila mutant BicD causes reduced larvae locomotion by impaired clathrin-mediated synaptic endocytosis in neuromuscular junctions. These data emphasize the relevance of BICD2 in synaptic-vesicle recycling and support the conclusion that BICD2 mutations cause congenital slowly progressive dominant SMA.
Main Text
Neurodegenerative or developmental disorders affecting motor neurons, their axons, or the presynapse at the neuromuscular junction (NMJ) produce a large spectrum of inherited diseases, including spinal muscular atrophies (SMAs), which partially overlap with hereditary motor neuropathies (HMNs), and amyotrophic lateral sclerosis (ALS).1–4 The term SMA (MIM 253300 for type I, MIM 253550 for type II, MIM 253400 for type III, and MIM 271150 for type IV) is used for the most frequent autosomal-recessive proximal SMA linked to chromosomal region 5q13.15 and as a general label for a group of non-5q SMAs characterized by anterior horn cell loss and progressive muscle weakness and atrophy without sensory neuron involvement.6 The underlying molecular defect of autosomal-recessive proximal SMA is the functional loss of survival of motor neuron 1 (SMN1), which mainly affects the maturation and functional maintenance of the presynapses of spinal α-motor neurons.5,7 On the basis of age of onset, pattern of muscle involvement, and inheritance, approximately 30 additional genetic entities for SMA and/or HMN have been described (World Federation of Neurology Research Committee and Online Mendelian Inheritance in Man). Among these entities, mutations in 12 genes have been identified to cause autosomal-dominant forms of SMA and/or HMN—these include GARS (MIM 600287),8 DCTN1 (MIM 601143),9 HSPB8 (MIM 608041),10 HSPB1 (MIM 602195),11 BSCL2 (MIM 606168),12 SETX (MIM 608465),13 VAPB (MIM 605704),14 HSPB3 (MIM 604624),15 TRPV4 (MIM 605427),16 DYNC1H1 (MIM 600112),17 REEP1 (MIM 609139),18 and SLC5A7 (MIM 608761).19 Mutations in each of these genes account for only small percentages of cases, so the molecular genetic basis is only known in fewer than 30% of individuals with dominant SMA and/or HMN.20 Many different processes—such as inhibition of microtubule-based dynein-mediated intracellular and axonal transport, inhibition of the ubiquitin-proteasome system, impaired RNA metabolism, protein aggregation or Ca2+ homeostasis, oxidative and nitrosative stress, redistribution of proteins from the nucleus to the cytoplasm, fragmentation of the Golgi apparatus, and dysfunction of mitochondria—underlie motor neuron degeneration or disturbed motor neuron development, as reviewed.6,20,21 Here, we identify bicaudal D homolog 2 (Drosophila) (BICD2 [MIM 609797]) mutations that cause autosomal-dominant SMA by affecting Golgi integrity. Our data emphasize the important role of BICD2, a protein known to be essential in the retrograde and anterograde transport along the microtubules, for the functional integrity of lower motor neurons.
Frijns and colleagues reported a Dutch family affected by an autosomal-dominant congenital benign SMA22 (family 1, Figure 1A), for which we identified the underlying genetic cause, presenting the initial fuse for this study. In this three-generation Dutch family, seven affected individuals have been reported with similar features of proximal and distal muscle weakness and atrophy of the lower limbs and congenital contractures of the ankles and feet. They presented with a waddling gait and were able to walk on toes, but not on heels. Muscle biopsies showed neurogenic changes. Serum creatine kinase (CK) activity was normal or marginally raised. Motor nerve conduction studies including speed and amplitude of motor nerve conduction velocity (NCV) and compound muscle action potential showed results in the normal range. Sensory NCVs were also normal or near normal. These findings are in favor of a motor neuron disease and argue against an axonal neuropathy.22 A recent examination of individuals II.6 and III.3, nearly 20 years later, revealed an almost unchanged phenotype, except for more severe muscle cramps, emphasizing the nonprogressive form of this disease.
Figure 1.
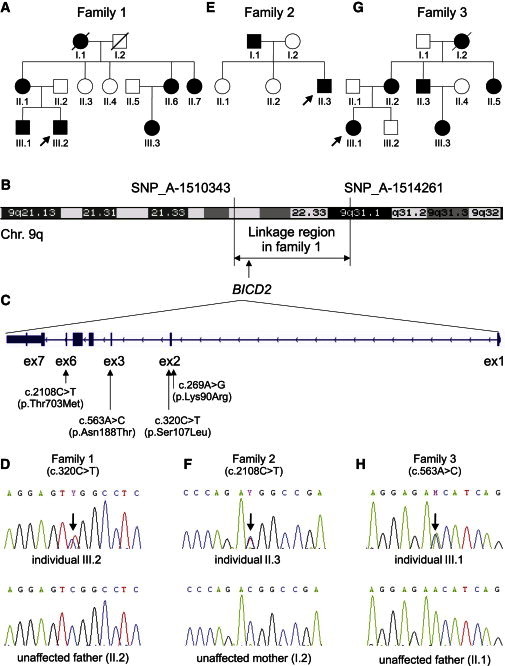
Identified BICD2 Mutations and Their Chromosomal Positions
(A, E, and G) Pedigrees of families 1 (A), 2 (E), and 3 (G).
(B) An ideogram of chromosome 9 shows the linked region and the flanking SNPs identified in family 1.
(C) An ideogram of BICD2 includes exon and intron structure and the identified variants.
(D, F, and H) A sequence of the region includes the mutation and the corresponding region of the unaffected parent in families 1 (D), 2 (F), and 3 (H).
For identifying the chromosomal location of the underlying disease-causing mutation, a genome-wide linkage analysis was performed in nine family members (family 1, Figures 1A and 1B), from whom written informed consent was obtained. Linkage was performed as described previously23 and identified a locus with a maximum multipoint LOD score of 2.065 between SNP_A-1510343 and SNP_A-1514261 on chromosome 9 (chr9: 94,440,951–104,432,543). This nearly 10 Mb chromosomal segment (Figure 1B) contains approximately 50 genes.
Subsequently, exome sequencing was performed in a single affected individual (III.2, family 1, Figure 1A) on a SOLiD4 sequencing platform from Life Technologies (Carlsbad, CA, USA). The exome of the proband was enriched according to the manufacturer’s protocol with the use of Agilent’s SureSelect Human All Exon v.2 Kit (50 Mb), which contains the exonic sequences of approximately 21,000 genes (Agilent Technologies, Santa Clara, CA, USA). LifeScope software v.2.1 from Life Technologies was used for mapping color-space reads along the UCSC Genome Browser (hg19) reference genome assembly. The DiBayes algorithm, with high-stringency calling, was used for single-nucleotide variant (SNV) calling. The small indel tool was used for detecting small insertions and deletions. Exome-sequencing data were filtered as described previously.24 Exome sequencing showed 50× median coverage. In total, 118,859,416 reads were obtained, and 96,800,227 of them could be mapped to the human genome (hg19). Variant calling resulted in a total of 27,190 variants. Filtering for variants located on chromosome 9 reduced this number to 1,064 variants. Further filtering was performed for obtaining nonsynonymous exonic and splice-site variants. After combined exclusion of known SNPs (dbSNP129) and of variants that had been identified previously and summarized in an in-house database,24 just five variants in five different genes remained (Tables S1 and S2, available online). Of these five genes, only BICD2 (RefSeq accession number NM_001003800.1) was located within the linkage region (Table S2 and Figures 1B and 1C). The detected variant (g.95491439C>T [c.320C>T]), localized within a CpG dinucleotide, affects a highly conserved serine residue (phyloP 3.65) and results in the amino acid change p.Ser107Leu. The effect of this amino acid change is predicted to be deleterious by SIFT25 and probably damaging by PolyPhen-226 (Table S3). These findings, together with the presence of variant c.320C>T in five tested affected individuals but in none of four tested unaffected relatives as shown by Sanger sequencing (Figure 1D), strongly support the view that this is the causative damaging mutation in this family.
To search for further BICD2 variants that might cause dominant SMA, we analyzed all seven exons by Sanger sequencing in 23 additional families affected by autosomal-dominant SMA. Informed consent was obtained from affected individuals and relatives, and the study was approved by the local ethical committee in Cologne. Heterozygous variants in BICD2 were identified in two additional families. First, in a previously described Canadian family (family 2, Figure 1E) affected by congenital autosomal-dominant distal SMA,27 both affected family members—father and son—carried the heterozygous missense variant, c.2108C>T (p.Thr703Met), localized within a CpG dinucleotide in exon 6 (Figures 1C and 1F). Second, we identified another Dutch family (family 3, Figure 1G) comprising five affected and four unaffected individuals, and all affected individuals, but none of the unaffected individuals, were heterozygous for variant c.563A>C (p.Asn188Thr) (Figures 1C and 1H). The Canadian p.Thr703Met substitution was located in the fifth coiled-coil domain (C-terminal part) of BICD2, whereas the Dutch p.Asn188Thr substitution was located in the second coiled-coil domain (N-terminal part) of BICD2 (Figure 2). Both amino acid positions (Thr703 and Asn188) are highly conserved throughout 46 vertebrate species (PhyloP 5.49 and 4.25, respectively) (UniProt and Figure 2), and the substitutions are predicted to be damaging by SIFT and PolyPhen-2 (Table S3). Moreover, the three variants (the two Dutch and one Canadian) were not found in the 1,094 genomes of the 1000 Genomes database, the 6,500 exomes of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (EVS), an in-house database comprising 1302 exomes, or dbSNP137.
Figure 2.
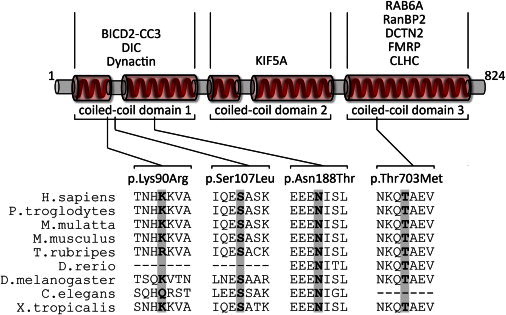
Schematic Drawing of BICD2 and Its Interaction Partners
The diagram depicts a simplified structure of human BICD2. Indicated are the five coiled-coil domains, which are typically combined as coiled-coil domains 1–3. Numbers indicate the respective amino acid residues. In the upper part of the diagram, known interaction partners of the respective coiled-coil domains are given. Please note that the interaction with FMRP is RNA mediated and the interaction between clathrin heavy chain (CLHC) and BICD2 has only been shown for Drosophila so far. In the lower part of the diagram, the four identified variants and a multispecies alignment of the affected amino acids are given. Protein sequences were aligned with blastp.
In addition, a rare SNV (c.269A>G [p.Lys90Arg]; rs61754130), localized in exon 2 of BICD2 was identified twice in our cohort of 23 independent SMA-affected families with dominant inheritance (Figure 1C). The minor allele frequency of this variant is 0.004 in the EVS and 0.0009 in dbSNP. We further performed segregation analysis in all available affected and unaffected members of these two families, which are both of German origin. The same variant was identified in one additional affected individual of one family but only in two out of four affected individuals of the second family, excluding the c.269A>G variant as the causative mutation (data not shown). Nevertheless, we cannot fully exclude a potential modifying role given that amino acid Lys90 is moderately conserved (Figure 2).
Furthermore, the entire coding region of the paralogous BICD1 was analyzed in those families in which no BICD2 mutation was found. No mutations were found in BICD1 either.
The phenotype described by Adams and colleagues for the Canadian family,27 harboring amino acid substitution p.Thr703Met, very much resembles that of the first Dutch family.22 Both father and son had congenital foot deformities and muscle weakness and atrophy of the lower limbs and mild proximal and hand intrinsic weakness. Knee and ankle tendon reflexes were absent. CK levels were normal, and both NCVs and amplitude of motor and sensory responses were normal. Both electromyography (EMG) and muscle biopsy showed chronic neurogenic changes.27 Both individuals were recently reexamined and showed no noticeable changes.
In the second Dutch family with the p.Asn188Thr substitution, two individuals (II.2 and III.1) belonging to a three-generation pedigree with six affected individuals (Figure 1G) were extensively clinically investigated. Together with her affected daughter, III.1, individual II.2 was seen for the first time at the age of 35 years. Individual II.2 had delayed motor development but did not mention muscle complaints, sensory disturbances, or exercise intolerance. Family history revealed a muscle disorder in two of her siblings and her mother. Neurological examination revealed mild atrophy of proximal limb muscles, muscle strength was mildly lowered (shoulder muscles scored a 4/5 on the Medical Research Council [MRC] scale for muscle strength), and she was unable to walk on her heels. Gowers’ sign was absent, but she had a waddling gait. Examination of sensory and cranial nerves and coordination were normal. She had generalized hyperreflexia with indifferent plantar responses. EMG showed a chronic neurogenic pattern. After 10 years, follow-up examination showed mild atrophy of deltoid muscles. Muscle tone was normal. Muscle strength was mildly diminished: MRC scale scores were 4/5 for shoulder abductors, 4/5 for biceps muscles, and 3/5 for tibial, peroneal, and extensor hallucis longus muscles. She still had a mild waddling gait, and Gowers’ sign was absent. Examination of cranial nerve function revealed a hoarse voice, and sensory function and coordination were normal. Tendon reflexes of the upper limbs were lowered, and knee tendon reflexes were exaggerated: there was a moderate crossed adductor response on the left knee with an absence of an ipsilateral response on the right and a minimal crossed adductor response on the right knee. The Achilles tendon reflexes were absent, and extensor responses were indifferent. Furthermore, she reported that her niece (III.3) was affected as well.
Her daughter (III.1) was seen for the first time at the age of 2 years and 11 months. The gestation period was complicated by oligohydramnios, and she was born with vacuum extraction. She was treated for congenital hip dysplasia from 4 to 7 months of age. She was able to sit without support from the age of 7.5 months and was able to walk without support at the age of 20 months. Neurological examination showed mild atrophy of proximal limb muscles, and muscle strength was mildly lowered (shoulder abductors scored a 4/5 on the MRC scale) with scapulae alatae and lumbar hyperlordosis. Cranial nerve function was normal. While walking, she showed a waddling gait and Gowers’ sign. Sensory function and coordination were normal. She had generalized hyperreflexia, more pronounced on the legs, and bilateral pathologic plantar responses. EMG was normal. Follow-up after 2 years (when she was almost 5 years old) showed a waddling gait and an inability to jump. Muscle strength of the arms was mildly affected: the bilateral proximal and distal grade was 4/5. Proximal muscle strength of the legs was 4/5, and distal was 2/5. Since early childhood, she has worn ankle-foot orthoses during the night to prevent calf muscle contractures (which were not present at examination). When she was 12 years and 9 months old, follow-up examination showed mild atrophy of deltoid muscles. Muscle tone was normal. Muscle strength was mildly diminished: MRS scale scores were 4/5 for shoulder abductors, 4/5 for biceps muscles, and 3/5 for tibial, peroneal, and extensor hallucis longus muscles. She still had a waddling gait, and Gowers’ sign was absent. Cranial nerve function and sensory function and coordination were normal. Tendon reflexes of the upper limbs were normal, knee tendon reflexes were increased, Achilles tendon reflexes were lowered, and extensor responses were plantar. She needed special education because of a learning disorder.
No clinical data were available from the affected grandmother. It has only been reported that she had muscle weakness. The other three affected individuals (II.3, II.5, and III.3) all had congenital hip dysplasia, and affected individual III.3 additionally had bilateral club feet. Affected individuals II.3 and II.5 were able to walk at 18 months of age but developed mild muscle atrophy and weakness in early childhood (especially of the proximal muscles of the legs) and hyperreflexia with bilateral indifferent plantar responses, whereas affected individual III.3 was unable to walk at the age of 18 months. She had mild atrophy of proximal limb muscles, and muscle strength was mildly reduced, no hyperreflexia was present, and plantar responses were normal. In all three affected individuals, EMG showed neurogenic changes. Muscle biopsy from the quadriceps of individuals II.3 and II.5 showed a neurogenic pattern.
BICD2 (MIM 609797) is homologous to Drosophila bicaudal D (BicD), which is evolutionarily conserved from flies to humans (UniProt). Although flies have one BicD gene, mammals have two paralogous genes: BICD1 (MIM 602204), which is localized in chromosomal region 12p11.2–p11.1 and whose encoded protein is mainly expressed in brain, skeletal muscle, and heart,28 and BICD2, which is localized in chromosomal region 9q22.3 and encodes a ubiquitously expressed protein. BICD2 is a cytoplasmic conserved motor-adaptor protein involved in anterograde and retrograde transport.29 BICD2, considered a golgin, has roles in the synaptic vesicle and membrane traffic and Golgi structure (reviewed in Goud and Gleeson30). Most importantly, third instar larvae of zygotic BicD mutants in Drosophila show a reduced rate of locomotion during active bouts of crawling by impaired clathrin-mediated synaptic-vesicle recycling.31 Panneuronal expression of BicD fully rescued larval locomotion and lethality of the mutants, whereas expression in the muscle did not, emphasizing its important role in the nervous system.
BICD2 is a highly versatile molecule with five coiled-coil domains (Figure 2). The N-terminal domain of BICD2 regulates the recruitment of dynein and promotes stable interaction between dynein and dynactin.32–34 The in vitro overexpression of the N-terminal domain of BICD2 has been reported to cause Golgi fragmentation.32–34 In line with these findings, transgenic mice expressing the N terminus of BICD2 (BICD2-N) show Golgi fragmentation, axonal neurofilament swelling, and reduced retrograde transport.35 Alterations in the dynein-dynactin motor complex are considered to be involved in neuron degeneration. Mutations in DCTN1 (MIM 601143), encoding the largest subunit (p150Glued) of dynactin, have been associated with a slowly progressive, autosomal-dominant form of lower motor neuron disease without sensory symptoms (HMN7B [MIM 607641]) and with ALS.9,36 After 10 months of age, heterozygous knockin mice of mutant DCTN1 (c.957C>T [p.Gly59Ser]) display motor-neuron-disease-like phenotypes, including excessive accumulation of cytoskeletal and synaptic-vesicle proteins at NMJs, loss of spinal motor neurons, increase in reactive astrogliosis, and shortening of gait.37 The central part of BICD2 interacts with another important motor protein, the neuronal kinesin heavy-chain protein KIF5A.29 Mutations in KIF5A (MIM 602821) cause hereditary spastic paraplegia (HSP) type 10 (SPG10 [MIM 604187]), an autosomal-dominant HSP.38,39 Knockout of Kif5a in mice causes reduced survival of motor neurons, which reveal a significant deficit in axonal and dendritic outgrowth and impaired anterograde axonal transport, whereas sensory neurons are spared.38,39 The C-terminal third of BICD2 mediates transport of a subset of cargos, including Golgi vesicles, lipid droplets, nuclei, and specific mRNAs.40,41 The C-terminal part of BICD2 directly interacts with dynactin subunit p50 dynamitin (DCTN2). The Drosophila BicD C terminus directly interacts with the clathrin heavy chain, which has been shown to facilitate and be essential to clathrin-mediated endocytosis at the NMJ level.31–34 Furthermore, BICD2 is targeted to the trans-Golgi network via the interaction between its C-terminal domain and the small GTPase RAB6A.32–34 Experimental evidence suggests that the C terminus inhibits the recruitment of the dynein-dynactin complex by the N terminus, implying a regulatory effect of both domains.32–34 Finally, BICD2 also has a role in the positioning of the nucleus and centrosomes in mitotic cells. During the G2 phase, the C terminus of BICD2 switches from the interacting partner RAB6A to RANBP2, a component of the nuclear pore. Interestingly, RANBP2 has been reported to also directly interact with KIF5B and KIF5C, supporting the notion that it is involved in microtubule motor recruitment.42 Depletion of BICD2 inhibits both dynein- and kinesin-1-dependent movements of the nucleus and cytoplasmic nucleopore complexes, demonstrating that BICD2 is essential for both retrograde and anterograde transport.29
Given that BICD2 plays an important role in Golgi dynamics and targeting,34 we investigated a possible effect of the identified BICD2 mutations on the integrity of the Golgi apparatus. For each variant, we generated a turbo GFP (tGFP)-BICD2 fusion protein by using the wild-type BICD2 cDNA and site-directed mutagenesis to introduce the respective mutations. All clones were Sanger sequenced for verification of the introduced mutation. HeLa cells were transiently transfected with each of the five tGFP-BICD2 plasmids (the wild-type, the three mutations, and the rare variant) and immunostained against Giantin, a conserved Golgi membrane marker,43 and with DAPI for visualization of the nucleus. The tGFP to which BICD2 was fused allowed us to easily distinguish transfected from nontransfected cells. Whereas the transient expression of the wild-type tGFP-BICD2 protein had no impact on Golgi integrity, the transfected cells with altered BICD2 showed very faint, few, or dispersed Giantin signals in the transfected cells compared to the surrounding nontransfected cells, suggesting Golgi fragmentation (Figure 3). Rare variant c.269A>G (p.Lys90Arg) showed only slightly fainter and more disorganized Golgi staining than did the control, so we cannot fully exclude a potential modifying effect (Figure 3).
Figure 3.
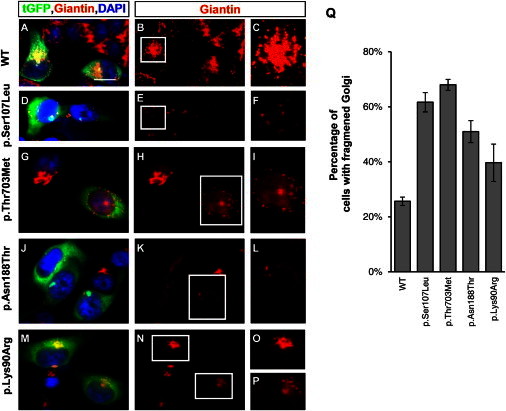
Effect of the Mutant BICD2 Alleles on the Golgi Apparatus
(A–P) HeLa cells were transfected with tGFP-BICD2 pCMV6-AC cDNA (either wild-type [A–C] or with BICD2 missense variants [D–P]). Cells were fixed and stained for a specific Golgi marker, Giantin (red) (1:100, Abcam), and DAPI (blue) (Life Technologies). Only transfected cells glowed green under the fluorescent microscope and allowed an easy comparison with untransfected cells, which reflected the wild-type situation. For better visibility, the Giantin staining alone is shown in the middle row and the inset in the right row, which represents an enlarged Golgi of a transfected cell. Scale bars represent 20 μm.
(A–C) The cells transfected with the wild-type construct show a condensed and abundant Golgi structure.
(D–P) Compared to wild-type or nontransfected cells, cells transfected with the three mutant cDNA constructs and the rare variant show a fragmented Golgi with a very diffuse and faint signal.
(Q) Quantification of cells with fragmented Golgi; 70–100 cells were counted for each experiment (n = 3) and are shown as the mean ± SD.
Next, the effect of the pathogenic BICD2 was investigated in primary fibroblast cells established from skin biopsies from a Canadian affected individual harboring the p.Thr703Met substitution and a Dutch affected individual harboring the p.Asn188Thr substitution. As shown by immunoblot analysis, BICD2 levels were lower in fibroblasts of affected individuals than in control fibroblasts by a so far unknown mechanism (Figures 4A and 4B). Immunostainings with BICD2 antibody and two different Golgi markers, p230 and MG160, revealed diffused BICD2 staining throughout the cytoplasm and an increased signal at the Golgi site in wild-type fibroblasts compared to affected fibroblasts, in which BICD2 appeared trapped within the fragmented Golgi. Both Golgi markers showed massive dispersion and fragmentation in the affected fibroblast cell lines (Figures 4D–4O and Figures S1A–S1F). Golgi fragmentation was more pronounced in the Canadian affected individuals with the p.Thr703Met substitution in the C-terminal coiled-coil domain than in the Dutch affected individual with the p.Asn188Thr substitution in the N-terminal coiled-coil domain (Figure 4C). These functional data correlate with the Canadian family’s more severe phenotype with contractures rather than the phenotype of the second Dutch family, in which no contractures were seen in the affected individuals.
Figure 4.
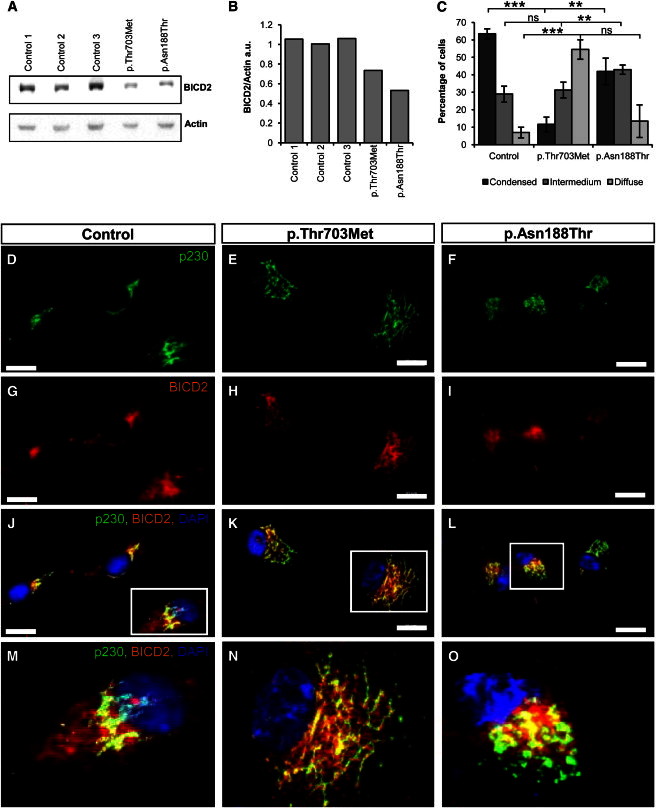
Mutations in BICD2 Cause Severe Golgi Fragmentation in Primary Fibroblasts
(A) Immunoblot analyses of primary-fibroblast proteins of one control cell line and two cell lines derived from affected individuals. The cells were stained with BICD2 and β-actin as a loading control.
(B) Quantification of the immunoblot shows reduced BICD2 levels in fibroblast lines isolated from affected individuals.
(C) Golgi structures, defined as condensed, intermedium, or diffuse in 100 cells of each cell line, were counted in three independent experiments and are given as the mean ± SD. Each quantification was done blindly by two independent persons. Statistical significance is given between controls and each of the two cell lines as ∗∗∗p < 0.001, ∗∗p < 0.01, or ∗p < 0.05. The following abbreviation is used: ns, not significant.
(D–O) Immunostaining of control (D, G, J, and M) and disease (E, F, H, I, K, L, N, and O) fibroblasts with antibodies against trans-Golgi marker p230 in green (1:100, BD Transduction Laboratories) and BICD2 in red (1:200, Sigma), as well as merged pictures, are shown. The nucleus is stained with DAPI in blue. (M), (N), and (O) show magnified insets of single cells from (J), (K), and (L), respectively. Note that compared to control fibroblasts, affected fibroblasts show strongly fragmented Golgi and BICD2 trapped in the Golgi. Scale bars represent 20 μm.
Hence, BICD2 mutations in humans, flies, and mice, as well as mutations in genes encoding some of the interaction partners (dynein, dynactin, and kinesins), are associated with motor neuron diseases. This strongly supports the view that the entire complex is crucial in proper motor neuron functioning. Moreover, the function of the survival of motor neuron (SMN) protein that when reduced causes proximal autosomal-recessive SMA has recently been linked to the Golgi network. SMN interacts with α-coatomer, which mediates vesicle trafficking between the Golgi compartments and regulates SMN granule secretion from the Golgi apparatus.44 Disruption of Golgi-mediated granule secretion reduces murine Smn levels in neuritis, suggesting that vesicular transport of Smn granules is mediated through the Golgi.45
In conclusion, our data reveal that mutations in BICD2 cause slowly progressive autosomal-dominant SMA with weakness and atrophy of proximal and distal muscles mainly of the legs. BICD2, a protein essential in dynein-dynactin microtubule transport, in clathrin-mediated synaptic-vesicular recycling, and in the Golgi network is thus crucial for the functional integrity of lower motor neurons. Interacting partners of BICD2 might represent good candidates for the still many unsolved cases of SMA and/or HMN.
Acknowledgments
We are very grateful to all participating affected families and their relatives. We would like to particularly thank Markus Storbeck for computational support and Karin Boss for critical reading of the manuscript. The research leading to these results received funding from the European Community’s Seventh Framework Program FP7/2007-2013 under grant agreement 223143 (project acronym TECHGENE) to H.S. and J.V. and under grant agreement 2012-305121 (project acronym NeurOmics) to B.W.
Contributor Information
Hans Scheffer, Email: h.scheffer@gen.umcn.nl.
Brunhilde Wirth, Email: brunhilde.wirth@uk-koeln.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
dbSNP build137, http://www.ncbi.nlm.nih.gov/snp
easyLINKAGE, http://nephrologie.uniklinikum-leipzig.de/nephrologie.site,postext,easylinkage,a_id,797.html
Human Gene Mutation Database (HGMD), http://www.hgmd.org/
Integrative Genomics Viewer (IGV), http://www.broadinstitute.org/igv
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
UCSC Genome Brower, http://www.genome.ucsc.edu
UniProt, http://www.uniprot.org
References
- 1.De Jonghe P., Auer-Grumbach M., Irobi J., Wagner K., Plecko B., Kennerson M., Zhu D., De Vriendt E., Van Gerwen V., Nicholson G. Autosomal dominant juvenile amyotrophic lateral sclerosis and distal hereditary motor neuronopathy with pyramidal tract signs: synonyms for the same disorder? Brain. 2002;125:1320–1325. doi: 10.1093/brain/awf127. [DOI] [PubMed] [Google Scholar]
- 2.Baets J., Deconinck T., De Vriendt E., Zimoń M., Yperzeele L., Van Hoorenbeeck K., Peeters K., Spiegel R., Parman Y., Ceulemans B. Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain. 2011;134:2664–2676. doi: 10.1093/brain/awr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zickler D., Kleckner N. Meiotic chromosomes: integrating structure and function. Annu. Rev. Genet. 1999;33:603–754. doi: 10.1146/annurev.genet.33.1.603. [DOI] [PubMed] [Google Scholar]
- 4.Kiernan M.C., Vucic S., Cheah B.C., Turner M.R., Eisen A., Hardiman O., Burrell J.R., Zoing M.C. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 5.Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 6.Wee C.D., Kong L., Sumner C.J. The genetics of spinal muscular atrophies. Curr. Opin. Neurol. 2010;23:450–458. doi: 10.1097/WCO.0b013e32833e1765. [DOI] [PubMed] [Google Scholar]
- 7.Torres-Benito L., Ruiz R., Tabares L. Synaptic defects in spinal muscular atrophy animal models. Dev. Neurobiol. 2012;72:126–133. doi: 10.1002/dneu.20912. [DOI] [PubMed] [Google Scholar]
- 8.Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puls I., Jonnakuty C., LaMonte B.H., Holzbaur E.L., Tokito M., Mann E., Floeter M.K., Bidus K., Drayna D., Oh S.J. Mutant dynactin in motor neuron disease. Nat. Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 10.Irobi J., Van Impe K., Seeman P., Jordanova A., Dierick I., Verpoorten N., Michalik A., De Vriendt E., Jacobs A., Van Gerwen V. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004;36:597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- 11.Evgrafov O.V., Mersiyanova I., Irobi J., Van Den Bosch L., Dierick I., Leung C.L., Schagina O., Verpoorten N., Van Impe K., Fedotov V. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- 12.Windpassinger C., Auer-Grumbach M., Irobi J., Patel H., Petek E., Hörl G., Malli R., Reed J.A., Dierick I., Verpoorten N. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 2004;36:271–276. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y.Z., Bennett C.L., Huynh H.M., Blair I.P., Puls I., Irobi J., Dierick I., Abel A., Kennerson M.L., Rabin B.A. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am. J. Hum. Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura A.L., Mitne-Neto M., Silva H.C., Richieri-Costa A., Middleton S., Cascio D., Kok F., Oliveira J.R., Gillingwater T., Webb J. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004;75:822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolb S.J., Snyder P.J., Poi E.J., Renard E.A., Bartlett A., Gu S., Sutton S., Arnold W.D., Freimer M.L., Lawson V.H. Mutant small heat shock protein B3 causes motor neuropathy: utility of a candidate gene approach. Neurology. 2010;74:502–506. doi: 10.1212/WNL.0b013e3181cef84a. [DOI] [PubMed] [Google Scholar]
- 16.Auer-Grumbach M., Olschewski A., Papić L., Kremer H., McEntagart M.E., Uhrig S., Fischer C., Fröhlich E., Bálint Z., Tang B. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat. Genet. 2010;42:160–164. doi: 10.1038/ng.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harms M.B., Ori-McKenney K.M., Scoto M., Tuck E.P., Bell S., Ma D., Masi S., Allred P., Al-Lozi M., Reilly M.M. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78:1714–1720. doi: 10.1212/WNL.0b013e3182556c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beetz C., Pieber T.R., Hertel N., Schabhüttl M., Fischer C., Trajanoski S., Graf E., Keiner S., Kurth I., Wieland T. Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type V. Am. J. Hum. Genet. 2012;91:139–145. doi: 10.1016/j.ajhg.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barwick K.E., Wright J., Al-Turki S., McEntagart M.M., Nair A., Chioza B., Al-Memar A., Modarres H., Reilly M.M., Dick K.J. Defective presynaptic choline transport underlies hereditary motor neuropathy. Am. J. Hum. Genet. 2012;91:1103–1107. doi: 10.1016/j.ajhg.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dierick I., Baets J., Irobi J., Jacobs A., De Vriendt E., Deconinck T., Merlini L., Van den Bergh P., Rasic V.M., Robberecht W. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study. Brain. 2008;131:1217–1227. doi: 10.1093/brain/awn029. [DOI] [PubMed] [Google Scholar]
- 21.Walker A.K., Atkin J.D. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life. 2011;63:754–763. doi: 10.1002/iub.520. [DOI] [PubMed] [Google Scholar]
- 22.Frijns C.J., Van Deutekom J., Frants R.R., Jennekens F.G. Dominant congenital benign spinal muscular atrophy. Muscle Nerve. 1994;17:192–197. doi: 10.1002/mus.880170210. [DOI] [PubMed] [Google Scholar]
- 23.Vermeer S., Hoischen A., Meijer R.P., Gilissen C., Neveling K., Wieskamp N., de Brouwer A., Koenig M., Anheim M., Assoum M. Targeted next-generation sequencing of a 12.5 Mb homozygous region reveals ANO10 mutations in patients with autosomal-recessive cerebellar ataxia. Am. J. Hum. Genet. 2010;87:813–819. doi: 10.1016/j.ajhg.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoischen A., van Bon B.W., Gilissen C., Arts P., van Lier B., Steehouwer M., de Vries P., de Reuver R., Wieskamp N., Mortier G. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 25.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams C., Suchowersky O., Lowry R.B. Congenital autosomal dominant distal spinal muscular atrophy. Neuromuscul. Disord. 1998;8:405–408. doi: 10.1016/s0960-8966(98)00042-x. [DOI] [PubMed] [Google Scholar]
- 28.Baens M., Marynen P. A human homologue (BICD1) of the Drosophila bicaudal-D gene. Genomics. 1997;45:601–606. doi: 10.1006/geno.1997.4971. [DOI] [PubMed] [Google Scholar]
- 29.Splinter D., Tanenbaum M.E., Lindqvist A., Jaarsma D., Flotho A., Yu K.L., Grigoriev I., Engelsma D., Haasdijk E.D., Keijzer N. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol. 2010;8:e1000350. doi: 10.1371/journal.pbio.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goud B., Gleeson P.A. TGN golgins, Rabs and cytoskeleton: regulating the Golgi trafficking highways. Trends Cell Biol. 2010;20:329–336. doi: 10.1016/j.tcb.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Li X., Kuromi H., Briggs L., Green D.B., Rocha J.J., Sweeney S.T., Bullock S.L. Bicaudal-D binds clathrin heavy chain to promote its transport and augments synaptic vesicle recycling. EMBO J. 2010;29:992–1006. doi: 10.1038/emboj.2009.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matanis T., Akhmanova A., Wulf P., Del Nery E., Weide T., Stepanova T., Galjart N., Grosveld F., Goud B., De Zeeuw C.I. Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat. Cell Biol. 2002;4:986–992. doi: 10.1038/ncb891. [DOI] [PubMed] [Google Scholar]
- 33.Splinter D., Razafsky D.S., Schlager M.A., Serra-Marques A., Grigoriev I., Demmers J., Keijzer N., Jiang K., Poser I., Hyman A.A. BICD2, dynactin, and LIS1 cooperate in regulating dynein recruitment to cellular structures. Mol. Biol. Cell. 2012;23:4226–4241. doi: 10.1091/mbc.E12-03-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoogenraad C.C., Akhmanova A., Howell S.A., Dortland B.R., De Zeeuw C.I., Willemsen R., Visser P., Grosveld F., Galjart N. Mammalian Golgi-associated Bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J. 2001;20:4041–4054. doi: 10.1093/emboj/20.15.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teuling E., van Dis V., Wulf P.S., Haasdijk E.D., Akhmanova A., Hoogenraad C.C., Jaarsma D. A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum. Mol. Genet. 2008;17:2849–2862. doi: 10.1093/hmg/ddn182. [DOI] [PubMed] [Google Scholar]
- 36.Münch C., Sedlmeier R., Meyer T., Homberg V., Sperfeld A.D., Kurt A., Prudlo J., Peraus G., Hanemann C.O., Stumm G., Ludolph A.C. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- 37.Lai C., Lin X., Chandran J., Shim H., Yang W.J., Cai H. The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J. Neurosci. 2007;27:13982–13990. doi: 10.1523/JNEUROSCI.4226-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karle K.N., Möckel D., Reid E., Schöls L. Axonal transport deficit in a KIF5A( -/- ) mouse model. Neurogenetics. 2012;13:169–179. doi: 10.1007/s10048-012-0324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia C.H., Roberts E.A., Her L.S., Liu X., Williams D.S., Cleveland D.W., Goldstein L.S. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 2003;161:55–66. doi: 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Claussen M., Suter B. BicD-dependent localization processes: from Drosophilia development to human cell biology. Ann. Anat. 2005;187:539–553. doi: 10.1016/j.aanat.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Dienstbier M., Li X. Bicaudal-D and its role in cargo sorting by microtubule-based motors. Biochem. Soc. Trans. 2009;37:1066–1071. doi: 10.1042/BST0371066. [DOI] [PubMed] [Google Scholar]
- 42.Cai Y., Singh B.B., Aslanukov A., Zhao H., Ferreira P.A. The docking of kinesins, KIF5B and KIF5C, to Ran-binding protein 2 (RanBP2) is mediated via a novel RanBP2 domain. J. Biol. Chem. 2001;276:41594–41602. doi: 10.1074/jbc.M104514200. [DOI] [PubMed] [Google Scholar]
- 43.Linstedt A.D., Hauri H.P. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell. 1993;4:679–693. doi: 10.1091/mbc.4.7.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peter C.J., Evans M., Thayanithy V., Taniguchi-Ishigaki N., Bach I., Kolpak A., Bassell G.J., Rossoll W., Lorson C.L., Bao Z.Z., Androphy E.J. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum. Mol. Genet. 2011;20:1701–1711. doi: 10.1093/hmg/ddr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ting C.H., Wen H.L., Liu H.C., Hsieh-Li H.M., Li H., Lin-Chao S. The spinal muscular atrophy disease protein SMN is linked to the Golgi network. PLoS ONE. 2012;7:e51826. doi: 10.1371/journal.pone.0051826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.