

Abstract
The G4C2 repeat expansion in C9orf72 is the most common known cause of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). We tested the hypothesis that the repeat expansion causes aberrant CpG methylation near the G4C2 repeat, which could be responsible for the downregulation of gene expression. We investigated the CpG methylation profile by two methods using genomic DNA from the blood of individuals with ALS (37 expansion carriers and 64 noncarriers), normal controls (n = 76), and family members of 7 ALS probands with the expansion. We report that hypermethylation of the CpG island 5′ of the G4C2 repeat is associated with the presence of the expansion (p < 0.0001). A higher degree of methylation was significantly correlated with a shorter disease duration (p < 0.01), associated with familial ALS (p = 0.009) and segregated with the expansion in 7 investigated families. Notably, we did not detect methylation for either normal or intermediate alleles (up to 43 repeats), bringing to question the current cutoff of 30 repeats for pathological alleles. Our study raises several important questions for the future investigation of large data sets, such as whether the degree of methylation corresponds to clinical presentation (ALS versus FTLD).
Main Text
Amyotrophic lateral sclerosis (ALS [MIM 612069]) and frontotemporal lobar degeneration (FTLD [MIM 600274]) constitute a neurodegenerative syndrome, with individual cases presenting along a clinico-pathological spectrum. Individuals with FTLD exhibit primary dementia commonly characterized by early behavioral and/or language changes, whereas ALS is characterized by the degeneration of motor neurons responsible for voluntary movements. Both syndromes may occur within the same family or the same person. The most common known genetic cause of ALS and FTLD is the recently discovered noncoding hexanucleotide (G4C2)n > 30 repeat expansion in the chromosome 9 open reading frame 72 gene (C9orf72 [MIM 614260]),1,2 which accounts for 24%–37% of familial and 6%–7% of sporadic cases in whites.3 This discovery was confirmed in multiple data sets, including ours;4–7 however, the disease mechanism related to the C9orf72 G4C2 repeat expansion remains unknown.
There are several hypotheses about the functional consequences of the repeat expansion. The toxic gain-of-function model is related either to the sequestering of RNA binding proteins by RNA foci consisting of pre-mRNA containing the repeat expansion1 or to the non-ATG-initiated translation from the repeat expansion in all reading frames leading to the aggregation of dipeptide-repeat proteins in neurons.8,9 Another model is a loss-of-function mechanism (haploinsufficiency), supported by observations that the expansion leads to ∼50% reduction of C9orf72 mRNA in frontal cortex and lymphoblasts derived from heterozygous mutation carriers.1,4 Finally, it is possible that all three mechanisms contribute to the disease.
Here we tested the hypothesis that the expansion may cause aberrant CpG methylation of C9orf72 leading to downregulation of its mRNA, as was shown for different C9orf72 transcripts in a few published expansion carriers.1,4 Notably, the G4C2 repeat is mapped to the promoter region of the NM_018325.3 transcript, and the promoter region of two other C9orf72 transcripts (NM_001256054.1 and NM_145005.5) overlaps a CpG island 5′ of the repeat (Figure S1 available online). Cytosine (C) methylation of CpG dinucleotides is an important epigenetic modification10 that could lead to gene expression silencing as a result of hypermethylation of CpG islands11,12 (defined as >200 bp regions with >50% CG content and >0.6 ratio of observed to expected number of CG-dinucleotides). Several genes with repeat expansions are known to have aberrant CpG methylation leading to downregulation of gene expression. For example, in individuals with Friedreich ataxia (MIM 229300), hypermethylation was detected upstream of the GAA expansion in intron 1 of FXN (MIM 606829), which leads to a deficit of FXN mRNA in blood, heart, and brain.13–15 DNA hypermethylation is also caused by a CGG expansion in the 5′ UTR of FMR1 (MIM 309550) responsible for fragile X mental retardation syndrome (MIM 300624).16–18 Moreover, the CTG expansion in the 3′ UTR of DMPK (MIM 605377) causes hypermethylation19 of the promoter of a downstream gene, reducing its transcription.20–22
To gain new insight into the C9orf72 disease mechanism, we investigated the CpG methylation profile around the G4C2 repeat by two methods using genomic DNA collected from our well-characterized Canadian ALS cohort and normal controls. Informed consent was obtained from all participants in accordance with the ethical review board. Study participants were diagnosed with ALS at the ALS Clinic of Sunnybrook Health Sciences Centre in Toronto according to established criteria.23–25 Expansion carriers were defined by the previously suggested 30-repeat cutoff.1,2 DNA from blood was available for 37 unrelated ALS expansion carriers (mainly identified in our previous study7) and 140 noncarriers (64 ALS cases and 76 controls >65 years old). Sample characteristics are presented in Table 1. The clinical parameters between ALS expansion carriers and noncarriers are similar, except that carriers have an earlier age of onset (p = 0.04) and higher percentage of familial cases (p = 2 × 10−6), which is a known association.4 In addition, we investigated blood DNA from available family members of seven ALS probands with the expansion (including four simplex cases) and three previously reported7 carriers of intermediate alleles (24, 32, 39 repeats) diagnosed with Parkinson disease (MIM 168600). DNA was also extracted from blood, frontal cortex, and cervical spinal cord for seven autopsy-confirmed expansion carriers, as well as from frontal cortex of 14 controls.
Table 1.
Sample Characteristics
|
Blood |
Brain (Frontal Cortex) |
|||||
|---|---|---|---|---|---|---|
| ALS Exp | ALS Nonexp | Control | ALS Exp | Control | ||
| Number of samples | 37 | 64 | 76 | 7 | 14 | |
| Age at sample collection | 60.5 ± 9.5 | 66.5 ± 9.3 | 73.0 ± 7.6 | 56.7 ± 7.8 | 78.1 ± 7.7 | |
| Age at onset | 58.6 ± 9.4 | 63.7 ± 11.4 | – | 55.3 ± 8.1 | – | |
| Female (frequency) | 15 (0.41) | 25 (0.39) | 48 (0.63) | 3 | 5 | |
| Family history ( frequency) | 18 (0.49) | 5 (0.08) | – | 5 | – | |
| Diagnosis | ALS ( frequency) | 32 (0.86) | 54 (0.84) | – | 4 | – |
| ALS-FTD ( frequency) | 5 (0.14) | 10 (0.16) | – | 3 | – | |
| Range of G4C2 repeats | Small allele | 2–11 | 2–8 | 2–8 | 2–10 | 2–5 |
| Larger allele | 35, exp | 2–22 | 2–19 | exp | 2–12 | |
Abbreviation is as follows: exp, expansion.
To assess the level of methylation we developed two assays. The basis of the methylation-sensitive restriction enzyme assay was adapted from previous reports26,27 (the experimental principle is shown in Figure S2). In brief, 100 ng of DNA was digested overnight at 37°C with 1 U methylation-sensitive restriction enzyme (HhaI, HpaII, or HpyCH4IV, New England Biolabs), followed by 10 min inactivation at 95°C. The enzyme digests unmethylated DNA, and therefore the following PCR would produce an amplification product only for methylated DNA. As an internal control, a duplicate reaction for each DNA sample was prepared without the enzyme. The digested and undigested DNA was amplified in parallel (Tm = 60.5°C) (Table S1). PCR products (1.5 μl) were resolved on a 1.5% agarose gel. To quantify the band intensity, black/white inverted gel images were analyzed by the Gel Analysis tool in Image J, and the observed methylation (OM) ratio was calculated based on the band intensities (PCR from digested DNA ÷ PCR from undigested DNA).
The direct bisulfite sequencing assay was adapted from previous reports.19,28,29 It is based on the conversion of unmethylated C to T by bisulfite treatment, while methylated C remain unchanged. Bisulfite treatment was performed with 100 ng of DNA by the Imprint DNA Modification kit (Sigma). The investigated region is shown in Figure S3. After conversion, the region 5′ of the G4C2 repeat was amplified by a seminested PCR (Table S1). Bisulfite-treated DNA (2 μl) was amplified by FastStart PCR Master Mix (Roche) by touchdown PCR (for primers BSP_1F, BSP_1R: Tm from 68°C to 58°C at a rate of −1°C/cycle and fixed at 58°C for 30 cycles; for primers BSP_2F, BSP_2R: Tm from 67°C to 57°C at a rate of −1°C/cycle and fixed at 57°C for 20 cycles; 3 min extension for each cycle). Methylation was detected by direct inspection of sequencing chromatograms and an integrated tool within Mutation Surveyor version 4.0 (Softgenetics). Only samples with >95% conversion rate of non-CpG C were included in the analyses. To avoid quantification errors caused by relative peak height measurements at each CpG, broad categories were used to score methylation level: unmethylated CpG (T peak) or methylated CpG (T/C double peaks). Of note, “C peak only” (indicating 100% methylation) was not observed in any sample. For each sample we obtained the total number of methylated CpG sites detected in the inspected region.
A linear regression analysis was performed between the OM ratio (HhaI assay) and number of methylated CpG sites (bisulfite sequencing assay) to calculate the correlation between the assays. A nonlinear regression analysis was used to evaluate the correlation between methylation level and disease duration. The goodness of fit measure (R2) was used to quantify these correlations. The p value was calculated by ANOVA to determine the significance of R2. Pearson’s or Spearman’s correlation coefficients were used to measure the correlation between independent variables: repeat size (<50 repeats), age, and methylation level. The nonparametric Mann-Whitney U test was used to compare continuous variables between two groups. The two-sided Pearson χ2 test or Fisher’s exact test (when expected value <5) were used to compare categorical variables. All analyses were performed with SPSS (v.20).
By using the UCSC database, we detected two predicted CpG islands immediately flanking the G4C2 repeat (Figure S1), which contain restriction sites for three methylation-sensitive enzymes (HhaI, HpaII, and HpyCH4IV). To assess the sensitivity of these enzymes for methylation detection, we used a premixed calibration standard DNA (EpigenDx) consisting of a low-methylated DNA (<5% methylation), a high-methylated DNA (>85% methylation), and a mixture series of both. The region 5′ of the G4C2 repeat (fragment #1) has two HhaI, one HpaII, and three HpyCH4IV sites (Figure S4A). Analysis of fragment #1 revealed that the HhaI assay is the most sensitive, because it is able to detect methylation in a mixture containing only 5% high-methylated DNA, whereas the other two enzymes start to detect methylation from 25% to 50% (Figure S4B). No methylation was observed for the DNA of a randomly selected nonexpansion carrier (Figure S4B). By measuring band intensity (four replicates), we obtained a standard curve with R2 = 0.96, indicating the reliability of the quantification by the HhaI assay (Figure S4C). Therefore, the following case-control study was conducted with the HhaI assay.
The same calibration DNA standards were used to assess the efficiency of the bisulfite sequencing assay. All non-CpG C were successfully converted to T (Figure S4D). As expected, only a few methylated C were detected in the low-methylated standard, and the assay was able to detect methylation of all CpG sites, even in the mixture with only 5% high-methylated DNA.
CpG islands on both sides of the G4C2 repeat were analyzed by the HhaI assay (Figure 1A). The region 5′ (but not 3′) of the repeat revealed evidence of hypermethylation in expansion carriers (Figure 1B). The bisulfite sequencing assay also revealed evidence of hypermethylation at the 5′ region (as presented below) but not the 3′ region, which was evaluated in the pilot ALS cohort of 32 expansion carriers and 16 noncarriers (data not shown). A similar methylation pattern was reported for Friedreich ataxia15 and myotonic dystrophy.19 It was suggested that an expanded repeat could act as a barrier inhibiting the spread of methylation downstream;15 however, the underlying mechanism is not well understood. Notably, the G4C2 repeat expansion itself could also be considered part of the CpG island and may be methylated as well, but currently available technologies do not allow the large expansion to be amplified by PCR or thoroughly sequenced.
Figure 1.
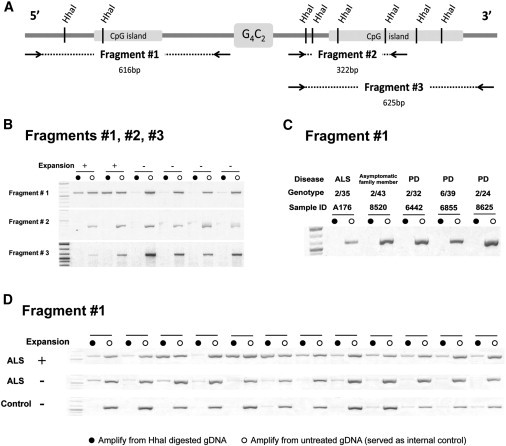
HhaI Assay
(A) A schematic presentation of the genomic region around the G4C2 repeat. Two predicted CpG islands located in the sequences flanking the repeat were covered by three fragments.
(B) DNA undigested and digested with HhaI was amplified for all three fragments and the resulting band intensity quantified. Only fragment #1 shows differential methylation between expansion carriers and noncarriers.
(C) Samples with intermediate size alleles (24–43 repeats) were not methylated upstream of the G4C2 repeat (fragment #1).
(D) Representative gel analysis of 12 ALS expansion carriers, 12 ALS noncarriers, and 12 controls (a higher methylation level was observed in expansion carriers versus noncarriers).
Next, methylation of the CpG island 5′ of the G4C2 repeat was assessed by both assays in the entire data set of 177 unrelated individuals. The OM ratio of the HhaI assay was significantly higher in the group of 37 ALS expansion carriers (mean ± SD = 0.52 ± 0.40) versus 64 ALS noncarriers (mean ± SD = 0.17 ± 0.09, Mann-Whitney U test p < 0.0001) or versus 76 controls (mean ± SD = 0.13 ± 0.07, Mann-Whitney U test p < 0.0001). Almost all samples without an expansion allele (138 out of 140) had an OM ratio below 0.3, which represents the methylation level of the low-methylated DNA standard (<5% methylation) in the HhaI assay standard curve. In contrast, 25 of 37 ALS expansion carriers had a ratio >0.3. A representative gel image of 12 samples from each group is shown in Figure 1D.
The bisulfite sequencing assay revealed the methylation status 5′ of the G4C2 repeat beyond the two CpG sites recognized by the HhaI assay. A total of 26 CpG sites were assessed (Figure S3). A representative sequence chromatogram is shown in Figure 2A. In the nonexpansion group, 97% of samples (n = 136) were unmethylated (number of methylated CpG sites = 0). In contrast, 73% of expansion carriers (n = 27) had a number of methylated CpG sites ≥1, and methylation was detected at each of the 26 CpG sites (Figure S5A). We categorized the different methylation levels based on the maximum number of methylated CpG observed in controls: 0, no methylation; 1–3, low methylation; and 4–26, high methylation (Table 2). In agreement with the HhaI assay, a significant difference in methylation level between the expansion and nonexpansion groups was observed (p < 0.0001). No difference was found between the two nonexpansion groups (control versus ALS, p = 0.625).
Figure 2.
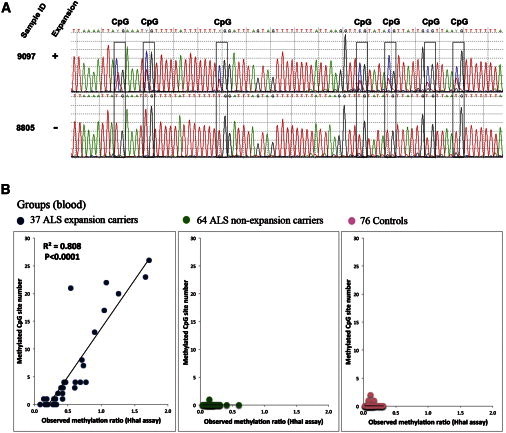
Correlation between HhaI Assay and the Bisulfite Sequencing Assay
(A) Representative chromatograms of a sequence containing seven CpG sites for an expansion carrier and a noncarrier.
(B) A scatter plot of the observed methylation ratio (HhaI assay) and number of methylated CpG sites (bisulfite sequencing) revealed a significant correlation between the two assays with blood DNA.
Table 2.
Methylation Level of Blood DNA from ALS Expansion Carriers, ALS Nonexpansion Carriers, and Controls
| Methylation Level (Number of Methylated CpG) |
ALS Exp |
ALS Nonexp |
Control |
|||
|---|---|---|---|---|---|---|
| N | Frequency | N | Frequency | N | Frequency | |
| No methylation (0) | 10 | 0.27 | 63 | 0.98 | 73 | 0.96 |
| Low methylation (1–3) | 12 | 0.32 | 1 | 0.02 | 3 | 0.04 |
| High methylation (4–26) | 15 | 0.41 | 0 | 0.00 | 0 | 0.00 |
| Total | 37 | 64 | 76 | |||
| pa: compared to ALS exp group | <0.0001 | <0.0001 | ||||
| pa: compared to ALS nonexp group | 0.625 | |||||
Abbreviation is as follows: exp, expansion.
2-sided Pearson χ2 test was used, df = 2. Fisher’s exact test was used (when expected value < 5).
A scatter plot of the OM ratio against the number of methylated CpG sites revealed a significant correlation between the two assays (p < 0.0001) (Figure 2B). Bisulfite sequencing analysis of the 26 CpG sites revealed that the two CpG sites recognized by the HhaI assay (CpG2 and CpG8) are among the most frequently methylated sites in expansion carriers (70% and 30%, respectively), which is the basis of the strong correlation between the assays (Figure S5A).
Notably, neither assay identified methylation in carriers with intermediate alleles (24, 32, 39, or 43 repeats; Figure 1C). Individuals without an expansion had a wide range of normal alleles (2–22 repeats) (Table 1), all of which had a low methylation level (methylated CpG sites < 3). Finally, the number of methylated CpG sites in expansion carriers and the size of their normal allele (2–11 repeats) were not correlated (Spearman’s correlation coefficient = 0.258, p = 0.124), indicating that the normal allele does not contribute to the methylation profile of expansion carriers.
The level of methylation 5′ of the G4C2 repeat did not correlate with age at time of examination (range: 39–80 years old; Spearman’s correlation coefficient = −0.015, p = 0.931) or age of onset (range: 37–78 years old; Spearman’s correlation coefficient = 0.041, p = 0.814). However, it was inversely associated with disease duration (n = 24; OM: R2 = 0.388, p = 0.001; number of methylated CpG sites: R2 = 0.303, p = 0.005). Moreover, methylation was also significantly associated with familial ALS (HhaI assay: Mann-Whitney U test p = 0.009; bisulfite sequencing assay: Mann-Whitney U test p = 0.026 and Pearson χ2 p = 0.036) but not with site of onset or diagnosis subcategory (ALS versus ALS-FTD). The study of seven families indicated that high methylation level segregates with the expansion (Table 3, Figure 3). The number of methylated CpG sites observed in expansion carriers was similar within each pedigree, but variable between families. High methylation levels were not observed in family members without an expansion, in contrast to their relatives with an expansion. For instance, in family PED 6 (Figure 3), individual #8259 (noncarrier, III-2) shares a 7-repeat allele with sibling #8261 (carrier, III-4) and inherited an 8-repeat allele from their parent #7387 (carrier, II-3) but does not have a high methylation level.
Table 3.
Methylation Levels within the ALS Pedigrees
| Pedigree | N | Number of Generations with Exp |
Observed Methylation Ratio (HhaI Assay) |
Number of Methylated CpG Sites of Each Pedigree (Bisulfite Sequencing) |
|
|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | Methylation Level | |||
| Expansion Carriers | |||||
| ped 1 | 4 | 1 | 0.37 ± 0.21 | 1 ± 1.2 | low |
| ped 3 | 2 | 2 | 0.44 ± 0.02 | 2 ± 1.0 | low |
| ped 6 | 4 | 2 | 0.83 ± 0.13 | 13 ± 3.6 | high |
| 1 (43 repeats) | 1 | 0.17 | 0 | no | |
| ped 8 | 2 | 1 | 0.38 ± 0.05 | 4 ± 1.5 | high |
| ped 13 | 1 | 1 | 0.9 | 13 | high |
| ped 16 | 2 | 1 | 1.05 ± 0.00 | 21 ± 4.0 | high |
| ped 17 | 2 | 1 | 0.72 ± 0.02 | 15 ± 7.5 | high |
| Nonexpansion Carriers | |||||
| All pedigrees | 8 | 0.21 ± 0.06 | 0 ± 0.6 | no | |
Abbreviation is as follows: exp, expansion.
Figure 3.
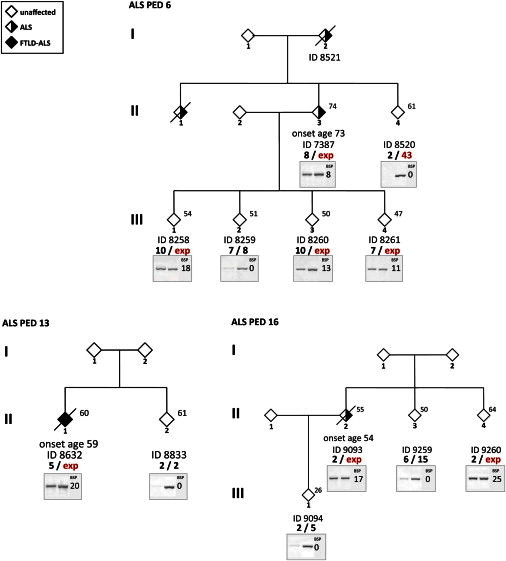
Methylation Results in Three Representative Families of Probands who Carry the C9orf72 Expansion
All available family members were studied to compare methylation levels. Individual ID and C9orf72 genotype are shown beneath the corresponding diamond. Arabic numbers indicate the repeat number and “exp” represents the expansion allele. Age at the time of examination is shown in the upper right corner. Age of onset is indicated above the ID number. Gender of the family members is masked to protect confidentiality. Black/white inverted gel images from the HhaI assay are shown below the genotype. The individual number of methylated CpG sites is shown on the right side of the gel image beneath “BSP,” which stands for bisulfite sequencing PCR.
Differential methylation across different tissues is known to be common in mammals,30 although concordance of methylation level across different tissues was seen in the GAA expansion causing Friedreich ataxia.15 According to GENEVESTIGATOR, C9orf72 is robustly expressed in different human tissues, including blood and brain. Therefore, we evaluated the methylation level in blood, frontal cortex, and cervical spinal cord available from seven individuals with ALS. The degree of methylation in frontal cortex of these individuals was higher than in DNA from control brain samples (n = 14) (Table S2). Importantly, methylation levels for DNA isolated from the three tissues for the same person were within a similar range by both assays (Figures S5B and S6). Significant correlation was observed between the different tissues in almost all pairwise combinations (Table S3). However, this observation remains to be validated in a larger sample set.
Available RNA samples (n = 8) were analyzed by an inventoried TaqMan gene expression assay targeting all known C9orf72 transcripts (Figure 4). Total blood RNA was extracted with the RNeasy Mini Kit (QIAGEN) and reverse transcribed to cDNA by the AffinityScript Multiple Temperature cDNA Synthesis Kit (Agilent Technologies). RNA integrity was checked on an Agilent 2100 Bioanalyzer (only samples with RIN > 7 were used). Real-time PCR was performed in triplicate for HPRT1 (MIM 308000) (Hs99999909_m1), UBC (MIM 191340) (Hs00824723_m1), B2M (MIM 109700) (Hs99999907_m1), and C9orf72 (Hs00376619_m1) (Applied Biosystems) and analyzed on an ABI Prism 7500 system (Life Technologies). Relative quantification was determined by qBASE PLUS software (Biogazelle) with the ddCt method after normalization to HPRT1, UBC, and B2M. Carriers of a hypermethylated allele with >50 repeats (n = 4) have reduced expression compared to noncarriers (n = 3). Notably, the carrier of an unmethylated allele with 43 repeats (ID#8520, II-4 in PED 6, Figure 3) has an expression level similar to noncarriers. These data support the hypothesis that hypermethylation may underlie the reduced C9orf72 expression.
Figure 4.

Relative Quantification of C9orf72 mRNA
All transcripts were detected by an inventoried TaqMan gene expression assay (Hs00376619_m1) and relative expression was obtained by normalizing to three reference genes (HPRT1, UBC, and B2M). Error bars represent the standard error of the triplicate reactions. Expansion carriers were shown to have reduced C9orf72 expression compared nonexpansion carriers, whereas ID #8520 with 43 repeats had a normal C9orf72 mRNA level. Notably, gene expression is inversely related to methylation level. Number of methylated CpG site is shown beneath each sample. “BSP” stands for bisulfite sequencing PCR.
It is intriguing that a high methylation level was significantly associated with familial ALS, whereas half of the expansion carriers (mainly sporadic cases) showed either low or no methylation. The repeat size itself could be an explanation for the wide range of methylation levels among expansion carriers. Indeed, a direct correlation between CpG methylation and repeat size was observed in Friedreich ataxia,31 in which the degree of methylation gradually increased along with the number of GAA repeats (from ∼200 repeats to ∼1,200 repeats32). However, assessment of the C9orf72 repeat number is technically challenging because of the high GC content and possible somatic instability. Repeat-primed PCR can only resolve the precise size for expansions of up to 50 repeats; although in a few published cases, the size of the expansion was estimated by Southern blot (up to 3,400 repeats in blood33 and 700–2,000 repeats in lymphoblasts1,34). Expansions on such a scale are much larger than the current cutoff for pathologic alleles (30 repeats). Hence, the repeat size is likely to be highly variable between expansion carriers, and whether these alleles have the same pathologic significance has to be carefully assessed in the future. Notably, we did not detect methylation 5′ of the repeat for either normal (2–22 repeats) or intermediate (up to 43 repeats) alleles, bringing to question the 30-repeat cutoff for pathological alleles. Future studies have to establish whether methylation level indeed mirrors repeat size, because differences in repeat size and methylation degree could be players in the highly heterogeneous clinical and neuropathological presentations observed in expansion carriers.35–37
Our initial results from seven families indicate that the level of methylation could depend on repeat size. Expansion carriers identified in each kindred are mostly from the same generation (Table 3, Figure 3), and the size of the expansion is likely to be more similar within the same pedigree than between different families. The observed degree of methylation reflected a similar tendency: high variability was observed between families, but a similar degree of methylation was detected among members of the same family with the exception of the asymptomatic individual ID#8520 (II-4 in PED 6, Figure 3) with an unmethylated intermediate allele (43 repeats). In contrast, the rest of the expansion carriers in PED6 carry a highly methylated expanded allele (>50 repeats). Gene expression analysis of C9orf72 further separates this 43-repeat carrier from family members with larger expansions (>50 repeats) by displaying a normal level of C9orf72 expression (Figure 4). So far, this pedigree is the only example that methylation level could reflect repeat size, and it would be important to evaluate more carriers of intermediate alleles. Furthermore, the significant association of hypermethylation with familial ALS could also indicate a correlation with repeat size. In repeat expansion diseases, longer alleles are more likely to undergo an expansion mutation than shorter ones,38 so familial cases could have an increased probability for their repeat to expand further and carry longer alleles than sporadic cases. In Friedreich ataxia and Huntington disease (MIM 143100), repeat length is known to relate to disease severity and age of onset.15,32,38–40 A hallmark of most repeat expansion disorders is genetic anticipation that reflects the tendency for larger expansions in subsequent generations. In C9orf72-linked disease, genetic anticipation was also suggested by a few studies.4,7
In conclusion, we conducted an epigenetic investigation into the functional consequence of the C9orf72 repeat expansion in ALS. Importantly, the hypermethylation was expansion specific in our data set, because it was not detected in any noncarriers. Moreover, a higher degree of methylation correlated with shorter disease duration, indicating that it may be associated with increased ALS severity. However, future replication studies would require the analysis of independent larger data sets to validate our findings. The obtained methylation data may be the basis for a future diagnostic approach to C9orf72 disease. By carefully comparing the CpG methylation profile between expansion carriers, future studies could determine whether disease phenotype is dependent on the degree or sites of methylation. For instance, it is tempting to speculate that the degree of methylation could be an important factor in modifying disease presentation (e.g., ALS versus FTLD).
Acknowledgments
This work was supported by the W. Garfield Weston Foundation and Ontario Research Fund (E.R., J.R.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GENEVESTIGATOR, https://www.genevestigator.com/gv/biomed.jsp
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., ITALSGEN Consortium A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rademakers R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. 2012;11:297–298. doi: 10.1016/S1474-4422(12)70046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gijselinck I., Van Langenhove T., van der Zee J., Sleegers K., Philtjens S., Kleinberger G., Janssens J., Bettens K., Van Cauwenberghe C., Pereson S. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 5.Sabatelli M., Conforti F.L., Zollino M., Mora G., Monsurrò M.R., Volanti P., Marinou K., Salvi F., Corbo M., Giannini F., ITALSGEN Consortium C9ORF72 hexanucleotide repeat expansions in the Italian sporadic ALS population. Neurobiol. Aging. 2012;33:1848. doi: 10.1016/j.neurobiolaging.2012.02.011. e15–e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith B.N., Newhouse S., Shatunov A., Vance C., Topp S., Johnson L., Miller J., Lee Y., Troakes C., Scott K.M. The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2013;21:102–108. doi: 10.1038/ejhg.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xi Z., Zinman L., Grinberg Y., Moreno D., Sato C., Bilbao J.M., Ghani M., Hernández I., Ruiz A., Boada M. Investigation of c9orf72 in 4 neurodegenerative disorders. Arch. Neurol. 2012;69:1583–1590. doi: 10.1001/archneurol.2012.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ash P.E., Bieniek K.F., Gendron T.F., Caulfield T., Lin W.L., Dejesus-Hernandez M., van Blitterswijk M.M., Jansen-West K., Paul J.W., 3rd, Rademakers R. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mori K., Weng S.M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 10.Klose R.J., Bird A.P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 12.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 13.Al-Mahdawi S., Pinto R.M., Ismail O., Varshney D., Lymperi S., Sandi C., Trabzuni D., Pook M. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 2008;17:735–746. doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- 14.Greene E., Mahishi L., Entezam A., Kumari D., Usdin K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 2007;35:3383–3390. doi: 10.1093/nar/gkm271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans-Galea M.V., Carrodus N., Rowley S.M., Corben L.A., Tai G., Saffery R., Galati J.C., Wong N.C., Craig J.M., Lynch D.R. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 2012;71:487–497. doi: 10.1002/ana.22671. [DOI] [PubMed] [Google Scholar]
- 16.Sutcliffe J.S., Nelson D.L., Zhang F., Pieretti M., Caskey C.T., Saxe D., Warren S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 17.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 18.Bell M.V., Hirst M.C., Nakahori Y., MacKinnon R.N., Roche A., Flint T.J., Jacobs P.A., Tommerup N., Tranebjaerg L., Froster-Iskenius U. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell. 1991;64:861–866. doi: 10.1016/0092-8674(91)90514-y. [DOI] [PubMed] [Google Scholar]
- 19.López Castel A., Nakamori M., Tomé S., Chitayat D., Gourdon G., Thornton C.A., Pearson C.E. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet. 2011;20:1–15. doi: 10.1093/hmg/ddq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klesert T.R., Otten A.D., Bird T.D., Tapscott S.J. Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat. Genet. 1997;16:402–406. doi: 10.1038/ng0897-402. [DOI] [PubMed] [Google Scholar]
- 21.Thornton C.A., Wymer J.P., Simmons Z., McClain C., Moxley R.T., 3rd Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet. 1997;16:407–409. doi: 10.1038/ng0897-407. [DOI] [PubMed] [Google Scholar]
- 22.Korade-Mirnics Z., Tarleton J., Servidei S., Casey R.R., Gennarelli M., Pegoraro E., Angelini C., Hoffman E.P. Myotonic dystrophy: tissue-specific effect of somatic CTG expansions on allele-specific DMAHP/SIX5 expression. Hum. Mol. Genet. 1999;8:1017–1023. doi: 10.1093/hmg/8.6.1017. [DOI] [PubMed] [Google Scholar]
- 23.Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J. Neurol. Neurosurg. Psychiatry. 1994;57:416–418. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks B.R., Miller R.G., Swash M., Munsat T.L., World Federation of Neurology Research Group on Motor Neuron Diseases El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 25.Hughes A.J., Daniel S.E., Lees A.J. Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology. 2001;57:1497–1499. doi: 10.1212/wnl.57.8.1497. [DOI] [PubMed] [Google Scholar]
- 26.Allen R.C., Zoghbi H.Y., Moseley A.B., Rosenblatt H.M., Belmont J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 27.Rogaev E.I., Lukiw W.J., Lavrushina O., Rogaeva E.A., St George-Hyslop P.H. The upstream promoter of the beta-amyloid precursor protein gene (APP) shows differential patterns of methylation in human brain. Genomics. 1994;22:340–347. doi: 10.1006/geno.1994.1393. [DOI] [PubMed] [Google Scholar]
- 28.Parrish R.R., Day J.J., Lubin F.D. Direct bisulfite sequencing for examination of DNA methylation with gene and nucleotide resolution from brain tissues. Curr. Protoc. Neurosci. 2012;Chapter 7 doi: 10.1002/0471142301.ns0724s60. Unit 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang M., Zhang Y., Fei J., Chang X., Fan W., Qian X., Zhang T., Lu D. Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab. Invest. 2010;90:282–290. doi: 10.1038/labinvest.2009.132. [DOI] [PubMed] [Google Scholar]
- 30.Nagase H., Ghosh S. Epigenetics: differential DNA methylation in mammalian somatic tissues. FEBS J. 2008;275:1617–1623. doi: 10.1111/j.1742-4658.2008.06330.x. [DOI] [PubMed] [Google Scholar]
- 31.Castaldo I., Pinelli M., Monticelli A., Acquaviva F., Giacchetti M., Filla A., Sacchetti S., Keller S., Avvedimento V.E., Chiariotti L., Cocozza S. DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet. 2008;45:808–812. doi: 10.1136/jmg.2008.058594. [DOI] [PubMed] [Google Scholar]
- 32.Filla A., De Michele G., Cavalcanti F., Pianese L., Monticelli A., Campanella G., Cocozza S. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 1996;59:554–560. [PMC free article] [PubMed] [Google Scholar]
- 33.Takada L.T., Pimentel M.L., Dejesus-Hernandez M., Fong J.C., Yokoyama J.S., Karydas A., Thibodeau M.P., Rutherford N.J., Baker M.C., Lomen-Hoerth C. Frontotemporal dementia in a Brazilian kindred with the c9orf72 mutation. Arch. Neurol. 2012;69:1149–1153. doi: 10.1001/archneurol.2012.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishiura H., Takahashi Y., Mitsui J., Yoshida S., Kihira T., Kokubo Y., Kuzuhara S., Ranum L.P., Tamaoki T., Ichikawa Y. C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii peninsula of Japan. Arch. Neurol. 2012;69:1154–1158. doi: 10.1001/archneurol.2012.1219. [DOI] [PubMed] [Google Scholar]
- 35.Murray M.E., DeJesus-Hernandez M., Rutherford N.J., Baker M., Duara R., Graff-Radford N.R., Wszolek Z.K., Ferman T.J., Josephs K.A., Boylan K.B. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122:673–690. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coon E.A., Daube J.R., Dejesus-Hernandez M., Adeli A., Savica R., Parisi J.E., Dickson D.W., Josephs K.A., Baker M.C., Johnson K.A. Clinical and electrophysiologic variability in amyotrophic lateral sclerosis within a kindred harboring the C9ORF72 repeat expansion. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013;14:132–137. doi: 10.3109/17482968.2012.724075. [DOI] [PubMed] [Google Scholar]
- 37.Stewart H., Rutherford N.J., Briemberg H., Krieger C., Cashman N., Fabros M., Baker M., Fok A., DeJesus-Hernandez M., Eisen A. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 2012;123:409–417. doi: 10.1007/s00401-011-0937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearson C.E., Nichol Edamura K., Cleary J.D. Repeat instability: mechanisms of dynamic mutations. Nat. Rev. Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 39.Lee J.M., Ramos E.M., Lee J.H., Gillis T., Mysore J.S., Hayden M.R., Warby S.C., Morrison P., Nance M., Ross C.A., PREDICT-HD study of the Huntington Study Group (HSG) REGISTRY study of the European Huntington’s Disease Network. HD-MAPS Study Group. COHORT study of the HSG CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78:690–695. doi: 10.1212/WNL.0b013e318249f683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tassone F., Adams J., Berry-Kravis E.M., Cohen S.S., Brusco A., Leehey M.A., Li L., Hagerman R.J., Hagerman P.J. CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS) Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2007;144B:566–569. doi: 10.1002/ajmg.b.30482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.