

Abstract
Infantile myofibromatosis (IM) is a disorder of mesenchymal proliferation characterized by the development of nonmetastasizing tumors in the skin, muscle, bone, and viscera. Occurrence within families across multiple generations is suggestive of an autosomal-dominant (AD) inheritance pattern, but autosomal-recessive (AR) modes of inheritance have also been proposed. We performed whole-exome sequencing (WES) in members of nine unrelated families clinically diagnosed with AD IM to identify the genetic origin of the disorder. In eight of the families, we identified one of two disease-causing mutations, c.1978C>A (p.Pro660Thr) and c.1681C>T (p.Arg561Cys), in PDGFRB. Intriguingly, one family did not have either of these PDGFRB mutations but all affected individuals had a c.4556T>C (p.Leu1519Pro) mutation in NOTCH3. Our studies suggest that mutations in PDGFRB are a cause of IM and highlight NOTCH3 as a candidate gene. Further studies of the crosstalk between PDGFRB and NOTCH pathways may offer new opportunities to identify mutations in other genes that result in IM and is a necessary first step toward understanding the mechanisms of both tumor growth and regression and its targeted treatment.
Main Text
Infantile myofibromatosis (IM [MIM 228550]) is one of the most common proliferative fibrous tumors of infancy and childhood. First described by Williams and Schrum1 and Stout,2 IM was further subcategorized by others into solitary, multiple, or generalized forms and shown to affect the skin, muscle, bone, and viscera.3,4 The term “infantile myofibromatosis” was recommended based on the fact that the cells have features of both differentiated fibroblasts and smooth muscle cells (myofibroblasts).5 Soft tissue lesions usually arise during childhood but can arise at any time during life and, intriguingly, can regress spontaneously. On the other hand, visceral lesions are associated with high morbidity and mortality.6 The mechanism(s) underlying tumor growth and regression are not known. Some have suggested tumor growth to be linked to angiogenic stimulation and regression.7 Indeed, in a single case report, regression of an intracardiac IM was achieved through use of interferon alpha-2b.8
The genetic etiology of IM is unknown and both autosomal-recessive (AR) and autosomal-dominant (AD) patterns of inheritance have been reported. Consanguinity in a number of pedigrees has been interpreted to be in accord with an AR pattern of inheritance.9–11 A large number of pedigrees, wherein affected individuals are identified across generations, are consistent with IM being an AD disease.12–19
After informed consent and Institutional Review Board approval from the Icahn School of Medicine of Mount Sinai and the corresponding institutions were obtained, blood samples were obtained from 32 affected individuals from 9 unrelated families with the diagnosis of IM and, where possible, unaffected family members (Figure 1). Clinical diagnoses were provided by the referring physicians. Genomic DNA was extracted with the Puregene kit according to the manufacturer’s protocol. Cell lines were established from tumor tissue that was removed from affected individuals as part of their medical care and which was considered pathologic waste. One unaffected and 11 affected family members, representing 9 unrelated kindreds, were selected for whole-exome sequencing at the Center for Applied Genomics at The Children’s Hospital of Philadelphia. Genomic DNA was isolated from a blood sample by standard methods and randomly sheared to 200–300 bp in size, followed by end-repair, A-tailing, and paired-end index adaptor ligation. Whole exomes were captured with the Agilent SureSelect Human All Exon V4+UTR kit (Agilent Technologies) according to the manufacturer’s protocol. The libraries were subsequently clustered on the cBOT instrument, multiplexing 4 samples per flow cell lane, and sequenced for 101 cycles with a paired-end mode on the Illumina HiSeq2000 according to the manufacturer’s instructions (Illumina). Base calling and index demultiplexing was performed with the Illumina CASAVA software (v.1.8.2).
Figure 1.
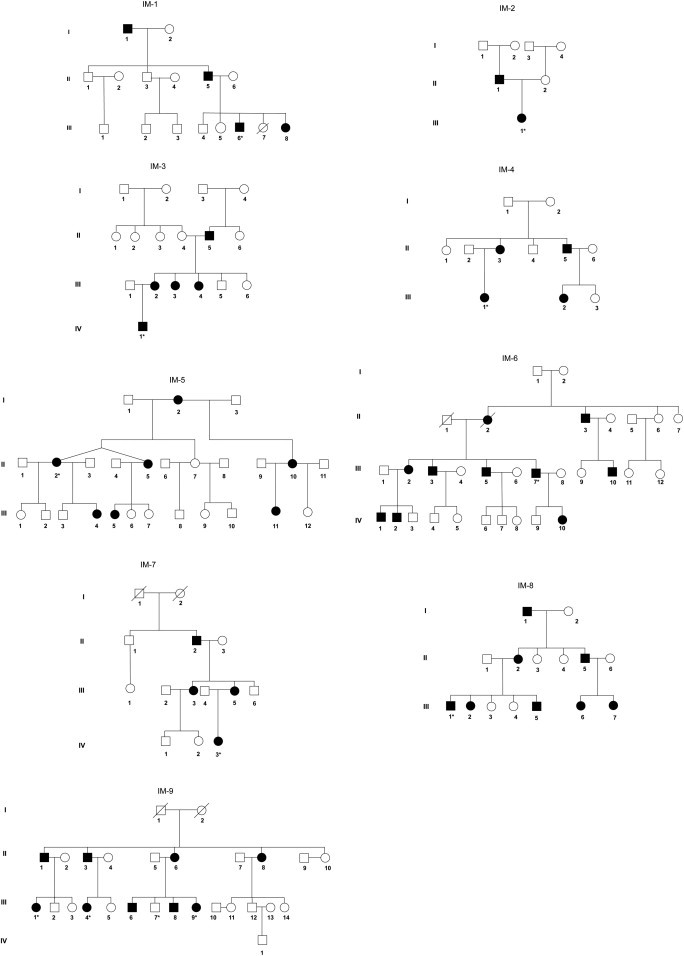
Pedigrees of Nine Unrelated IM Families
The inheritance pattern in all the families used in this study was consistent with autosomal-dominant transmission. Five families have been previously reported: IM-1,14 IM-2,15 IM-6,14 IM-7,16 IM-8.17 Asterisk indicates that these samples were whole-exome sequenced.
Sequencing reads were aligned to the human reference genome (UCSC hg19) with Burrows-Wheeler Aligner (BWA, v.0.6.2).20 Optical and PCR duplicates were marked and removed with Picard (v.1.73). Local realignment of reads containing indel sites and base quality score recalibration (BQSR) were performed with the Genome Analysis Tool Kit (GATK, v.2.3).21 Single-nucleotide variation (SNV) and small indels were called with GATK UnifiedGenotyper. Variants were marked as potential sequencing artifacts if the filters on the following annotations were evaluated to be true: (1) for SNVs, DP < 10, QD < 2.0, MQ < 40.0, FS > 60.0, HaplotypeScore > 13.0, MQRankSum < −12.5, ReadPosRankSum < −8.0; and (2) for small indels, DP < 10, QD < 2.0, ReadPosRankSum < −20.0, InbreedingCoeff < −0.8, FS > 200.0. The kinship coefficient was calculated for each sample via KING22 to confirm reported relationships and identify cryptic relationships among samples. ANNOVAR23 and SnpEff (v.2.0.5)24 were used for annotating variants. Human Gene Mutation Database (HGMD)25 was used for annotating known genes and mutations for human inherited diseases. Prediction scores from SIFT,26 Polyphen2,27 LRT,28 and MutationTaster,29 along with conservation scores PhyloP30 and GERP++,31 for every potential nonsynonymous SNV in the human genome were retrieved from dbNSFP (database for nonsynonymous SNPs’ functional predictions).32 SNVs and indels were selected as potential pathogenic variants if they met all the following criteria: (1) heterozygous; (2) not previously described or rare (minor allele frequency [MAF] < 0.5%) in a control cohort of more than 9,000 control individuals (1000 Genomes Project, April 2012 release), 6,503 exomes from NHLBI GO Exome Sequencing Project (ESP6500SI), and 1,200 in-house whole exomes; (3) nonsynonymous, or splice acceptor and donor site SNVs, or frameshift coding indels (NS/SS/I); (4) predicted to be deleterious by at least three prediction methods, e.g., SIFT, PolyPhen2, MutationTaster, and LRT; and (5) conserved PhyloP score and GERP++ score > 2.0. Variants were also analyzed by the Ingenuity Variant Analysis web-based application.
On average, 9.7 Gb of sequences were produced for each sample, 97% of the reads were mappable to the human reference genome (hg19), and 94% of targeted exome had at least 10× depth of coverage. The mean depth of coverage was 74-fold. A total of 195,651 SNVs and 20,700 indels were identified, of which 178,991 SNVs (91%) and 17,238 indels (83%) were reported in dbSNP135. On average, 82,855 SNVs and 11,882 indels were called per sample. We applied the filtering strategy to focus on a subset of potentially pathogenic variants.33 Variants were filtered by mode of inheritance, variant quality, conservation, predicted deleterious scores, and allele frequency in the public and in-house whole exomes. Two missense variants in PDGFRB (MIM 173410; RefSeq accession number NM_002609.3) were present in eight members in eight families. No PDGFRB mutations were identified in family IM-9 (Table S1 available online). Sanger sequencing of all available family members, affected and unaffected, in the eight families revealed that the two PDGFRB variants segregated appropriately with disease status (Figure 2). In family IM-9, in which no PDGFRB mutations were identified, we exome sequenced two other affected and one unaffected individual from this kindred. Variants in NOTCH3 (MIM 600276; NM_000435.2) and PET112 (MIM 603645; NM_004564.2) were found in all three affected members but not in the unaffected family member (Table 1). Sanger sequencing of 16 family members, consisting of 9 affected and 7 unaffected individuals, revealed that only the NOTCH3 mutation c.4556T>C (p.Leu1519Pro) segregated appropriately with affected status (Figure 2). Given the unexpected finding of candidate disease-causing mutations in a second gene, we re-examined the histologic findings in a soft tissue tumor isolated from this family and also generated a cell line from affected tissue.34 Histopathologic analysis was consistent with the diagnosis of IM, and staining with α-SMA further demonstrated the tumor’s myofibroblastic nature (Figure 3).
Figure 2.
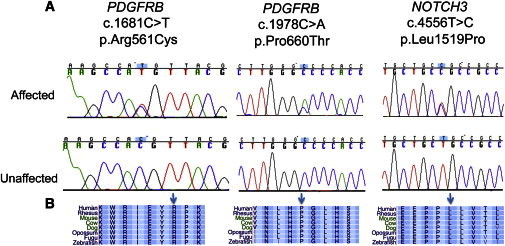
Mutations in PDGFRB and NOTCH3
(A) Representative sequence chromatograms for each of the different mutations identified.
(B) Conservation of the mutations and the surrounding region in vertebrates. Arrows indicate the positions of the mutated alleles.
Table 1.
Rare Variants in PDGFRB and NOTCH3 Identified in Nine IM Families from WES
| Gene (MIM) | Genomic Location (hg19) (RefSeq) | Exon | Family | cDNA | Protein | MAF in 1000 Genomes Project or ESP6500SI |
|---|---|---|---|---|---|---|
| PDGFRB (173410) | chr5: 149,503,858 (NM_002609.3) | 14 | IM-1 | c.1978C>A | Pro660Thr | 0.000077 |
| PDGFRB (173410) | chr5: 149,505,134 (NM_002609.3) | 12 | IM-2–IM-8 | c.1681C>T | Arg561Cys | – |
| NOTCH3 (600276) | chr19: 15,285,059 (NM_000435.2) | 25 | IM-9 | c.4556T>C | Leu1519Pro | – |
Figure 3.
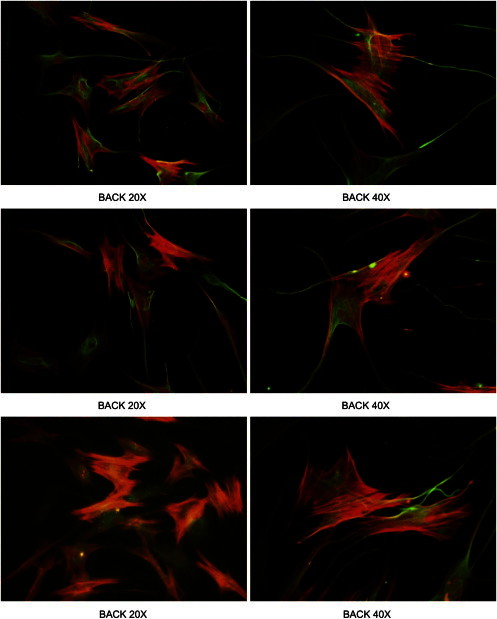
Tumor Cell Lines Derived from Affected Individuals Demonstrate a Myofibroblastic Phenotype
Vimentin (green) and a-SMA (red) staining of tumor cell lines from members of family IM-9. Cells were cultured from a soft-tissue tumor excised from an affected area on the affected individual’s back as part of their care. Three paired views at 20× (left column) and 40× (right column) are shown.
All three rare missense variants in both genes were predicted to be damaging with high probability according to the prediction algorithms LRT, MutationTaster, Polyphen2, and SIFT and they were located in highly conserved exonic regions. In PDGFRB, we identified a heterozygous missense variant in exon 14, c.1978C>A (p.Pro660Thr). It is located in the tyrosine kinase domain of the protein. Interestingly, the variant was present in the ESP6500SI data set with a MAF of 0.000077. It was reported in dbSNP135 (rs144050370) but was not found in the 1000 Genomes Project, in the catalog of somatic mutations in cancer (COSMIC v.63), nor in a database of approximately 1,200 in-house sequenced whole exomes. The second PDGFRB variant is a heterozygous missense variant in exon 12, c.1681C>T (p.Arg561Cys). It is not present in the publically available databases nor in approximately 9,000 public and in-house sequenced whole-exome data sets. For family IM-9, the NOTCH3 variant c.4556T>C (p.Leu1519Pro) predicts a heterozygous missense variant in exon 25. It is a newly described variant, not present in public databases and in-house whole exomes. It is located in the protein’s highly conserved hetero-dimerization domain.
In our current study, two missense mutations in PDGFRB were identified in eight IM families. PDGFRB, located on 5q32, encodes the platelet-derived growth factor receptor-β. It is a cell surface tyrosine kinase receptor for members of the platelet-derived growth factor family (PDGF A, B, C, and D), which are mitogens for cells of mesenchymal origin. Activation of the receptor leads to its dimerization, autophosphorylation of tyrosine residues, and activation of downstream signaling pathways, inducing cellular proliferation, differentiation, survival, and migration. PDGFRB is expressed in neurons, plexus choroideus, vascular smooth muscle cells (VSMCs), and pericytes. PDGFRB signal transduction is required for proliferation and migration of a subset of VSMCs. PDGFRB signaling has been well established in early hematopoiesis and blood vessel formation.35 Enhanced PDGF-PDGFR signaling is a hallmark in a variety of diseases, including cancers, atherosclerosis, pulmonary fibrosis, and restenosis. Recently, a missense mutation, c.1973T>C (p.Leu658Pro) in PDGFRB, was reported to be an identified cause of idiopathic basal ganglia calcification (IBGC [MIM 615007]).36
One novel missense mutation, c.4556T>C (Leu1519Pro), in NOTCH3 was identified as the most probable causative mutation for one IM family. NOTCH3 encodes the third discovered human homolog of the Drosophila melanogaster type I membrane protein notch. Notch signaling allows cells to coordinate fate decisions in metazoan development. Notch signals are highly pleiotropic, dictating cellular fates in a way that depends on cellular context. NOTCH3 is primarily expressed in adult arterial vascular smooth muscle cells (VSMCs) in large conduit, pulmonary, and systemic resistance arteries. Mutations in NOTCH3 have also been identified as the underlying cause of cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL [MIM 125310]).37 The NOTCH3 IM family members are notable for possessing multiple, recurrent soft tissue lesions and have no reported clinical history consistent with a diagnosis of CADASIL. The majority of reported CADASIL-associated mutations affect amino acids that are located in the epidermal growth factor-like (EGF-like) domain in the extracellular domain of the protein (exons 2–24). Recently, Fouillade et al. reported a heterozygous missense mutation (c.4544T>C [p.Leu1515Pro]) in exon 25, a highly conserved hetero-dimerization domain of Notch3, in an affected individual with cerebral small vessel disease but lacking typical deposits and Notch3 accumulation.38 Biochemical analysis suggests that the c.4544T>C (p.Leu1515Pro) mutation renders Notch3 hyperactive through destabilization of the heterodimer. The mutation c.4556T>C (p.Leu1519Pro) identified in an IM family was located close to the Leu1515Pro substitution.
Of particular interest, in trying to understand how mutations in two different genes, PDGFRB and NOTCH3, could result in the same disease, a possible mechanistic link was recently provided. Specifically, Jin et al. demonstrated that PDGFRB was a previously unrecognized and immediate NOTCH3 target gene.39 PDGFRB expression was upregulated by NOTCH3 ligand induction or by activated forms of the NOTCH3 receptor. The availability of established tumor cell lines from affected individuals will allow us to directly explore this mechanistic link. Importantly, if these two signaling pathways are linked and the IM disease-causing mutations in either PDGFRB or NOTCH3 are demonstrated to be activating, theoretically, inhibition of PDGFRB or NOTCH3 would result in a targeted therapeutic strategy.
In conclusion, our study suggests that PDGFRB mutations are a cause of autosomal-dominant IM, a genetically heterogeneous disease with incomplete penetrance and variable expressivity. These studies have also identified a single family with a germline NOTCH3 mutation. Study of the PDGFRB/NOTCH3 pathways will offer new opportunities to identify other IM mutations and/or genes and to understand the mechanisms of both tumor growth and regression in IM.
Acknowledgments
We thank all the families that participated in this study. This work was supported by an Institutional Development Award to the Center for Applied Genomics from The Children’s Hospital of Philadelphia, a donation from Adele and Daniel Kubert (to H.H.), and the Levitt Family Foundation (to J.A.M.).
Contributor Information
John A. Martignetti, Email: john.martignetti@mssm.edu.
Hakon Hakonarson, Email: hakonarson@email.chop.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Ingenuity Variant Analysis, http://www.ingenuity.com/products/variant-analysis
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The dbSNP accession numbers for the PDGFRB and NOTCH3 variants reported in this paper are rs367543286 and rs367543285, respectively.
References
- 1.Williams J.O., Schrum D. Congenital fibrosarcoma; report of a case in a newborn infant. AMA Arch. Pathol. 1951;51:548–552. [PubMed] [Google Scholar]
- 2.Stout A.P. Juvenile fibromatoses. Cancer. 1954;7:953–978. doi: 10.1002/1097-0142(195409)7:5<953::aid-cncr2820070520>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 3.Kauffman S.L., Stout A.P. Congenital mesenchymal tumors. Cancer. 1965;18:460–476. doi: 10.1002/1097-0142(196504)18:4<460::aid-cncr2820180410>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 4.Enzinger F.M. Fibrous hamartoma of infancy. Cancer. 1965;18:241–248. doi: 10.1002/1097-0142(196502)18:2<241::aid-cncr2820180216>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 5.Chung E.B., Enzinger F.M. Infantile myofibromatosis. Cancer. 1981;48:1807–1818. doi: 10.1002/1097-0142(19811015)48:8<1807::aid-cncr2820480818>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 6.Wiswell T.E., Davis J., Cunningham B.E., Solenberger R., Thomas P.J. Infantile myofibromatosis: the most common fibrous tumor of infancy. J. Pediatr. Surg. 1988;23:315–318. doi: 10.1016/s0022-3468(88)80196-9. [DOI] [PubMed] [Google Scholar]
- 7.Leaute-Labreze C., Labarthe M.P., Blanc J.F., Sanyas P., Dosquet C., Taieb A. Self-healing generalized infantile myofibromatosis with elevated urinary bFGF. Pediatr. Dermatol. 2001;18:305–307. doi: 10.1046/j.1525-1470.2001.01933.x. [DOI] [PubMed] [Google Scholar]
- 8.Auriti C., Kieran M.W., Deb G., Devito R., Pasquini L., Danhaive O. Remission of infantile generalized myofibromatosis after interferon alpha therapy. J. Pediatr. Hematol. Oncol. 2008;30:179–181. doi: 10.1097/MPH.0b013e31815e62bb. [DOI] [PubMed] [Google Scholar]
- 9.Baird P.A., Worth A.J. Congenital generalized fibromatosis: an autosomal recessive condition? Clin. Genet. 1976;9:488–494. doi: 10.1111/j.1399-0004.1976.tb01602.x. [DOI] [PubMed] [Google Scholar]
- 10.Salamah M.M., Hammoudi S.M., Sadi A.R. Infantile myofibromatosis. J. Pediatr. Surg. 1988;23:975–977. doi: 10.1016/s0022-3468(88)80398-1. [DOI] [PubMed] [Google Scholar]
- 11.Narchi H. Four half-siblings with infantile myofibromatosis: a case for autosomal-recessive inheritance. Clin. Genet. 2001;59:134–135. doi: 10.1034/j.1399-0004.2001.590213.x. [DOI] [PubMed] [Google Scholar]
- 12.Bartlett R.C., Otis R.D., Laakso A.O. Multiple congenital neoplasms of soft tissues. Report of 4 cases in 1 family. Cancer. 1961;14:913–920. doi: 10.1002/1097-0142(196109/10)14:5<913::aid-cncr2820140503>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 13.Pfluger V.H., Kolb R., Mayr W.R. Kongenitale Polyfibromatose: Klinische und genetische Untersuchungen. Wiener klinishe Wochenshrift. 1976;88:92–94. [PubMed] [Google Scholar]
- 14.Zand D.J., Huff D., Everman D., Russell K., Saitta S., McDonald-McGinn D., Zackai E.H. Autosomal dominant inheritance of infantile myofibromatosis. Am. J. Med. Genet. A. 2004;126A:261–266. doi: 10.1002/ajmg.a.20598. [DOI] [PubMed] [Google Scholar]
- 15.de Montpréville V.T., Zemoura L., Vaksmann G., Lecourt-Tierny G., Planché C., Dulmet E. Endocardial location of familial myofibromatosis revealed by cerebral embolization: cardiac counterpart of the frequent intravascular growth of the disease? Virchows Arch. 2004;444:300–303. doi: 10.1007/s00428-003-0938-4. [DOI] [PubMed] [Google Scholar]
- 16.Jennings T.A., Duray P.H., Collins F.S., Sabetta J., Enzinger F.M. Infantile myofibromatosis. Evidence for an autosomal-dominant disorder. Am. J. Surg. Pathol. 1984;8:529–538. [PubMed] [Google Scholar]
- 17.Ikediobi N.I., Iyengar V., Hwang L., Collins W.E., Metry D.W. Infantile myofibromatosis: support for autosomal dominant inheritance. J. Am. Acad. Dermatol. 2003;49(2, Suppl Case Reports):S148–S150. doi: 10.1067/mjd.2003.333. [DOI] [PubMed] [Google Scholar]
- 18.Smith A., Orchard D. Infantile myofibromatosis: two families supporting autosomal dominant inheritance. Australas. J. Dermatol. 2011;52:214–217. doi: 10.1111/j.1440-0960.2011.00730.x. [DOI] [PubMed] [Google Scholar]
- 19.Kulkarni K., Desai S., Grundy P., Sergi C. Infantile myofibromatosis: report on a family with autosomal dominant inheritance and variable penetrance. J. Pediatr. Surg. 2012;47:2312–2315. doi: 10.1016/j.jpedsurg.2012.09.046. [DOI] [PubMed] [Google Scholar]
- 20.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cingolani P., Platts A., Wang L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stenson P.D., Mort M., Ball E.V., Howells K., Phillips A.D., Thomas N.S., Cooper D.N. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 27.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chun S., Fay J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 30.Siepel, A., Pollard, K., and Haussler, D. (2006). New methods for detecting lineage-specific selection. Proceedings of the 10th International Conference on Research in Computational Molecular Biology (RECOMB 2006). pp. 190–205.
- 31.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput. Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X., Jian X., Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L., Pedroja B.S., Meyers E.E., Garcia A.L., Twining S.S., Bernstein A.M. Degradation of internalized αvβ5 integrin is controlled by uPAR bound uPA: effect on β1 integrin activity and α-SMA stress fiber assembly. PLoS ONE. 2012;7:e33915. doi: 10.1371/journal.pone.0033915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demoulin J.B., Montano-Almendras C.P. Platelet-derived growth factors and their receptors in normal and malignant hematopoiesis. Am. J. Blood. Res. 2012;2:44–56. [PMC free article] [PubMed] [Google Scholar]
- 36.Nicolas G., Pottier C., Maltête D., Coutant S., Rovelet-Lecrux A., Legallic S., Rousseau S., Vaschalde Y., Guyant-Maréchal L., Augustin J. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology. 2013;80:181–187. doi: 10.1212/WNL.0b013e31827ccf34. [DOI] [PubMed] [Google Scholar]
- 37.Joutel A., Corpechot C., Ducros A., Vahedi K., Chabriat H., Mouton P., Alamowitch S., Domenga V., Cécillion M., Marechal E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 38.Fouillade C., Chabriat H., Riant F., Mine M., Arnoud M., Magy L., Bousser M.G., Tournier-Lasserve E., Joutel A. Activating NOTCH3 mutation in a patient with small-vessel-disease of the brain. Hum. Mutat. 2008;29:452. doi: 10.1002/humu.9527. [DOI] [PubMed] [Google Scholar]
- 39.Jin S., Hansson E.M., Tikka S., Lanner F., Sahlgren C., Farnebo F., Baumann M., Kalimo H., Lendahl U. Notch signaling regulates platelet-derived growth factor receptor-beta expression in vascular smooth muscle cells. Circ. Res. 2008;102:1483–1491. doi: 10.1161/CIRCRESAHA.107.167965. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.