

Abstract
The most common form of spinal muscular atrophy (SMA) is a recessive disorder caused by deleterious SMN1 mutations in 5q13, whereas the genetic etiologies of non-5q SMA are very heterogeneous and largely remain to be elucidated. In a Bulgarian family affected by autosomal-dominant proximal SMA, we performed genome-wide linkage analysis and whole-exome sequencing and found a heterozygous de novo c.320C>T (p.Ser107Leu) mutation in bicaudal D homolog 2 (Drosophila) (BICD2). Further analysis of BICD2 in a cohort of 119 individuals with non-5q SMA identified a second de novo BICD2 mutation, c.2321A>G (p.Glu774Gly), in a simplex case. Detailed clinical and electrophysiological investigations revealed that both families are affected by a very similar disease course, characterized by early childhood onset, predominant involvement of lower extremities, and very slow disease progression. The amino acid substitutions are located in two interaction domains of BICD2, an adaptor protein linking the dynein molecular motor with its cargo. Our immunoprecipitation and localization experiments in HeLa and SH-SY5Y cells and affected individuals’ lymphoblasts demonstrated that p.Ser107Leu causes increased dynein binding and thus leads to accumulation of BICD2 at the microtubule-organizing complex and Golgi fragmentation. In addition, the altered protein had a reduced colocalization with RAB6A, a regulator of vesicle trafficking between the Golgi and the endoplasmic reticulum. The interaction between p.Glu744Gly altered BICD2 and RAB6A was impaired, which also led to their reduced colocalization. Our study identifies BICD2 mutations as a cause of non-5q linked SMA and highlights the importance of dynein-mediated motility in motor neuron function in humans.
Main Text
Inherited spinal muscular atrophies (SMAs) form a diverse group of disorders characterized by muscle weakness and atrophy caused by the degeneration of anterior horn cells.1,2 Although the most common form of SMA is an autosomal-recessive condition associated with loss-of-function mutations in SMN1 (MIM 600354) in chromosomal region 5q13,3 rare families with dominant inheritance have been reported.4–8 The non-5q SMAs are genetically and clinically heterogeneous disorders classified by their pattern of inheritance and distribution of weakness (distal or proximal).8 The forms with predominantly distal weakness have a clinical overlap with the distal hereditary motor neuropathies,3,9–15 whereas the proximal forms are caused by mutations in VAPB (MIM 605704),5 LMNA (MIM 150330),16 and TRPV4 (MIM 605427)6 or are linked to unresolved loci.4,7,17 Furthermore, several clinical entities remain without a molecular genetic diagnosis.8
We studied a four-generation Bulgarian family (with Turkish ethnic background) afflicted with autosomal-dominant proximal SMA (Figure 1A). Disease status in the affected individuals was determined by clinical examination and electrophysiological evaluation. All clinical findings are summarized in Table 1.
Figure 1.
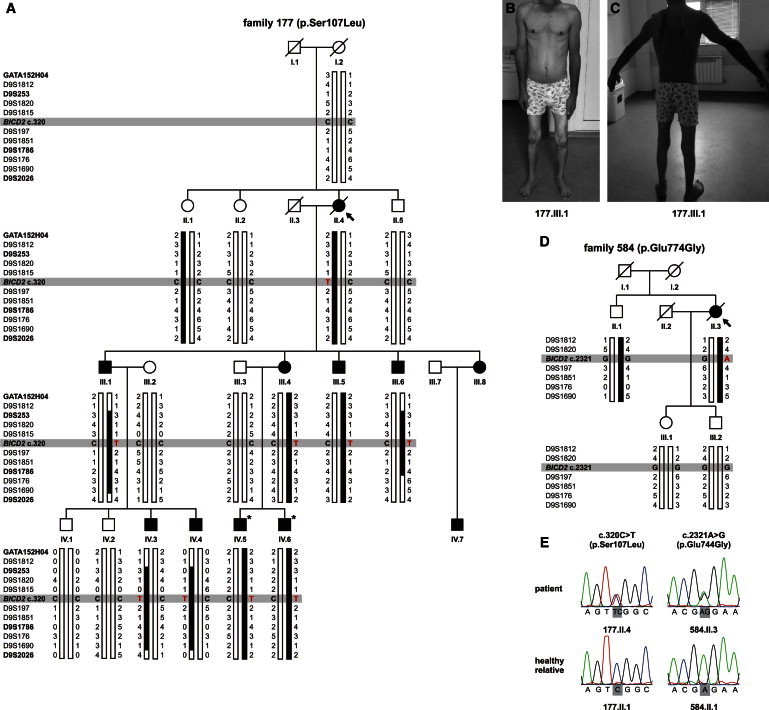
Pedigrees, Clinical Features, and Electropherograms of Individuals with BICD2 Mutations
(A and D) Pedigree structure and haplotype analysis of families 177 (A) and 584 (D). The probands are denoted by an arrow. Squares and circles symbolize males and females, respectively, and symbols with diagonal black lines represent deceased individuals. Filled symbols denote family members affected by SMA, and open symbols indicate unaffected individuals. Individuals with an asterisk were selected for exome sequencing. The disease-segregating haplotype on chromosome 9q is indicated in black. Short-tandem-repeat (STR) markers are shown in order from p-ter (top) to q-ter (bottom). Markers of the genome-wide linkage panel are represented in bold.
(B and C) The clinical features of person 177.III.1 show hypotrophy of proximal and distal muscles of the lower limbs (B) and scapular winging (C).
(E) The electropherograms of heterozygous BICD2 mutations c.320C>T (p.Ser107Leu) and c.2321A>G (p.Glu774Gly).
Table 1.
Clinical and Electrophysiological Features of the Individuals with BICD2 Mutations
|
Affected Individual |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 177.II.4 | 177.III.1 | 177.III.4 | 177.III.5 | 177.III.6 | 177.III.8 | 177.IV.3 | 177.IV.4 | 177.IV.5 | 177.IV.6 | 177.IV.7 | 584.II.3 | |
| Age of onset (years) | 2 | 2 | 5 | 6 | 4 | 4 | 1 | 1 | 5 | 2 | 4 | 2 |
| Age at assessment (years) | 48 | 44 | 30 | 24 | 24 | 29 | 6 | 5 | 6 | 3 | 5 | 42 |
| Waddling gait | + | + | + | + | + | + | + | + | + | + | + | + |
| CPK level | ND | 1,110 | 178 | ND | ND | ND | 120 | 51 | 123 | ND | ND | ND |
| Fasciculations | − | + | − | − | − | − | − | − | − | − | − | + |
| Muscle Weakness (MRC Scale) | ||||||||||||
| Proximal muscles in LLs | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | + | 4 | 3 |
| Distal muscles in LLs | 5 | 4 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | − | 5 | 4 |
| Axial muscles | 4 | 4 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | − | 5 | 3 |
| Reflexes | ||||||||||||
| Patellar | − | +/− | +/− | + | +/− | +/− | +/− | +/− | +/− | ND | +/− | − |
| Achilles | − | − | − | +/− | +/− | +/− | +/− | +/− | +/− | ND | +/− | − |
| Deformities and Posture | ||||||||||||
| Scapular winging | + | + | − | + | + | + | + | + | + | + | + | + |
| Lumbal hyperlordosis | − | + | − | + | + | − | + | + | + | + | + | + |
| Hypotrophies | ||||||||||||
| Proximal muscles in LLs | + | + | − | + | + | + | + | − | + | − | − | + |
| Distal muscles in LLs | − | + | − | + | + | − | + | − | − | − | − | + |
| Nerve Conduction Studies and EMG | ||||||||||||
| MCV of the nervous peroneus | ND | 43.5 | ND | ND | ND | ND | 54.8 | 58.3 | 55.1 | ND | ND | 45.0 |
| Anterior horn involvement | ND | + | + | ND | ND | ND | + | + | + | ND | ND | + |
Normal CPK range: 20–200. The following abbreviations are used: −, absent; +, present; +/−, moderately reduced; MRC, Medical Research Council; LL, lower limb; CPK, creatine phosphokinase; EMG, electromyography; MCV, motor conduction velocity; and ND, not determined.
The onset age varied between 1 and 6 years (mean 3.17 ± 1.70 years), and individuals presented with delayed motor milestones, such as walking (in persons 177.III.1, 177.IV.3, and 177.IV.4), difficulties in getting upright from a squatting position (Gowers’ sign) and in climbing stairs, a waddling gait, and slow running. The leg weakness did not progress significantly over time, given that affected individuals still remained ambulatory even into the fifth decade.
Upon neurologic examination, muscle weakness was limited to the lower extremities and showed predominant involvement in the proximal leg muscles (Figures 1B and 1C). Distal muscle weakness in the legs (score 4/5 on the Medical Research Council [MRC] scale for muscle strength) was found in one of the oldest family members (177.III.1). Axial muscle involvement, with difficulties in getting up from a lying position, was present upon disease progression. Bulbar muscles were spared in all affected individuals. Other muscles, including those of the face and upper extremities, showed normal strength. Patellar tendon reflexes were depressed in nine persons, whereas Achilles hyporeflexia or areflexia was found in all of them. Reflexes in the upper limbs were preserved. Fasciculations were observed in the proximal muscles of the upper limbs in person 177.III.1. Skeletal deformities (lumbal hyperlordosis and scapular winging) were present in most affected individuals. Symmetric wasting was most prominent in the hip muscles, whereas distal leg muscles seemed atrophic in the older family members. Sensation was normal for all modalities. Serum creatine phosphokinase (CPK) levels were mostly normal.
We performed electromyography (EMG) studies while maintaining skin temperature over the first dorsal interosseous muscle at >30.5°C on a Toennies NeuroScreen System (Jaeger GnbH). Sensory and motor nerve conduction studies showed normal conduction velocities and amplitudes for the upper and lower extremities (Figures S1A and S1B, available online). EMG showed predominantly large-amplitude and long-duration motor unit potentials in the muscles of the lower and upper extremities, in keeping with anterior horn cell damage (Figures S1C and S1D).
All study participants or their legal representatives provided written informed consent prior to enrollment. Local institutional review boards approved this study. Genomic DNA was isolated from peripheral blood according to standard procedures.
We performed genome-wide multipoint parametric linkage analysis on family 177 with Allegro v.1.2c in the easyLinkage software18 by using an in-house-developed panel of 436 polymorphic short-tandem-repeat (STR) markers under an autosomal-dominant model, equal male and female recombination rates and allele frequencies, and a 0.0001 disease frequency. Because the disease was presumed to have originated de novo in the proband, the disease status of her unaffected siblings and parents was put as “unknown.” The initial analysis showed only one suggestive linkage peak with a LOD ≥ 2.00 on chromosome 9q (LOD = 2.71) (Figure S2A). After fine mapping with additional STR markers, the disease-segregating locus could be delineated to a 12.7 Mb region containing 132 distinct genes between D9S1812 and D9S176 (Figure 1A and Figure S2B).
The TargetSeq Exome Enrichment System was used for library preparation, capture, and enrichment of about 45.1 Mb of coding regions from two affected brothers (177.IV.5 and 177.IV.6). Fragment reads (75 + 35 bp) were sequenced on a SOLiD 5500xl instrument. Mapping to the human reference genome (GRCh37) and variant calling were performed with the CLC Genomics Workbench. Subsequent annotation and filtering were executed with GenomeComb.19
We extracted heterozygous, novel variants (defined as a frequency ≤ 1% in public databases such as dbSNP137 and 1000 Genomes Project and our in-house collection of genomes) that were shared between both affected siblings, were located within the linkage interval, and had an impact on the protein level (nonsynonymous variations and splice-site changes). The analysis revealed that the linkage region contained 50 shared variations, of which only one nonsynonymous variant was novel: c.320C>T (p.Ser107Leu) in bicaudal D homolog 2 (Drosophila) (BICD2 [MIM 609797; RefSeq accession number NM_015250.3]). This missense mutation cosegregated with disease in the family, as shown by bidirectional sequencing with the BigDye Terminator v.3.1 Cycle Sequencing kit on an ABI3730xl DNA Analyzer (Applied Biosystems), and was, as presumed, a de novo event in the disease founder, 177.II.4 (Figures 1A and 1E). It was absent in a cohort of 289 ancestry-matched controls (51 Bulgarian Turks, 51 Bulgarians, and 187 Turks). Interestingly, the c.320C>T BICD2 mutation is positioned within a CpG dinucleotide. Given that an estimated 80% of CpG cytosines are methylated in human cells,20 the c.320 position could as such be a potential mutational “hotspot,” prone to C>T transitions through spontaneous deamination of 5-methylcytosine.21,22
In order to obtain further genetic evidence of BICD2-dependent pathogenicity, we performed an unbiased screen of all coding exons and exon-intron boundaries of BICD2 (primers are available upon request) in a cohort of 119 unrelated Bulgarian simplex cases who presented with clinical and EMG features of progressive anterior horn involvement and who had been excluded for SMN1 deletions. In 113 (95%) individuals, the clinical picture was dominated by proximal muscle weakness, whereas in 6 (5%) persons, distal muscles were mostly affected. The subjects with proximal SMA were further classified as having type 1 (27.4%), type 2 (14.1%), type 3 (55.8%), or type 4 (2.7%). In this cohort, we identified a second heterozygous missense mutation in BICD2, c.2321A>G (p.Glu774Gly), in a Bulgarian simplex case diagnosed with proximal SMA (Figures 1D and 1E). Haplotype analysis suggested that this mutation also originated de novo in the proband (584.II.3) (Figure 1D). The variant was absent in 304 control individuals (108 Bulgarians, 49 Bulgarian Turks, and 147 Turks). In silico prediction of the functional effect of both mutations with PolyPhen-223 and MutationTaster24 showed that they target highly conserved nucleotide and amino acid residues and are therefore very likely to be deleterious.
The clinical course of person 584.II.3, who harbors the BICD2 c.2321A>G (p.Glu774Gly) mutation, was extremely similar to that of individuals with the c.320C>T (p.Ser107Leu) mutation. This person was initially diagnosed with the Kugelberg-Welander type of SMA (MIM 253400). She presented with delayed motor milestones, a waddling gait, Gowers’ sign, and proximal upper-limb fasciculations. Neurological examination showed predominantly proximal muscle weakness (3/5 on the MRC scale) and some distal muscle involvement, comparable to that of an age-matched affected individual in family 177 (177.III.1). EMG analysis showed anterior horn involvement.
BICD2, an evolutionary conserved motor-adaptor protein, is implicated in dynein-mediated transport as a molecular linker between the dynein motor and various cargos.25–28 According to tissue expression databases (GeneCards), mammalian BICD2 is widely present. We confirmed this finding via immunoblot analysis of BICD2 levels in a range of mouse tissues (Figure S3). In addition, we could demonstrate a high presence of BICD2 in the spinal cord, the afflicted tissue in SMA.
BICD2 consists of three highly conserved coiled-coil regions, of which the N-terminal domain binds to the dynein motor,25 whereas the C-terminal coiled-coil domain interacts with various cargos, such as RAB6A (Figure 2A).26 The BICD2 N terminus on its own has very strong dynein-recruiting activity and can chronically impair dynein-dynactin function, thus disturbing retrograde trafficking and causing cellular abnormalities such as Golgi fragmentation and neurofilament swellings.25,27,29 Full-length BICD2 does not display such perturbing activities, presumably because the C terminus regulates motor-complex binding and inhibits the N terminus in the native state until after cargo loading.25
Figure 2.
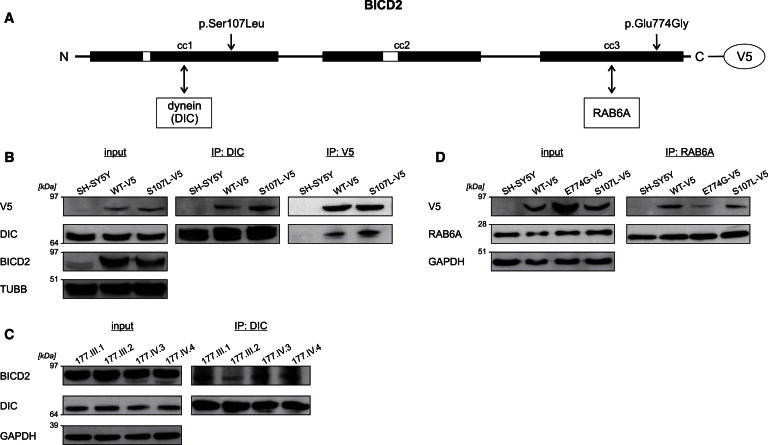
Coimmunoprecipitation of BICD2 and Its Interactors Dynein and RAB6A
(A) Schematic representation of the structure of BICD2. The Ser107 residue is positioned in the N-terminal region that interacts with dynein, whereas the Glu774 amino acid lies within the C-terminal domain that interacts with RAB6A.
(B) Lysates of SH-SY5Y cells stably expressing BICD2-V5 wild-type and altered proteins were used for immunoprecipitation (IP) with antibodies against dynein intermediate chain 1 (DIC) and V5. The p.Ser107Leu altered BICD2-V5 coprecipitated more with DIC than did the wild-type protein in reciprocal experiments. TUBB was used for showing equal loading of the lysates.
(C) Lysates of lymphoblasts from members of family 177 were immunoprecipitated with DIC antibody and show an increased BICD2 load in the samples of the three affected individuals compared to the healthy relative (177.III.2). GAPDH was used for showing equal loading of the lysates.
(D) Immunoprecipitation (IP) of stable SH-SY5Y cell extracts with RAB6A antibody demonstrates that there is less interaction between RAB6A and p.Glu774Gly altered BICD2-V5 than between RAB6A and the wild-type. The amount of coprecipitated p.Ser107Leu BICD2-V5 was comparable to that of the wild-type. GAPDH was used for showing equal loading of the lysates.
Interestingly, the targeted residues are positioned in two different interaction domains of BICD2. Whereas the p.Ser107Leu substitution lies within the N-terminal dynein-binding region, the p.Glu774Gly alteration is located in the C-terminal cargo-loading domain (Figure 2A). To study the functional effect of these amino acid substitutions on BICD2 binding properties, we performed coimmunoprecipitation (coIP) and colocalization studies in cellular models. To this end, BICD2 expression constructs in pLenti6/V5-DEST or pLenti6/EGFP-DEST were generated by Gateway cloning with full-length human BICD2 cDNA from the IMAGE Human cDNA clone 6169195 (Thermo Fischer Scientific). Mutations were introduced into the human transcript by site-directed mutagenesis, and stable SH-SY5Y cell lines were created by lentiviral transduction as described.30
For the immunoprecipitation experiments, human neuroblastoma (SH-SY5Y) cells stably expressing wild-type or altered BICD2 proteins were grown in modified Eagle’s medium (Invitrogen) under blasticidin selection. Cell lysates were incubated with Anti-V5 Agarose (Sigma-Aldrich) or Protein G Sepharose 4 Fast Flow beads (GE Healthcare) with monoclonal mouse dynein intermediate chain 1 (DIC) antibody (Abcam) or polyclonal rabbit RAB6 antibody (Cell Signaling) according to standard procedures.
We detected enhanced coprecipitation of the p.Ser107Leu altered protein, compared to wild-type BICD2, by endogenous DIC pulldown. In a reciprocal experiment pulling down BICD2-V5, higher amounts of DIC coprecipitated with p.Ser107Leu BICD2 than with the wild-type protein (Figure 2B). To explore whether this altered N-terminal BICD2 interaction is relevant for the pathophysiology in humans, we further analyzed lymphoblast cultures derived from affected individuals with the same p.Ser107Leu substitution. Consistent with our findings in the BICD2 cellular model, coIP of BICD2 with DIC showed their enhanced interaction in protein extracts of three affected persons compared to a healthy family member (Figure 2C).
The p.Glu774Gly alteration, on the other hand, is localized in the BICD2 C-terminal domain, known to directly interact with RAB6A, a regulator of Golgi-ER trafficking.26 Pulldown of endogenous RAB6A protein coimmunoprecipitated less p.Glu774Gly protein than wild-type BICD2 (Figure 2D). The interaction efficiency between p.Ser107Leu altered BICD2 and RAB6A was comparable to that between the wild-type protein and RAB6A (Figure 2D).
To investigate whether these alterations in binding capacity affect the subcellular localization of BICD2, we performed immunocytochemistry experiments in neuronal (SH-SY5Y) and nonneuronal (HeLa) cell lines. HeLa cells were first transiently transfected with EGFP-tagged wild-type or altered BICD2 constructs with the use of Lipofectamine LTX reagent (Life Technologies). Immunostaining was performed with various antibodies (see below), and cells were imaged with a LSM700 confocal microscope (Zeiss) with identical acquisition parameters.
In wild-type-expressing HeLa cells, BICD2-EGFP signal largely overlapped with the RAB6A signal (polyclonal rabbit RAB6A antibody, Cell Signaling), as reported before.26 In line with its reduced coIP, the p.Glu774Gly altered BICD2, compared to the wild-type protein, showed a decreased colocalization with RAB6A, as illustrated by line intensity plots and quantification of fluorescence correlation (Figure 3). Intriguingly, the p.Ser107Leu altered BICD2 also showed significantly reduced colocalization with RAB6A-positive regions, albeit less pronounced than the other altered protein. In addition, only cells expressing p.Ser107Leu showed an intense punctual signal in the perinuclear region (Figure 3, see arrow). Further analysis demonstrated that this signal colocalized with the microtubule-organizing center (MTOC, as visualized with polyclonal rabbit γ-tubulin, Abcam), to which dynein motor transport is directed (Figure 4). Interestingly, previous studies with a truncated BICD2-N protein showed enhanced targeting of this BICD2 fragment toward the MTOC.27 This targeting was associated with Golgi fragmentation both in vitro and in vivo.25,29 To test whether p.Ser107Leu also caused this peculiar phenotype, we stained our stable transgenic SH-SY5Y lines with the Golgi marker Giantin (polyclonal rabbit Giantin antibody, Covance). Indeed, cells expressing p.Ser107Leu BICD2 presented with a disrupted Golgi morphology. This phenotype was not present in the wild-type or p.Glu774Gly-expressing cells (Figure 5), despite comparable levels of both altered proteins (data not shown).
Figure 3.
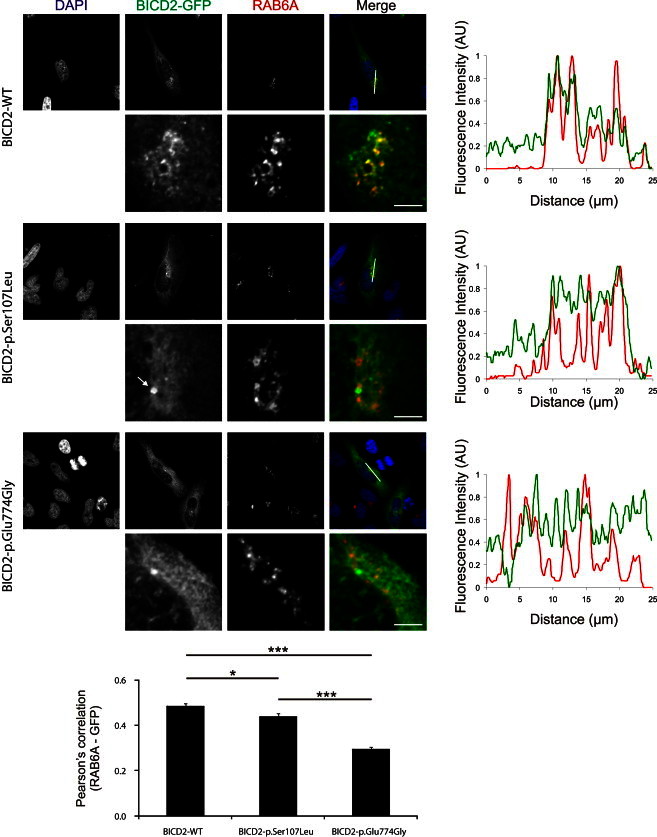
Colocalization of BICD2 and RAB6A Is Reduced by the SMA-Causing BICD2 Alterations
HeLa cells expressing BICD2-EGFP (green) were stained for RAB6A (red). p.Glu774Gly altered BICD2-EGFP is decreased in RAB6A-positive regions. Also, the p.Ser107Leu altered protein shows reduced colocalization with RAB6A, but not to the same extent as p.Glu774Gly. In addition, the p.Ser107Leu altered BICD2 shows an intense punctual staining in the perinuclear region (arrow). Graphs and error bars represent the mean and SEM. The Pearson’s correlation coefficient (calculated with the ImageJ Coloc2 plugin) is determined in 62, 62, and 61 cells expressing wild-type, p.Ser107Leu, and p.Glu774Gly proteins, respectively, from three independent transfection and immunostaining experiments. ∗p < 0.05, ∗∗∗p < 0.001. The scale bar represents 5 μm.
Figure 4.
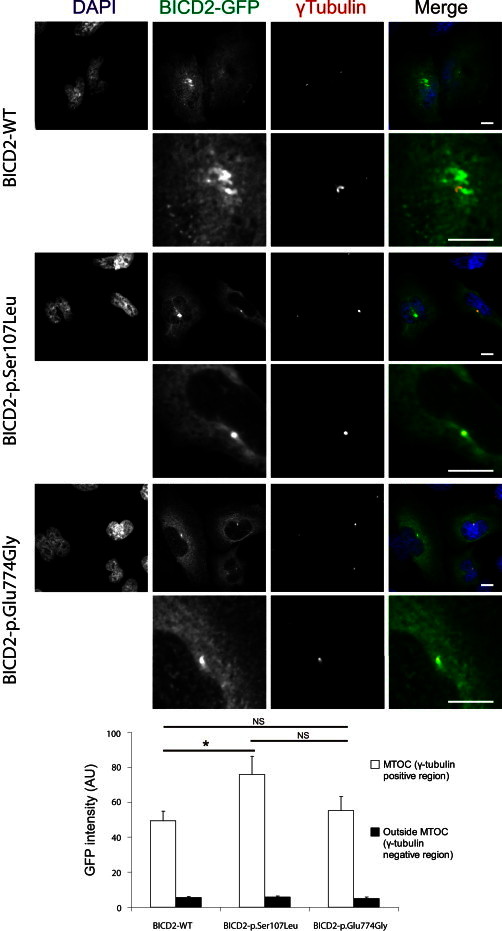
The p.Ser107Leu Substitution in the Dynein Binding Domain Induces BICD2 Accumulation at the MTOC
Compared to wild-type and p.Glu774Gly altered BICD2-EGFP, p.Ser107Leu altered BICD2-EGFP (green) shows increased localization at the MTOC (visualized with γ-tubulin, red). The graph and error bars represent the mean and SEM of mean EGFP intensity at the MTOC (white bars) and in the rest of the cell (black bars) measured in 34, 37, and 17 HeLa cells expressing wild-type, p.Ser107Leu, and p.Glu774Gly proteins, respectively. ∗p < 0.05. The scale bar represents 10 μm. The following abbreviations is used: NS, not significant.
Figure 5.
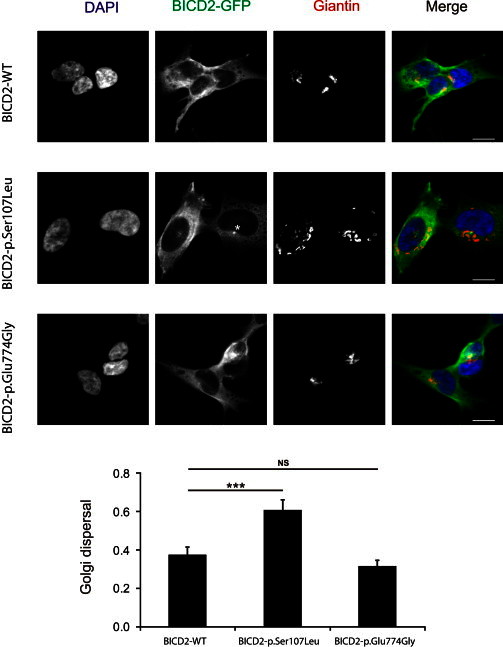
p.Ser107Leu Altered BICD2 Leads to Golgi Fragmentation
SH-SY5Y cells stably expressing BICD2-EGFP (green) were immunostained with the Golgi marker Giantin (red). Compared to wild-type and p.Glu774Gly mutant cells, cells with p.Ser107Leu altered BICD2 show a dispersed Golgi apparatus. In addition, in p.Ser107Leu mutant cells, a perinuclear accumulation of BICD2 is apparent (asterisk). Golgi dispersal is represented as a ratio between the minimal rectangle area around the Giantin-positive structures (segmented in ImageJ by automatic intensity thresholding) and the total area of the cell. The graph and error bars represent the mean and SEM of Golgi dispersal measured in 131, 103, and 104 SH-SY5Y cells expressing wild-type, p.Ser107Leu, and p.Glu774Gly proteins, respectively. ∗∗∗p < 0.001. The scale bar represents 5 μm. The following abbreviation is used: NS, not significant.
Our study links genetic defects in BICD2 with proximal SMA. We identified two disease-causing alterations that are located in different interaction domains and that have differential effects on BICD2 binding properties. Nevertheless, the clinical presentation of SMA is very similar in both families. The disease, predominantly involving the lower extremities, was always recognized in early childhood as a result of delayed motor milestones or proximal leg weakness. Although scapular winging was a typical feature in our affected individuals, strength of the upper extremities was preserved. Upon disease progression, mild involvement of distal leg and axial muscles was found. However, the clinical histories we obtained suggest static or very slowly progressive weakness, given that independent ambulation was preserved even in the fifth decade. Nerve conduction and EMG studies were consistent with anterior horn loss and normal motor conduction velocities.
Unlike in other previously described families affected by dominant SMA characterized by early onset and lower-extremity predominance, artrogryposis and severe contractures were not present in the affected individuals in our study.31–33 Their phenotype seems mild and is more similar to the 14q32-linked SMA,7 which only affects the lower limbs. Although upper-limb weakness is frequently found in autosomal-dominant SMA,16 in our affected individuals, scapular winging and fasciculations were the only symptoms of upper-limb involvement.
The BICD2-associated clinical phenotype overlaps with that of the most common autosomal-recessive 5q-linked SMA. Although disease onset was early in the affected individuals in our study, their phenotype is milder in terms of degree of progression, preservation of strength in the upper limbs, and lack of respiratory involvement.1,2 Therefore, our findings suggest that screening of BICD2 should be considered in individuals diagnosed with sporadic proximal SMA, for which SMN1 mutations were excluded. In our cohort, the frequency of the BICD2 mutation approximated 1.7%.
Our functional studies provide evidence that both the p.Ser107Leu and p.Glu774Gly BICD2 amino acid substitutions affect the binding properties and subcellular localization of this motor adaptor. Both alterations consistently impaired the colocalization of BICD2 with RAB6A, a known regulator of Golgi-ER trafficking. The reduced colocalization of the p.Glu774Gly altered BICD2 is in accordance with its compromised binding to RAB6A. Although the binding capacity of the p.Ser107Leu altered protein was preserved, it also showed decreased colocalization with RAB6A-positive structures. This, however, might be explained by the stronger dynein binding and targeting to the MTOC. By its sequestration, BICD2 will be withdrawn from its cellular destination, thus impairing RAB6A targeting.
Interestingly, RAB6A might be implicated in the trafficking of SMN, the causal protein for the most common form of SMA. The dynamic transport of SMN-containing granules in the neurites of motor-neuron-like cells was recently demonstrated to be mediated by the COPI vesicle-protein complex.34,35 Depletion of COPI results in reduced SMN levels in the neurites and causes growth-cone defects, mimicking SMN deficiency in neuronal cells.34 Excitingly, the trafficking of COPI-coated vesicles is mediated by the BICD2 binding partner RAB6A.36 Taken together, these findings provide a mechanistic link among BICD2, RAB6A, and motor neuron survival.
Cells expressing p.Ser107Leu showed Golgi fragmentation. Because this phenotype was not present in p.Glu774Gly-expressing cells and this hallmark was not associated with neuronal degeneration in a mouse model expressing a truncated BICD2 N terminus,29 the significance of this finding is currently undefined.
Our combined genetic and functional findings demonstrate that BICD2 function is essential for motor neuron physiology. This study enlarges the genetic heterogeneity and clinical spectrum of hereditary SMA and has important implications for future clinical and molecular diagnostics of this disease.
Acknowledgments
We are grateful to the study participants and their families for their cooperation. We thank the VIB Genetic Service Facility for sequencing and cell-maintenance support, the VIB Department of Molecular Genetics Centralized Service Facility for exome sequencing, P. De Rijk for bioinformatics support, S. Bichev for technical assistance, and C. Van Broeckhoven for helpful discussions. This work was supported in part by the University of Antwerp (IWS BOF 2008 23064 to A.J.), the Fund for Scientific Research-Flanders (to A.J. and V.T.), the Medical Foundation Queen Elisabeth (to V.T.), the Association Belge Contre les Maladies Neuromusculaires (to A.J. and V.T.), the Research Fund of Medical University-Sofia (to A.J.), and the FP7 EU Neuromics network (to V.T.). K.P., T.G., and M.Z. are supported by a PhD fellowship from the Fund for Scientific Research-Flanders. E.Y. receives a PhD fellowship from the Institute for Science and Technology-Flanders.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
CLC Genomics Workbench, http://www.clcbio.com/products/clc-genomics-workbench
GenomeComb, http://genomecomb.sourceforge.net
GeneCards, http://www.genecards.org
MutationTaster, http://www.mutationtaster.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2
References
- 1.Dubowitz V. Ramblings in the history of spinal muscular atrophy. Neuromuscul. Disord. 2009;19:69–73. doi: 10.1016/j.nmd.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 2.Kolb S.J., Kissel J.T. Spinal muscular atrophy: a timely review. Arch. Neurol. 2011;68:979–984. doi: 10.1001/archneurol.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viollet L., Barois A., Rebeiz J.G., Rifai Z., Burlet P., Zarhrate M., Vial E., Dessainte M., Estournet B., Kleinknecht B. Mapping of autosomal recessive chronic distal spinal muscular atrophy to chromosome 11q13. Ann. Neurol. 2002;51:585–592. doi: 10.1002/ana.10182. [DOI] [PubMed] [Google Scholar]
- 4.Takashima H., Nakagawa M., Suehara M., Saito M., Saito A., Kanzato N., Matsuzaki T., Hirata K., Terwilliger J.D., Osame M. Gene for hereditary motor and sensory neuropathy (proximal dominant form) mapped to 3q13.1. Neuromuscul. Disord. 1999;9:368–371. doi: 10.1016/s0960-8966(99)00021-8. [DOI] [PubMed] [Google Scholar]
- 5.Nishimura A.L., Mitne-Neto M., Silva H.C., Richieri-Costa A., Middleton S., Cascio D., Kok F., Oliveira J.R., Gillingwater T., Webb J. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004;75:822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng H.X., Chen W., Hong S.T., Boycott K.M., Gorrie G.H., Siddique N., Yang Y., Fecto F., Shi Y., Zhai H. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harms M.B., Allred P., Gardner R.J., Jr., Fernandes Filho J.A., Florence J., Pestronk A., Al-Lozi M., Baloh R.H. Dominant spinal muscular atrophy with lower extremity predominance: linkage to 14q32. Neurology. 2010;75:539–546. doi: 10.1212/WNL.0b013e3181ec800c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darras B.T. Non-5q spinal muscular atrophies: the alphanumeric soup thickens. Neurology. 2011;77:312–314. doi: 10.1212/WNL.0b013e3182267bd8. [DOI] [PubMed] [Google Scholar]
- 9.Christodoulou K., Zamba E., Tsingis M., Mubaidin A., Horani K., Abu-Sheik S., El-Khateeb M., Kyriacou K., Kyriakides T., Al-Qudah A.K., Middleton L. A novel form of distal hereditary motor neuronopathy maps to chromosome 9p21.1-p12. Ann. Neurol. 2000;48:877–884. [PubMed] [Google Scholar]
- 10.Grohmann K., Varon R., Stolz P., Schuelke M., Janetzki C., Bertini E., Bushby K., Muntoni F., Ouvrier R., Van Maldergem L. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Ann. Neurol. 2003;54:719–724. doi: 10.1002/ana.10755. [DOI] [PubMed] [Google Scholar]
- 11.McEntagart M., Norton N., Williams H., Teare M.D., Dunstan M., Baker P., Houlden H., Reilly M., Wood N., Harper P.S. Localization of the gene for distal hereditary motor neuronopathy VII (dHMN-VII) to chromosome 2q14. Am. J. Hum. Genet. 2001;68:1270–1276. doi: 10.1086/320122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puls I., Jonnakuty C., LaMonte B.H., Holzbaur E.L., Tokito M., Mann E., Floeter M.K., Bidus K., Drayna D., Oh S.J. Mutant dynactin in motor neuron disease. Nat. Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 13.Evgrafov O.V., Mersiyanova I., Irobi J., Van Den Bosch L., Dierick I., Leung C.L., Schagina O., Verpoorten N., Van Impe K., Fedotov V. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- 14.Gopinath S., Blair I.P., Kennerson M.L., Durnall J.C., Nicholson G.A. A novel locus for distal motor neuron degeneration maps to chromosome 7q34-q36. Hum. Genet. 2007;121:559–564. doi: 10.1007/s00439-007-0348-9. [DOI] [PubMed] [Google Scholar]
- 15.Maystadt I., Rezsöhazy R., Barkats M., Duque S., Vannuffel P., Remacle S., Lambert B., Najimi M., Sokal E., Munnich A. The nuclear factor kappaB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset. Am. J. Hum. Genet. 2007;81:67–76. doi: 10.1086/518900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudnik-Schöneborn S., Botzenhart E., Eggermann T., Senderek J., Schoser B.G., Schröder R., Wehnert M., Wirth B., Zerres K. Mutations of the LMNA gene can mimic autosomal dominant proximal spinal muscular atrophy. Neurogenetics. 2007;8:137–142. doi: 10.1007/s10048-006-0070-0. [DOI] [PubMed] [Google Scholar]
- 17.Penttilä S., Jokela M., Hackman P., Maija Saukkonen A., Toivanen J., Udd B. Autosomal dominant late-onset spinal motor neuronopathy is linked to a new locus on chromosome 22q11.2-q13.2. Eur. J. Hum. Genet. 2012;20:1193–1196. doi: 10.1038/ejhg.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindner T.H., Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–407. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 19.Reumers J., De Rijk P., Zhao H., Liekens A., Smeets D., Cleary J., Van Loo P., Van Den Bossche M., Catthoor K., Sabbe B. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 2012;30:61–68. doi: 10.1038/nbt.2053. [DOI] [PubMed] [Google Scholar]
- 20.Ehrlich M., Gama-Sosa M.A., Huang L.H., Midgett R.M., Kuo K.C., McCune R.A., Gehrke C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–2721. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper D.N., Youssoufian H. The CpG dinucleotide and human genetic disease. Hum. Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 22.Coulondre C., Miller J.H., Farabaugh P.J., Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- 23.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 25.Hoogenraad C.C., Akhmanova A., Howell S.A., Dortland B.R., De Zeeuw C.I., Willemsen R., Visser P., Grosveld F., Galjart N. Mammalian Golgi-associated Bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J. 2001;20:4041–4054. doi: 10.1093/emboj/20.15.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matanis T., Akhmanova A., Wulf P., Del Nery E., Weide T., Stepanova T., Galjart N., Grosveld F., Goud B., De Zeeuw C.I. Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat. Cell Biol. 2002;4:986–992. doi: 10.1038/ncb891. [DOI] [PubMed] [Google Scholar]
- 27.Hoogenraad C.C., Wulf P., Schiefermeier N., Stepanova T., Galjart N., Small J.V., Grosveld F., de Zeeuw C.I., Akhmanova A. Bicaudal D induces selective dynein-mediated microtubule minus end-directed transport. EMBO J. 2003;22:6004–6015. doi: 10.1093/emboj/cdg592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Splinter D., Tanenbaum M.E., Lindqvist A., Jaarsma D., Flotho A., Yu K.L., Grigoriev I., Engelsma D., Haasdijk E.D., Keijzer N. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol. 2010;8:e1000350. doi: 10.1371/journal.pbio.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teuling E., van Dis V., Wulf P.S., Haasdijk E.D., Akhmanova A., Hoogenraad C.C., Jaarsma D. A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum. Mol. Genet. 2008;17:2849–2862. doi: 10.1093/hmg/ddn182. [DOI] [PubMed] [Google Scholar]
- 30.Salmon P., Trono D. Production and titration of lentiviral vectors. Curr. Protoc. Neurosci. 2006;37:4.21.1–4.21.24. doi: 10.1002/0471142301.ns0421s37. [DOI] [PubMed] [Google Scholar]
- 31.Frijns C.J., Van Deutekom J., Frants R.R., Jennekens F.G. Dominant congenital benign spinal muscular atrophy. Muscle Nerve. 1994;17:192–197. doi: 10.1002/mus.880170210. [DOI] [PubMed] [Google Scholar]
- 32.Mercuri E., Messina S., Kinali M., Cini C., Longman C., Battini R., Cioni G., Muntoni F. Congenital form of spinal muscular atrophy predominantly affecting the lower limbs: a clinical and muscle MRI study. Neuromuscul. Disord. 2004;14:125–129. doi: 10.1016/j.nmd.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Reddel S., Ouvrier R.A., Nicholson G., Dierick I., Irobi J., Timmerman V., Ryan M.M. Autosomal dominant congenital spinal muscular atrophy—a possible developmental deficiency of motor neurones? Neuromuscul. Disord. 2008;18:530–535. doi: 10.1016/j.nmd.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 34.Peter C.J., Evans M., Thayanithy V., Taniguchi-Ishigaki N., Bach I., Kolpak A., Bassell G.J., Rossoll W., Lorson C.L., Bao Z.Z., Androphy E.J. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum. Mol. Genet. 2011;20:1701–1711. doi: 10.1093/hmg/ddr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ting C.H., Wen H.L., Liu H.C., Hsieh-Li H.M., Li H., Lin-Chao S. The spinal muscular atrophy disease protein SMN is linked to the Golgi network. PLoS ONE. 2012;7:e51826. doi: 10.1371/journal.pone.0051826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Storrie B., Micaroni M., Morgan G.P., Jones N., Kamykowski J.A., Wilkins N., Pan T.H., Marsh B.J. Electron tomography reveals Rab6 is essential to the trafficking of trans-Golgi clathrin and COPI-coated vesicles and the maintenance of Golgi cisternal number. Traffic. 2012;13:727–744. doi: 10.1111/j.1600-0854.2012.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.