

Abstract
The first catalytic, enantioselective carbosulfenylation of alkenes with an aromatic nucleophile is described, using a BINAM-based selenophosphoramide catalyst. E-Alkyl- and aryl-substituted alkenes afforded tetrahydronaphthalenes with complete diastereospecificity, and generally high enantiomeric ratios.
The reaction of sulfur(II) electrophiles with alkenes1 to afford sulfenofunctionalized products has been thoroughly studied since the 1960’s primarily in the context of investigating the chemistry of thiiranium ions.2 These reactive intermediates can be generated by a variety of different sulfenylating reagents (Scheme 1). In addition to the wide range of nucleophiles that have been employed in intermolecular sulfenofunctionalization reactions, the intramolecular capture (sulfenocyclization reactions) can be effected with sulfenylating agents to form carbocycles and heterocycles. Moreover, sulfenium initiated cyclizations have also been successful with polyolefins5 and electron rich arenes.6 Several examples of nucleophilic opening of chiral thiiranium ions are reported. However, these species are generated by anchimerically-assisted ionization of enantiomerically enriched hydroxy sulfides formed from asymmetric dihydroxylation of alkenes.7 These enantiomerically enriched thiiranium ions have been captured with various nucleophiles in inter- and intramolecular fashion.8 However, only two reports of direct, enantioselective methyl-sulfenylation reactions have been reported, both employing stoichiometric reagents.9
Scheme 1.

As part of a broadly based program to apply the concept of Lewis base activation of Lewis acids10 to the reactions of main group elements, we have investigated the chemistry of selenium (II)11 and sulfur (II)12 electrophiles. From extensive preparative and mechanistic studies, we have discovered that the functionalization of isolated alkenes with N-phenylsulfenylphthalimide, 1, is susceptible to catalysis by Lewis bases and the intermediate thiiranium ions are stable12a and can be captured enantiospecifically with a variety of heteroatom nucleophiles. 12b These insights led to the development of the first, catalytic, enantioselective sulfenoetherification of unactivated double bonds using 1, MsOH and a chiral selenophosphoramide as the Lewis base.12c In continuation of these studies we sought to expand the scope of the asymmetric sulfenofunctionalization13–15 to include carbocyclizations with aromatic nucleophiles. Although such transformations are known, no examples of catalytic, enantioselective cyclizations are on record.
In view of the conditions reported for the sulfenoetherification, the initial conditions for the sulfenocarbocyclization of alkene 2a employed 1 (1.0 equiv), MsOH (1.0 equiv), and Lewis base (10 mol %) in CDCl3 at −20 °C. A number of chiral phosphoramides were tested to develop an asymmetric variant of the intramolecular sulfenocarbocyclization. The initial survey of Lewis bases was carried out with E/Z mixtures of the starting material 2a. The trans/cis ratio of the diastereomeric products 3a was determined by 1H NMR spectroscopic analysis of the purified material and the enantiomeric composition of the products was determined by chiral stationary phase, supercritical fluid chromatographic (CSP-SFC) analysis (Table 1). The exclusive formation of trans- and cis-1,2-disubstituted-1,2,3,4-tetrahydronaphthalenes from E- and Z-olefins respectively is in accordance with the anticipated diastereospecificity characteristic of other seleno-11 and thiofunctionalizations. 12 Chiral Lewis base (R)-4a afforded cyclized products 3a with moderate enantioselectivity for the major product, trans-3a (Table 1, entry 1). However, the minor, cis-3a was produced in racemic form.
Table 1.
Survey of Chiral Lewis Bases to Sulfenocarbocyclization of 2a.a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | E/Zb | acid (equiv) | cat | mol, % |
X | trans/cisc | er,d trans |
er,d cis |
| 1e | 62:38 | MeSO3H (1.0) | (R)-4a | 10 | 62:38 | 32:68 | 51:49 | |
| 2e | 96:4 | MeSO3H (1.0) | (R)-4b | 10 |  |
96:4 | 11:89 | 49:51 |
| 3e | 94:6 | MeSO3H (1.0) | (S)-4c | 10 | 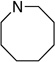 |
94:6 | 86:14 | 59:41 |
| 4e | 94:6 | MeSO3H (1.0) | (S)-4d | 10 | N(i-Pr)2 | 94:6 | 83:17 | 61:39 |
| 5e | 96:4 | MeSO3H (1.0) | (S)-4e | 10 | N(i-Bu)2 | 96:4 | 95:5 | 32:68 |
| 6e | 95:5 | MeSO3H (1.0) | (S)-4f | 10 | N(i-amyl)2 | 95:5 | 88:12 | 64:36 |
| 7e | 62:38 | MeSO3H (1.0) | (R)-4g | 10 | N(Et)2 | 62:38 | 15:85 | 47:53 |
| 8e | 95:5 | MeSO3H (1.0) | (S)-4h | 10 | N(n-Bu)2 | 95:5 | 92:8 | 66:34 |
| 9f | 100:0 | EtSO3H (0.75) | (S)-4e | 10 | N(i-Bu)2 | 100:0 | 96:4 | - |
| 10f | 100:0 | EtSO3H (0.75) | (S)-4e | 5 | N(i-Bu)2 | 100:0 | 71:29 | - |
| 11f | 100:0 | EtSO3H (0.75) | (S)-4e | 2 | N(i-Bu)2 | 100:0 | 67:33 | - |
Reaction conditions: 2a (0.12 mmol), 1 (0.12 mmol), RSO3H (0.12 mmol), CDCl3 (0.2 M), −20 °C.
E/Z ratio of the starting material was determined by 1H NMR spectroscopic analysis.
the E/Z ratio and trans/cis ratio of the cyclization products was determined by 1H NMR spectroscopic analysis.
the enantiomeric ratio was determined by CSP-SFC analysis.
Reactions were completed in 24 h.
Reactions were completed in 48 h.
Increasing the size of the ring to the azepane catalyst (R)-4b led to higher enantioselectivity for trans-3a but gave cis-3a in racemic form (entry 2). A further increase in the size of the ring with azocane catalyst (R)-4c achieved slightly reduced enantioselectivity in the formation of trans-3a (entry 3). cis-3a was still obtained with moderate enantioselectivity. Use of acyclic, branched and secondary external amines with diisopropylamino substituted selenophosphoramide (R)-4d afforded reasonable enantioselectivity for trans-3a (entry 4). Evaluation of other catalysts prepared from branched secondary amines allowed the identification of the diisobutyl-derived catalyst (S)-4e, which afforded excellent enantioselectivity for trans-3a (entry 5). Further extension to the diisoamylamine derived catalyst (S)-4f afforded results similar to the azepane catalyst (R)-4b for trans-3a and a slight improvement for cis-3a (entry 6). The use selenophosphoramide catalyst (R)-4g and (S)-4h derived from linear dialkylamines afforded excellent enantioselectivities for trans-3a (entry 7 and 8) but with lower enantioselection than (S)-4e bearing a diisobutylamino group.
With a highly enantioselective Lewis base catalyst in hand, the exploration of the diversity of the substituent patterns on the alkene as well as the nucleophilicity of the aromatic residue needed for effective cyclization were investigated. Surprisingly, however, upon scaling the reactions to 1.0 mmol, the enantioselectivities decreased. Consequently, the critical reaction parameters had to be revaluated, in particular the acid source and loading.16 After extensive optimization it was found that EtSO3H (0.75 equiv) served effectively and reproducibly as the Brønsted acid for cyclization. Using 10 mol % of catalyst (S)-4e, EtSO3H (0.75 equiv) substrate (E)-2a substrate afforded excellent enantioselection of trans-3a (96:4) with high conversion (Table 1, entry 9). Lowering the loading of (S)-4e to 5 and 2 mol % afforded enantiomeric ratios of 71:29 and 67:33 respectively (entries 10 and 11).
At this point, the exploration of the generality of this transformation was investigated. Gratifyingly, application of the optimized conditions to a large variety of E-alkyl- and aryl-substituted alkenes afforded the cyclized products in moderate to excellent yields (50–92%) with good to excellent enantioselectivities and as single (trans) diastereomer (Table 2). For example, under these conditions E-methyl-substituted alkene (E)-2a formed trans-3a in excellent yield and enantioselectivity to furnish cyclized products trans-3b - trans-3d in moderate yields but very good enantiomeric ratios (entries 3–5). Unactivated E-alkenes bearing linear, β-branched, and alicyclic substituents afforded excellent levels of enantioselectivity and reacted with similar rates. The length of the alkyl chain on the alkene did not influence the rate or enantioselectivity of the reaction. However, the presence of a chlorine atom on the alkyl chain ((E)-2c) decreased the rate of the reaction while maintaining high level of enantioselectivity. Cyclopropyl substrate (E)-2e bearing branching at the allylic position led to a good yield and enantioselectivity (entry 6) whereas common cycloalkyls (5- and 6-membered) gave lower yields and enantioselectivities (entries 7–8). If the alkene contained a tert-butyl substituent, no cyclization occurred. Cyclization of substrates 2h and (E)-2i containing geminally-disubstituted alkenes allowed formation of quaternary carbon centers in good yields. Trisubstituted alkenes were more reactive than disubstituted E-alkenes, but the enantioselectivities were lower (entries 9 and 10). Interestingly, byproducts resulting from proton initiated cyclization reaction were formed in 2–8% yield with trisubstituted alkenes.18
Table 2.
Cyclization with Alkyl-substituted Alkenes.17
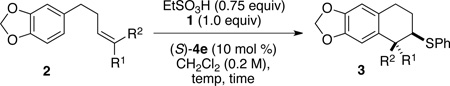 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | substrate | R1 | R2 | product | time, d |
temp, °C |
era,b | yield, %c |
| 1 | (E)-2a | CH3 | H | trans-3a | 3 | −20 | 97:3 | 92 |
| 2 | (Z)-2a | H | CH3 | cis-3a | 1 | 0 | 52:48 | 91 |
| 3 | (E)-2b | n-C5H11 | H | trans-3b | 3 | −20 | 96:4 | 73 |
| 4 | (E)-2c | (CH2)3Cl | H | trans-3c | 6 | 20 | 94:6 | 63 |
| 5 | (E)-2d | i-Bu | H | trans-3d | 3 | −20 | 96:4 | 77 |
| 6 | (E)-2e | cyclopropyl | H | trans-3e | 3 | −20 | 95:5 | 88 |
| 7 | (E)-2f | cyclopentyl | H | trans-3f | 3 | 0 | 82:18 | 50d |
| 8 | (E)-2g | cyclohexyl | H | trans-3g | 3 | 0 | 85:15 | 70e |
| 9 | 2h | CH3 | CH3 | 3h | 1 | −20 | 80:20 | 90f |
| 10 | (E)-2i | C6H5 | CH3 | trans-3i | 2 | −20 | 71:29 | 82g |
| 11 | (E)-2j | C6H5 | H | trans-3j | 2 | −20 | 94:6 | 90 |
| 11 | (E)-2k | 4-CH3C6H4 | H | trans-3k | 2 | −20 | 94:6 | 86 |
| 13 | (E)-2l | 4-CH3OC6H4 | H | trans-3l | 1 | −20 | 92:8 | 86 |
| 14 | (E)-2m | 2-CH3C6H4 | H | trans-3m | 3.5 | −20 | 92:8 | 82h |
| 15 | (E)-2n | 2-naphthyl | H | trans-3n | 2 | −20 | 89:11 | 61i |
| 16 | (E)-2o | 4-NC-C6H4 | H | trans-3o | 6 | 20 | 89:11 | 83 |
| 17 | (E)-2p | 4-CF3C6H4 | H | trans-3p | 3 | 20 | 92:8 | 85 |
The enantiomeric ratio was determined by CSP-SFC analysis.
The absolute configurations of the products were assigned by comparison of their CD spectra with trans-3b.
Yields of analytically pure products.
Unreacted starting material was recovered (15%).
Unreacted starting material was recovered (16%).
An 8% yield of protiocyclization product was isolated.
A 2% yield of protiocyclization product was was isolated.
Unreacted starting material was recovered (4%).
Unreacted starting material was recovered (36%).
The next stage of the investigation focused on the scope of (E)-aryl-substituted alkenes that could participate in the cyclization (Table 2). Substrates (E)-2j, (E)-2k, and (E)-2l bearing no substituents or electrondonating substituents afforded cyclization products in good yields and enantioselectivities (entries 11–13). As was observed with trisubstituted alkenes, increasing the electronic density of the double bond led to faster cyclization. However, substrate (E)-2m bearing a methyl group in the ortho position reacted slowly, but with good enantioselectivity (entry 14). Substrate (E)-2n bearing a 2-naphthyl substituent afforded good selectivity but in lower yield (entry 15). Finally, alkenes (E)-2o and (E)-2p bearing electron-deficient aromatic groups furnished nearly identical enantioselectivities in very slow reactions (entries 16 and 17). The very slow cyclization (even at room temperature) of E-styrenes bearing electron– withdrawing substituents could be ascribed to the electron-deficient nature of the alkene, which would slow the formation of the thiiranium species.
The final exploration of substrate scope was carried out with less monosubstituted aryl groups. Substrates bearing a methoxy group in the 3-position with respect to the tethered alkene afforded a mixture of cyclized products 6–9, with both n-pentyl ((E)-5b) and phenyl ((E)-5j) substituents (Scheme 2). With (E)-5b, a 3:1 ratio of 6/7 was produced in good yields and enantioselectivities (entry 1). However, substrate (E)-5j bearing a phenyl substituent afforded a 1:1 ratio of 8/9, but good enantioselectivity was still achieved for both constitutional isomers.
Scheme 2.

In conclusion, the first catalytic, asymmetric carbosulfenylation of olefins has been accomplished by using a cocatalytic system of a Brønsted acid (EtSO3H) and a chiral Lewis base (S)-4e. This method enables efficient access to enantioenriched trans-tetrahydronaphthalenes with complete diastereospecificity, generally high enantiomeric ratios and a broad substrate scope with Eolefins. Formation of quaternary centers was achieved in moderate enantioselectivities. Further investigations
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the National Institutes of Health for generous financial support (R01 GM85235).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Full experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.de la Mare PBD, Bolton R. In: Electrophilic Additions to Unsaturated Systems. 2nd Ed. de la Mare PBD, Bolton R, editors. Vol. 9. Amsterdam: Elsevier; 1982. pp. 198–246. [Google Scholar]
- 2.(a) Schmid GH. Supplement A: The chemistry of double bonded functional groups. 1989;Vol. Volume 2(Part 1):679–731. [Google Scholar]; (b) Schmid GH, Garratt DG. The chemistry of double-bonded functional groups Part 2. London: John Wiley & Sons; 1977. pp. 725–912. [Google Scholar]; (c) Lucchini V, Modena G, Pasquato L. Gazz. Chim. Ital. 1997;127(4):177–188. [Google Scholar]; (d) Rayner CM. In: Organosulfur Chemistry: Synthetic Aspects. Page P, editor. London: Academic Press; 1995. pp. 89–131. [Google Scholar]; (e) Modena G, Pasquato L, Lucchini V. Phosphorus, Sulfur Silicon Relat. Elem. 1994;95-6(1–4):265–282. [Google Scholar]; (f) Capozzi G, Modena G, Pasquato L. In: The Chemistry of Sulphenic Acids and Their Derivatives. Patai S, editor. West Sussex: John Wiley & Sons Ltd; 1990. pp. 403–516. [Google Scholar]; (g) Harring SR, Edstrom ED, Livinghouse T. Adv. Heterocyclic Nat. Prod. Synth. 1992;2:299–376. [Google Scholar]; (h) Capozzi G, Modena G. In: Organic Sulfur Chemistry: Theoretical and Experimental Advances. Bernardi F, Csizmadia IG, Mangini A, editors. Vol. 19. Amsterdam: Elsevier; 1985. pp. 246–298. [Google Scholar]
- 3.(a) Drabowicz J, Lyzwa P, Mikolajczyk M. In: The Chemistry of Sulphenic Acids and Their Derivatives. Patai S, editor. West Sussex: John Wiley & Sons Ltd; 1990. pp. 187–220. [Google Scholar]; (b) Drabowicz J, Kielbasinski P, Mikolajczyk M. In: The Chemistry of Sulphenic Acids and Their Derivatives. Patai S, editor. West Sussex: John Wiley & Sons Ltd; 1990. pp. 221–292. [Google Scholar]
- 4.Hogg DR. In: The Chemistry of Sulphenic Acids and Their Derivatives. Patai S, editor. West Sussex: John Wiley & Sons Ltd; 1990. pp. 361–402. [Google Scholar]
- 5.(a) Moore JT, Soldi C, Fettinger JC, Shaw JT. Chem. Sci. 2013:292–296. doi: 10.1039/C2SC21405A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Edstrom ED, Livinghouse T. J. Org. Chem. 1987;52:949–951. [Google Scholar]; (c) Masaki Y, Hashimoto K, Sakuma K, Kaji K. Tetrahedron Lett. 1982:1481–1484. [Google Scholar]
- 6.Edstrom ED, Livinghouse T. J. Am. Chem. Soc. 1986;108:1344–1346. [Google Scholar]
- 7.(a) T. Toshimitsu A, Hirosawa C, Tamao K. Synlett. 1996:465–467. [Google Scholar]; (b) Toshimitsu A, Abe H, Hirosawa C, Tamao K. J. Chem. Soc. Perkin Trans. 1. 1994:3465–3471. [Google Scholar]; (c) Toshimitsu A, Hirosawa C, Tamao K. Tetrahedron. 1994;50:8997–9008. [Google Scholar]; (d) Toshimitsu A, Hirosawa C, Tanimoto S. Chem. Lett. 1992:239–242. [Google Scholar]; (e) Toshimitsu A, Abe H, Hirosawa C, Tanimoto S. J. Chem. Soc. Chem. Commun. 1992:284–285. [Google Scholar]; (f) Toshimitsu A, Hirosawa C, Tanimoto S. Tetrahedron Lett. 1991;32:4317–4320. [Google Scholar]
- 8.Branchaud BP, Blanchette HS. Tetrahedron Lett. 2002;43:351–353. [Google Scholar]
- 9.(a) Archer NJ, Rayner CM, Bell D, Miller D. Synlett. 1994:617–619. [Google Scholar]; (b) Pasquato L, Modena G. Chem. Commun. 1999:1469–1470. [Google Scholar]
- 10.Denmark SE, Beutner GL. Angew. Chem. Int. Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 11.(a) Denmark SE, Kalyani D, Collins WR. J. Am. Chem. Soc. 2010;132:15752–15765. doi: 10.1021/ja106837b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Denmark SE, Collins WR. Org. Lett. 2007;9:3801–3804. doi: 10.1021/ol701617d. [DOI] [PubMed] [Google Scholar]
- 12.(a) Denmark SE, Collins WR, Cullen MD. J. Am. Chem. Soc. 2009;131:3490–3491. doi: 10.1021/ja900187y. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Vogler T. Chem. Eur. J. 2009;15:11737–11745. doi: 10.1002/chem.200901377. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Kornfilt DJP, Vogler T. J. Am. Chem. Soc. 2011;133:15308–15311. doi: 10.1021/ja2064395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enantioselective sulfenylation of activated alkenes derived from aldehydes, ketones and amides has also been reported recently. Aldehydes: Zhao GL, Rios R, Vesely J, Eriksson L, Córdova A. Angew. Chem. Int. Ed. 2008;47:8468–8472. doi: 10.1002/anie.200802335. Marigo M, Wabnitz TC, Fielenbach D, Jorgensen KA. Angew. Chem. Int. Ed. 2005;44:794–797. doi: 10.1002/anie.200462101. Ketones: Lin A, Fang L, Zhu X, Zhu C, Cheng Y. Adv. Synth. Catal. 2011;353:545–549. Polaske NW, Dukey R, Nichol GS, Olenyuk B. Tetrahedron: Asymmetry. 2009;20:2742–2750. doi: 10.1016/j.tetasy.2009.10.037. Fang L, Lin A, Hu H, Zhu C. Chem. Eur. J. 2009;15:7039–7043. doi: 10.1002/chem.200901099. Sobhani S, Fielenbach D, Marigo M, Wabnitz TC, Jorgensen KJ. Chem. Eur. J. 2005;11:5689–5694. doi: 10.1002/chem.200500512. Indolones: Han Z, Chen W, Dong S, Yang C, Liu H, Pan Y, Yan L, Jiang Z. Org. Lett. 2012;14:4670–4673. doi: 10.1021/ol3021176. Li X, Liu C, Xue X-S, Cheng J-P. Org. Lett. 2012;14:4374–4377. doi: 10.1021/ol301833f. Wang C, Yang X, Loh CCJ, Raabe G, Enders D. Chem. Eur. J. 2012;18:11531–11535. doi: 10.1002/chem.201201262. Cai Y, Li J, Chen W, Xie M, Liu X, Lin L, Feng X. Org. Lett. 2012;14:2726–2929. doi: 10.1021/ol3009446.
- 14.For a report of catalytic, enantioselective oxysulfenylation of unactivated alkenes under chiral Brønsted acid catalysis see: Guan H, Wang H, Huang D, Shi Y. Tetrahedron. 2012;68:2728–2735.
- 15.For catalytic desymmetrization of meso thiiranium ions see: Hamilton GL, Kanai T, Toste FD. J. Am. Chem. Soc. 2008;110:14984–14986. doi: 10.1021/ja806431d. Lin S, Jacobsen EN. Nature: Chemistry. 2012;4:817–824. doi: 10.1038/nchem.1450.
- 16.A complete disquisition of these studies and the mechanistic implications of the role of the Brønsted acid will be disclosed in a full account of this work.
- 17.The relative configuration of tetrahydronaphthalenes was confirmed by correlation of the 1H NMR chemical shifts of HC(3) with known products trans-2a and cis-2a.8,18 The absolute configuration of (−)-trans-3k was determined to be (1R,2R) by X-ray crystallographic analysis of the derived sulfone trans-12. The crystallographic coordinates of trans-12 have been deposited with the Cambridge Crystallographic Data Centre deposition no. 913987. These data can be obtained free of charge via from the Cambridge Crystallographic Data Centre Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033 via www.ccdc.cam.ac.uk/conts/retrieving.html or deposit@ccdc.cam.ac.uk).
- 18.See the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.